

Abstract
Despite a variety of novel therapeutic agents, such as bortezomib, thalidomide and topotecan, multiple myeloma (MM) remains an incurable disease, thus the development of new chemotherapeutical agents is of high priority. We found HM910, a novel camptothecin (CPT) derivative, exhibited potent inhibition of MM cell growth in vitro and in xenografts of nude mice. Mechanistically, HM910 reduced the mitochondrial transmembrane potential (ΔΨm) via an increase in reactive oxygen species (ROS), which eventually resulting in the release of cytochrome c and the activation of mitochondrial-dependent apoptotic pathway. On the other hand, HM910 significantly triggered cell cycle arrest in G1 phase via downregulating the expressions of cyclin dependent kinase (CDK) 4 and 6, resulting in down-regulation of cyclin D1. Therefore, HM910 maybe a promising candidate for treating MM patients and is currently in phase I clinical trial in China.
Keywords: Multiple myeloma, HM910, camptothecin derivative, cell cycle, apoptosis
Introduction
Multiple myeloma (MM) is a hematologic malignancy, characterized by the accumulation of vicious monoclonal plasma cells in the bone marrow (BM) [1]. The presence of monoclonal immunoglobulin (Ig) in the urine or serum gives rise to osteolytic bone lesions, renal disease, and immunodeficiency. Generally, MM occurs in older patients with a median age of 65–70 years at initial diagnosis, and onset is frequently preceded by a premalignant disease, monoclonal gammopathy of undetermined significance (MGUS) [2,3]. Both genetic and environmental factors play important roles in the progression from MGUS to malignant MM [4]. Emerging evidence confirms that progression to MM is closely associated with changes in the bone marrow (BM) microenvironment, including increased angiogenesis and suppressed immune response [5]. In the treatment of MM, conventional chemotherapy shows limited efficacy in disease-free survival (DFS) and overall survival (OS). Novel therapeutic agents, such as proteasome inhibitors (bortezomib) and immunomodulatory drugs (thalidomide and lenalidomide) followed by autologous stem cell transplantation (ASCT) have significantly improved the short- and long-term outcomes of the patients with multiple myeloma. In spite of these advances, MM remains an incurable disease and patients commonly relapse [6]. Therefore, new therapeutic agents that can notably prolong the OS and DFS of the patients with MM are actively being sought.
Camptothecin (CPT) represents an important class of useful oncotherapeutic agents which show a broad spectrum of antitumor activity by inhibiting the activity of DNA topoisomerase I (Topo I) in various cancers, including lung, ovarian, breast, pancreas, stomach cancer and leukemia [7]. Topo I inhibitors usually act as interfacial inhibitors by inserting into the DNA base pairs to producing collisions between the replication and transcription forks [8], leading to double strand DNA breaks and cell death [9]. However, CPT is limited in its treatment applications, due to its low solubility and instability of the active lactone form [10]. To overcome these drawbacks, two strategies have been put forward. One is to synthesize water-soluble analogues including pro-drugs and derivatives of CPT. Topotecan and irinotecan, both of which are natural CPT derivatives, have been approved by the US Food and Drug Administration (FDA) to treat ovarian and colon cancer, respectively. Various reports suggested that the addition of topotecan to high-dose chemotherapy regimen for both pre-treatment, recurrent and refractory multiple myeloma patients, has been a novel application of the agent [11,12]. Taken together, camptothecin derivatives should be further investigated as treatment options for the patients with MM.
Cyclin dependent kinases (CDKs), which are responsible for cell cycle progression, have been a focus of tumor target therapies. It has been reported that CDK4/6, and their binding partner cyclin D, play crucial roles in the cell cycle check point between G0 and G1 phase, and have been implicated in cancer progression [13]. CDK inhibitors hinder the proliferation of tumor cells via down-regulation of CDK activities. A variety of CDK inhibitors (CDKIs) are currently in development and various stages of clinical trial. Flavopiridol (alvocidi) and CYC-202, which are regarded as pan-CDK inhibitors, exhibit global inhibitory activities to CDK1, CDK2, CDK4 and CDK7/CDK9. PD0332991, a CDK4 and CDK6 inhibitor, and P276-00, a CDK4 inhibitor, are currently in clinical trials [14]. MM generally leads to a poor outcome because of the ability of self-renewal and high proliferation [15]. Research indicates MM cells can enter the cell cycle through the up-regulation of cyclin D1, 2, CDK4 and 6 [16,17]. Therefore, anti-proliferative therapies aimed at breaking down the cellular networks which control cell cycle progression may be the key to more effective treatments in the patients with multiple myeloma [18].
Mitochondria have been suggested to play a key role in the regulation of apoptosis. Mitochondrial dysfunctions including loss of mitochondrial membrane potential (ΔΨm), permeability transition, and release of cytochrome c from mitochondria into cytosol are likely to cause apoptosis. Recent studies have indicated that some anticancer agents [e.g. Adriamycin (ADR) and topotecan] induce apoptosis partly via the generation of ROS and the disruption of redox homeostasis which linked to loss of ΔΨm [19,20].
HM910, a new CPT derivative, was synthesized by Fangsheng Pharmaceuticals, Inc. (Changsha, China) and is in phase I clinical trials in China. Here, we found that HM910, which has a chemical structure similar to topotecan, exhibited potential anticancer activity against MM in vitro and in vivo. In addition, HM910 induced apoptosis via the activation of mitochondrial-dependent apoptotic pathway. More importantly, HM910 induced cell cycle arrest in G1 phase via downregulating the expressions of CDK4 and 6, resulting in down-regulation of cyclin D1. Our findings are novel, encouraging further study of HM910 as a novel chemotherapeutical agent for MM in clinic.
Materials and methods
Chemicals and agents
RPMI-1640 and Dulbecco modified Eagle’s medium (DMEM) were purchased from Gibco BRL (Gaithersburg, MD, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Rhodamine123 (Rh123), and RNase A were purchased from Sigma (St. Louis, MO, USA). ApopNexin FITC Apoptosis Detection Kit was purchased from BestBio Biotechnology (Beyotime, Shanghai, China). 2’,7’-dichlorofluorescin diacetate (DCFH-DA), NAC, nuclear and cytoplasmic protein extraction kit and BCA Protein Assay Kit were purchased from Beyotime Biotechnology (Beyotime). Propidium iodide (PI) was obtained from MP Biomedicals, U.S.A. Antibodies against Bcl-2 (sc-7382), Bcl-xl (sc-492), and c-myc (sc-8000) were obtained from Santa Cruz Biotechnology, Inc. (California, USA). Antibodies against CDK2 (#2546), CDK4 (#12790), CDK6 (#3136), cleaved caspase-9 (#7237), -3 (#9664), poly ADP-ribose polymerase (PARP) (#5625), Bad (#9295), Bax (#2772), Cyclin D1 (#2922), and Cytochrome c (#4272) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). An antibody against Bim (1036-1) was obtained from Epitomics, Inc. (California, USA). An antibody against β-actin (AP0060) was purchased from Bioworld technology co., Ltd, USA. Antibodies against glyceraldehydes-3-phosphate dehydrogenase, anti-mouse IgG-horseradish peroxidase, and anti-rabbit IgG-horseradish peroxidase were purchased from KangCheng Biotechnology, Guangzhou, China. Other routine laboratory reagents were obtained from commercial sources, and were of analytical grade. The camptothecin derivative, HM910 (purity > 99%), was provided by Fangsheng Pharmaceuticals, Inc. (Changsha, China) (Figure 1A).
Figure 1.
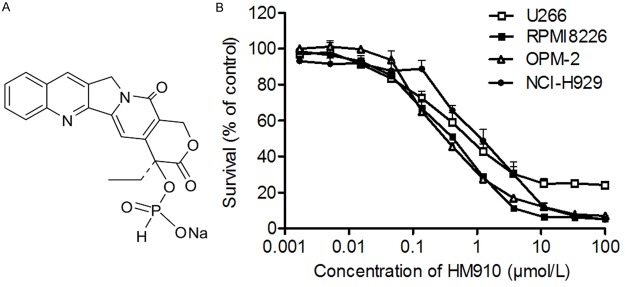
HM910 inhibited cell growth in a variety of MM cells. A. HM910 chemical structure. B. HM910 cytotoxicity in MM RPMI8226, OPM-2, NCI-H929, and U266 cells. Cytotoxicity was measured by MTT assay. The cells were exposed to a full concentration range of HM910 (0-100 µmol/L), for 72 h. Data are presented as mean ± SD, using triplicate samples from a minimum of three independent experiments.
Cell lines and culture conditions
The following cell lines were cultured in DMEM or RPMI-1640, containing 100 units/mL penicillin, 100 units/mL streptomycin, and 10% fetal bovine serum, at 37°C, in a humidified atmosphere, at 5% CO2. The human multiple myeloma cell lines U266, OPM-2 and NCI-H929 were obtained from Bioleaf Biotech Co., Ltd, Shanghai, China. RPMI8226 cells were provided by surgical lab of the first affiliated hospital, Sun Yat-Sen University, Guangzhou, China.
Drug
Topotecan and HM910 (Fangsheng Pharmaceuticals, Inc.) compounds were mixed with mannitol (in a ratio of 1:1.2 to increase the solubility), and solubilized in normal saline (NS) at the appropriate concentrations before injection. Topotecan (batch number 1006A) applied to MM xenografts in nude mice was obtained from GlaxoSmithKline (GSK).
Animals and nude mouse xenograft model
A first batch of athymic nude mice (BALB/c-nu), 5 to 6 weeks old and weighing 16 to 18 g, were obtained from the Center of Experimental Animals, Sun Yat-Sen University, Guangzhou, China. A second batch of athymic nude mice, 6 to 7 weeks old and weighing 18 to 24 g, were purchased from Medical Experimental Animal Center of Guangdong Province, Guangzhou, China. All animals received sterilized food and water. All experiments were carried out in accordance with the guidelines on animal care and experiments of laboratory animals (Center of Experimental Animals, Sun Yat-Sen University), and were approved by the ethics committee for animal experiments.
NCI-H929 cells grown in vitro were harvested and implanted s.c. under the shoulder in the nude mice. When 48 h after inoculation, the mice were randomized into five groups and treated with various regimens: (a) saline (q4d × 2); (b) topotecan (2 or 4 mg/kg, i.p., q4d × 2); (c) HM910 (35 mg/kg, i.p., q4d × 2); (d) HM910 (25 mg/kg, i.p., q4d × 2); and (e) HM910 (18 mg/kg, i.p., q4d × 2). Caliper measurements of the longest perpendicular tumor diameters were performed on alternate days to estimate the tumor volume, using the following formula, representing the three-dimensional volume of an ellipse: V = 4π/3 × (width/2)2 × (length/2) [21]. Animals were sacrificed when tumors reached 2 cm3. Survival was evaluated from the first day of treatment until death [22]. The inhibition rate (IR) was calculated according to the following formula [23]:
IR (%) = (1-Mean tumor weight of experiment group)/Mean tumor weight of control group × 100
Cytotoxicity assay
Cells were harvested during the logarithmic growth phase, and seeded in 96-well plates, at a density of 5 × 103/well, in a final volume of 190 μL per well. After 2 h incubation, HM910 (10 μL) at full-range concentrations (0-100 µmol/L) was added to the 96-well plates. Cells were incubated for 68 h, and then MTT (20 μL) (5 mg/mL stock solution of saline) was added to each well for 4 h. Subsequently, the supernatant was removed, and MTT crystals were solubilized with DMSO (120 μL) in each well. Thereafter, cell viability was measured using a 550 microplate reader (Bio-Rad, Hercules, CA, USA), at 540 nm, with 655 nm as a reference filter [24]. The IC50 was calculated from survival curves using the Bliss method [25]. Percent cell survival was calculated using the following formula: survival (%) = [(mean experimental absorbance)/(mean control absorbance)] × 100.
Cell cycle analysis
Multiple myeloma cells were incubated with or without HM910 (2.5 μM) for 24 h. Then cells were harvested and washed twice with PBS, then fixed in ethanol (70%), overnight, at 4°C, and then resuspended in PBS (500 µL) containing Triton X-100 (0.12%), EDTA (0.12 mmol/L), and RNase A (100 µg/mL). Next, propidium iodide (50 µg/mL) was added to cell suspension for 30 min, at 4°C, in dark. The cell cycle was determined by flow cytometry using a Coulter EPICS XL-MCL. Data were analyzed using the Phoenix flow system.
Real-time quantitative PCR
After treatment with HM910 or topotecan, total RNA was isolated from cell cultures with Trizol Reagent (Molecular Research Center, Cincinnati, USA) according to the manufacture’s instruction. Next, cDNA synthesis was performed with reverse transcription kits (Promega Corp.). The primers used for real-time quantitative PCR were 5’-CACCCGTGGTTGTTACACTC-3’ (forward) and 5’-AACTGGTCGGCTTCAGAGTT-3’ (reverse) for CDK4; 5’-GAAGGTGAAGGTCGGAGTC-3’ (forward) and R: 5’-GAAGATGGTGATGGGATTTC-3’ (reverse) for GAPDH, respectively. Real-time quantitative PCR was performed with Real-time PCR kit (Biomics Biotechnologies Co.,Ltd) according to the directions. The geometric mean of GAPDH was used as an internal control to normalize expression level. PCR conditions for CDK4 were predenaturation at 94°C for 3 min, denaturation at 94°C for 15 sec, annealing at 60°C for 34 sec, extension at 72°C for 15 sec and, after a total of 40 cycles, extension at 72°C for 10 min [26]. Relative quantification of CDK4 was performed using the 2-ΔΔCt method [27]. Results are based on an average of three replicate reactions per sample, and analyzed using SPSS software.
CDK4 protein half-life analysis
To further understand the mechanism underlying HM910 mediated the down-regulation of CDK4 protein activity, NCI-H929 cells were pre-incubated with cycloheximide (10 mg/mL) (CHX) for 2 h. Then, cells were treated with HM910 (5 µmol/L). Every six hours, proteins were extracted from cells and CDK4 was detected by Western blot. GAPDH was used as a loading control.
Measurement of apoptosis
Annexin V and propidium iodide (PI) staining was performed using the BestBio Annexin V- FITC Apoptosis Detection Kit. Myeloma cells (5 × 105) were seeded in six-well plates. After the treatment with the concentration of HM910 (2.5, 5 and 10 μmol/L), both floating and attached cells were collected, washed with ice-cold PBS twice, and resuspended in 1 × binding buffer (400 μL), samples were then incubated with Annexin V-FITC (5 μL) for 15 min and PI (10 μL) for 5 min, on ice, in the dark. Next, viable, apoptotic, and necrotic cells were analyzed by flow cytometry (Becton Dickinson), and data was analyzed by Cell-Quest software. A minimum of 10,000 cells were analyzed per sample. Percent apoptosis (%) = [(number of apoptotic cells) / (number of total cells observed)] × 100.
Reactive oxygen species generation
To measure ROS levels induced by HM910 in MM cells, we used DCFH-DA assay. DCFH-DA is a fluorescent dye that penetrates the cell membrane and is enzymatically hydrolyzed by intracellular esterases to non-fluorescent DCFH [28]. After NCI-H929 cells were exposed to HM910 (2.5, 5 and 10 μmol/L) or topotecan (0.5, 1 and - 2 μmol/L), for 24 h, 5 × 105 cells were harvested, washed once with ice-cold serum-free medium, and incubated with DCFH-DA (10 mmol/L), at 37°C, for 30 min, in the dark. Next, cells were washed twice and resuspended in serum-free medium (1 mL). The ROS generation was determined by FACS Calibur flow cytometer, at the excitation wavelength of 488 nm and emission wavelength of 530 nm. The data of 2’,7’-dichlorofluorescin diacetate fluorescence intensity was evaluated by CellQuest software, and expressed as mean fluorescence intensity. The assay was repeated a minimum of three independent experiments, and 10,000 cells per sample were used.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was assessed by the retention of Rho123, a membrane-permeable, fluorescent, cationic dye that is selectively taken up by mitochondria, and is proportional to the mitochondrial membrane potential [29]. After a 24 h incubation in normal medium plus HM910 or topotecan treatment, cells were incubated in serum-free medium containing Rho123 (0.1 μg/mL), in the dark, at 37°C, for 30 min. Cells were harvested and washed with PBS twice, then analyzed by flow cytometry, with excitation and emission wavelengths of 480 and 530 nm, respectively. The assay was carried out in triplicate and the results were expressed as the mean ±SD.
Western blot analysis
After NCI-H929 cells were exposed to HM910 (2.5, 5 and 10 μmol/L) or topotecan (0.5, 1 and 2 μmol/L), for 24 h, respectively, whole cells were harvested and washed twice with ice cold PBS. Next, the cell pellet was vortexed, and 1 × lysis buffer [Tris-HCl (50 mmol/L, pH6.8), glycerol (10%), SDS (2%), bromophenol blue (0.25%), and DTT (0.1 mol/L]) was added, 100 mL/5 × 106 cells. Samples were heated, 100°C for 10 min, lysates were then centrifuged at 12,000 rpm for 10 min, and the supernatant was collected [30]. The protein concentration was determined using BCA assay followed the instruction. To prepare cytoplasmic fraction of cytochrome c, cells were lysed using a nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China), and the assay was performed according to the manufacturer’s instructions. The lysates were ultra-centrifuged at 12,000 rpm, for 10 min, at 4°C. Clear supernatant was collected as the cytoplasmic fraction. The cytoplasmic fraction protein concentration was measured using Bradford’s reagent (Bio-Rad). An equal amount of protein was separated on 10%-12% SDS-PAGE gels and transferred onto polyvinylidene difluoride membrane (Pall). The nonspecific binding sites were blocked with TBST buffer [NaCl (150 mmol/L), Tris-HCl (20 mmol/L, pH7.4), and Tween 20 (0.4%, v/v)] containing nonfat dry milk (5%) for 2 h. The membranes were incubated overnight, at 4°C, with specific primary antibodies (at a 1:1000 dilution). Next, the membranes were washed three times with TBST buffer, and incubated at room temperature for 2 h with horseradish peroxidase-conjugated secondary antibody (at a 1:5000 dilution). Membranes were then washed three times with TBST buffer. The immunoblots were visualized using an enhanced Phototope-Horseradish Peroxidase Detection Kit, purchased from Cell Signaling Technology, and film was developed using a Kodak medical X-ray processor.
Statistical analysis
Results were analyzed using a t-test or one-way ANOVA, and SPSS 19.0 software. Data were presented as mean ± SD using a minimum of triplicate determinations. *, P < 0.05 was indicative of significant difference and **, P < 0.01 was indicative of very significant difference.
Results
HM910 inhibited cell growth in a variety of MM cells
The cytotoxicity of HM910 was measured by MTT assay in different MM cells. As shown in Figure 1B, HM910 strongly reduced the viability in human multiple myeloma cells, in a concentration-dependent manner. The IC50 values of HM910 were 0.33 ± 0.02, 0.38 ± 0.03, 0.81 ± 0.23, and 1.21 ± 0.27 μmol/L in OPM-2, RPMI8226, U266 and NCI-H929 cells, respectively. These suggest HM910 potentially inhibits MM cell growth in vitro.
HM910 significantly inhibited tumor growth in vivo
Based on the in vitro findings, we investigated the ability of HM910 to inhibit MM cell growth in the nude mouse xenograft model with NCI-H929 cells. The results demonstrated that HM910 significantly inhibited the growth of xenografts (Figure 2 and Table 1). Importantly, no mortality and significant decrease in body weight were observed at the experimental doses of HM910. This suggests the toxicity of HM910 is tolerable. Collectively, HM910 potentially inhibited the growth of multiple myeloma cells in vitro and in vivo.
Figure 2.
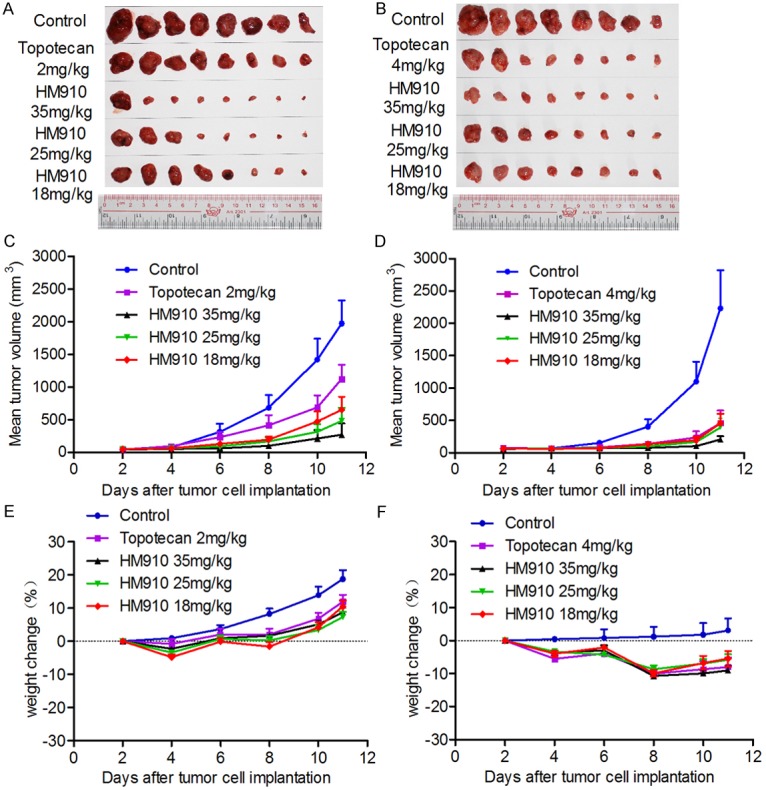
HM910 significantly inhibited tumor growth in vivo. Antitumor effects of HM910 in a NCI-H929 xenograft model in athymic nude mice. A & B. Tumor size. The picture was taken on the 11th day after implantation. The various treatments were as follows: (a) saline (q4d × 2); (b) topotecan (2 or 4 mg/kg, i.p., q4d × 2); (c) HM910 (35 mg/kg, i.p., q4d × 2); (d) HM910 (25 mg/kg, i.p., q4d × 2); and (e) HM910 (18 mg/kg, i.p., q4d × 2). C & D. Changes in tumor volume with time after tumor cell inoculation. Points, mean tumor volume for each group of 8 mice after implantation; bars, SEM. E & F. Changes in weight with time after tumor cell inoculation.
Table 1.
Effect of HM910 on inhibiting multiple myeloma cell NCI-H929 growth in vivo
| Group | Pre-experiment | Post-experiment | Tumor weight (g) | Inhibition ratio (%) | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| n | Weight (g) | n | Weight (g) | |||
| Control | 8 | 17.40 ± 0.70 | 8 | 20.60 ± 1.40 | 1.49 ± 0.89 | - |
| Topotecan 2 mg/kg | 8 | 17.20 ± 1.00 | 8 | 19.20 ± 1.30 | 0.71 ± 0.44* | 51.97 |
| HM910 35 mg/kg | 8 | 17.30 ± 0.70 | 8 | 18.80 ± 0.60 | 0.18 ± 0.36* | 88.18 |
| HM910 25 mg/kg | 8 | 17.30 ± 0.70 | 8 | 18.50 ± 0.70 | 0.27 ± 0.35* | 81.98 |
| HM910 18 mg/kg | 8 | 17.40 ± 0.80 | 8 | 19.20 ± 0.90 | 0.41 ± 0.34* | 72.67 |
| Control | 8 | 20.90 ± 1.10 | 8 | 21.50 ± 2.20 | 1.38 ± 0.91 | - |
| Topotecan 4 mg/kg | 8 | 20.70 ± 1.30 | 8 | 19.00 ± 2.20 | 0.31 ± 0.38* | 77.28 |
| HM910 35 mg/kg | 8 | 19.90 ± 0.80 | 8 | 17.30 ± 1.00 | 0.12 ± 0.08* | 90.93 |
| HM910 25 mg/kg | 8 | 20.60 ± 1.20 | 8 | 19.30 ± 1.20 | 0.26 ± 0.25* | 81.24 |
| HM910 18 mg/kg | 8 | 20.40 ± 0.90 | 8 | 19.30 ± 1.50 | 0.27 ± 0.20* | 80.64 |
P < 0.05 vs the control Group 1.
Means ± SD. n = 8.
HM910 induced cell cycle arrest in G1 phase in MM cells
To explore the mode of action, NCI-H929 cells were exposed to HM910 for 24h and then cell cycle was analyzed. HM910 resulted in an increased number of cells in G1 phase (Figure 3). Furthermore, we observed that G1 phase related proteins, such as CDK4, CDK6, c-myc, and cyclin D1 expression levels decreased in the presence of HM910 (Figure 3C). Taken together, HM910 could induce cell cycle G1 phase arrest which was associated with down-regulation of CDK4, CDK6, c-myc, and cyclin D1 expression.
Figure 3.

HM910 induced cell cycle arrest in G1 phase in MM cells. A. Cells were stained with PBS containing Triton X-100 (0.12%), EDTA (0.12 mmol/L), RNase A (100 µg/mL) and propidium iodide (50 µg/mL), and the cell cycle stages of MM cells were determined by flow cytometry. Representative results were exhibited, similar results were obtained from two other independent trials. B. Data shown as means ± SD, from triplicate independent determinations. C. HM910 or topotecan had effect on CDK2, CDK4, CDK6, cyclin D1 and c-myc protein levels determined by Western bloting. D. Gray value analysis of CDKs and C-myc determined by Image J. β-actin was used as a loading control. Data shown as means ± SD using triplicate independent determinations.
To further understand the mechanism of downregulation of CDK4 expression by HM910, we examined the mRNA expression level and degradation of CDK4 in the presence or absence of HM910. The results showed that HM910 could shorten the half-life of CDK4 from 38.6 to 15.7 h (Figure 4A, 4B), but had no influence on CDK4 mRNA level (Figure 4C). These suggested that HM910 downregulated CDK4 expression level via promoting its degradation.
Figure 4.
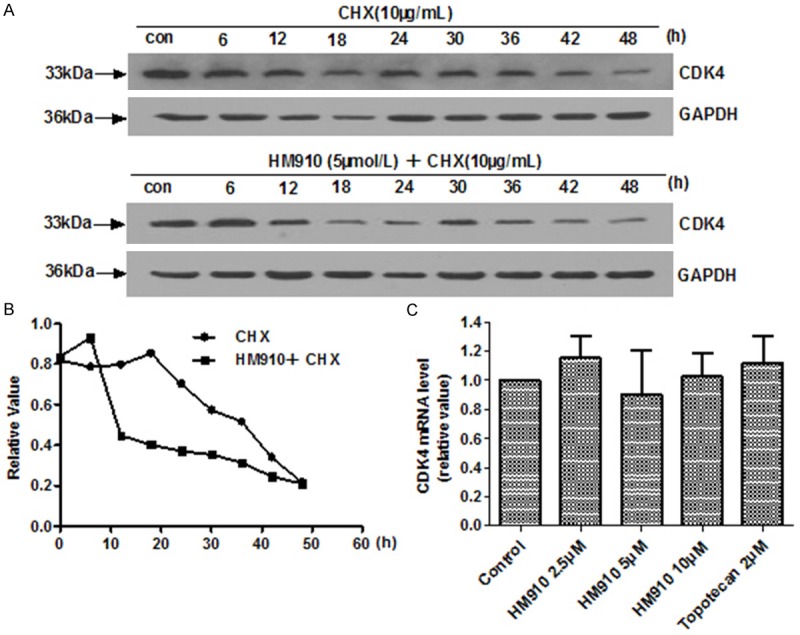
HM910 deceased CDK4 protein levels via a shortened protein half-life. A. Protein turn-over assay of CDK4 after HM910 treatment. NCI-H929 cells were pre-incubated with CHX (10 mg/mL), for 2 h. Next, NCI-H929 cells were treated with HM910 (5 μmol/L). Every six hours, cells were harvested and protein levels assessed using Western blotting. GAPDH was used as a loading control. B. Gray value analysis of CDK4 protein turn-over was determined by Image J. The CDK4 protein half-life was carried out by densitometric analysis. CDK4 protein half-life decreased from 38.6 to 15.7 h. C. The mRNA levels of CDK4 as determined by Real-time quantitative PCR. GAPDH expression was used to normalize data.
HM910 induced MM cell apoptosis
Apoptosis is a physiological process that functions as an essential mechanism of tissue homeostasis and is regarded as the preferred way to eliminate unwanted cells. Compounds able to induce apoptosis is generally considered to be potential antitumor agent for cancer treatment [31]. To identify the role of HM910 in MM cell apoptosis, we used Annexin V-FITC and propidium iodide staining to evaluate cell apoptosis by flow cytometry. The results indicated a significant increase of apoptosis rate after HM910 treatment in a concentration- and time-dependent manner in MM cells (Figure 5).
Figure 5.
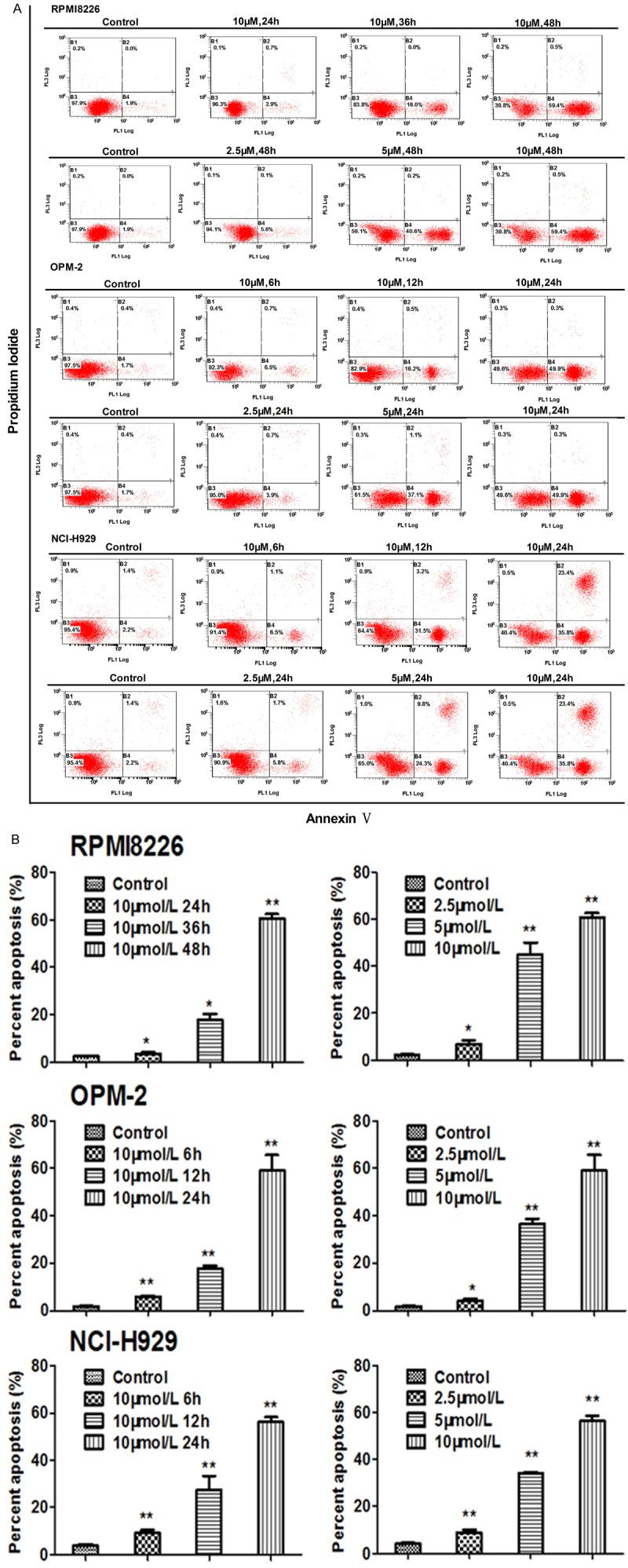
HM910 induced MM cell apoptosis. A. Apoptosis analysis was performed using Annexin V/PI double staining. After cells were exposed to HM910 (2.5, 5 and 10 μmol/L), the attached and detached cells were collected. Following staining with Annexin V and PI, cells were subjected to flow cytometry analysis. B. Percentage of apoptosis. Early apoptotic cell population increased in HM910 treated myeloma cells, in a time- and dose-dependent manner. Data are presented as mean ± SD from three independent experiments (*, P < 0.05; **, P < 0.01).
It is known that some cytotoxic agents such as topotecan can increase the levels of reactive oxygen species (ROS) and initiated cell apoptosis [20]. 2’,7’-dichloroflorescein diacetate (DCFH-DA), a fluorescent probe, was used to detect ROS level. Increased ROS levels were observed in NCI-H929 cells after treatment with HM910 or topotecan (Figure 6A and 6B). Furthermore, the loss of mitochondrial membrane potential (ΔΨm) is an important event of apoptosis via ROS-mediated mitochondrial dependent pathway [32]. It was shown that the Rho123 fluorescence intensity in the NCI-H929 cells treated by HM910 or topotecan decreased in a concentration-dependent manner (Figure 6C and 6D). On the other hand, NAC, an anti-oxidant, could inhibit cell apoptosis induced by HM910 (Figure 6E). The data suggested that mitochondrial dysfunction was involved in the apoptosis induced by HM910. The release of cytochrome c from the mitochondria to cytosol is the limiting factor in mitochondrial pathway and the mitochondrial dysfunction has been suggested to cause the release of cytochrome c. Subsequently, it causes apoptosis by the activation of caspase-9 in the presence of Apaf-1 and in turn results in the activation of downstream caspase-3, which can cleave PARP [33]. On the other hand, Bcl-2 protein family is increasingly believed to have relationship with mitochondrial dysfunctions [34]. To elucidate the interactions between the above apoptotic proteins, we detected the cytochrome c in cytosol, caspases, and other apoptotic related proteins of whole-cell lysates by Western blot after NCI-H929 cells were exposed to HM910 or topotecan for 24 h, respectively. The up-regulation of Bad and Bim, as well as downregulation of Bcl-2 and Bcl-xl were observed in NCI-H929 cells treated with HM910 or topotecan (Figure 7A-C). In fact, a concentration-dependent increase of cytochrome c in cytosol, and an activated caspase 9, 3 and a cleaved PARP were also exhibited in NCI-H929 cells treated with HM910 or topotecan (Figure 7D-F). The data demonstrate the cell apoptosis induced by HM910 was associated with the activation of mitochondrial-dependent apoptotic pathway.
Figure 6.
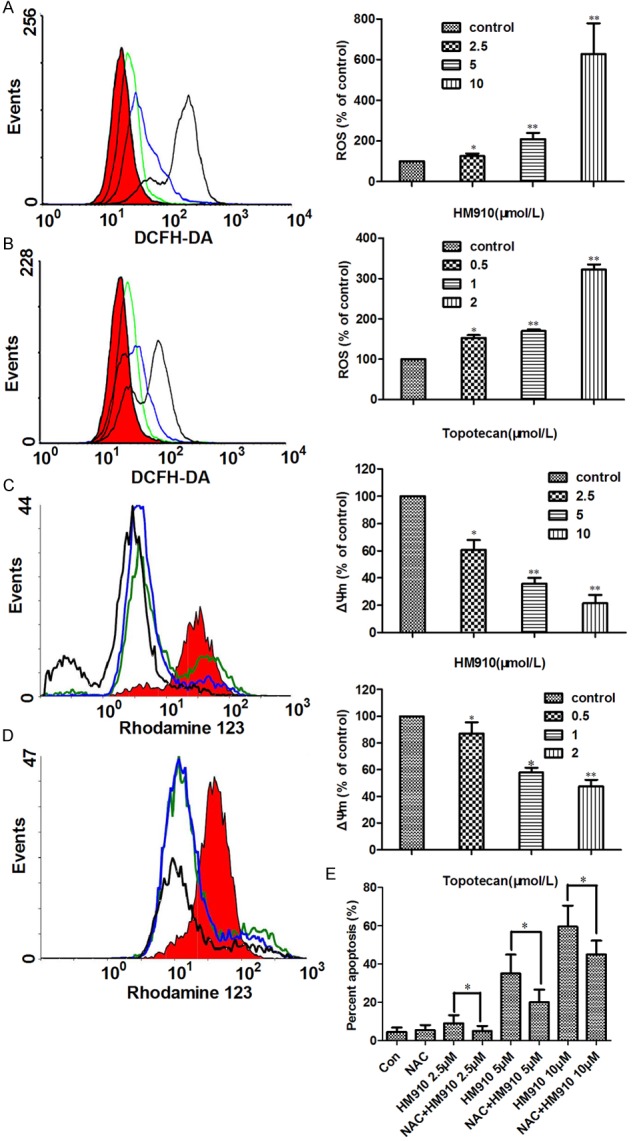
HM910 increased reactive oxygen species (ROS) generation, and induced loss of mitochondrial membrane potential (ΔΨm) in NCI-H929 cells. A & B. Both HM910 and topotecan induced significant increases in ROS generation, in NCI-H929 cells. Representative results (left) are shown, similar results were obtained in two other independent trials. Data (right) shown are means ± SD for independent determinations in triplicate. C & D. Both HM910 and topotecan reduced mitochondrial transmembrane potential. Representative results (left) are shown, similar results were obtained in two other independent trials. Data (right) are presented as mean ± SD from three independent experiments (*, P < 0.05; **, P < 0.01). E. NAC, an anti-oxidant, could inhibit cell apoptosis induced by HM910.
Figure 7.

HM910 induced MM cell apoptosis through mitochondrial dependent pathway. A. Apoptosis related changes in protein levels. In both HM910 and topotecan treatment groups expression of Bcl-2 and Bcl-xl were down regulated, and Bad and Bim protein levels were up regulated, however, no change Bax protein levels was observed. B and C. The averaged relative expression levels of Bcl-2 protein family in NCI-H929 cells in the presence of increasing concentrations of HM910 or topotecan. Values are presented as mean ± SD calculated from three independent experiments (*, P < 0.05; **, P < 0.01). D. Changes in mitochondrial dependent apoptotic pathway related proteins. Cytochrome c released from mitochondria to cytoplasm increased, as did activation of caspases 9, 3 and PARP cleavage were observed in a concentration dependent manner. E and F. The averaged relative expression levels of caspase in NCI-H929 cells in the presence of increasing concentrations of HM910 or topotecan. Representative blots from three independent experiments are shown (*, P < 0.05; **, P < 0.01).
Discussion
Multiple myeloma is a plasma cell malignancy. Chemotherapy can prolong patients’ OS and PFS. However, relapse is common due to traditional drug resistance [35]. Therefore, it is imperative to develop novel treatment agents for MM.
Topotecan, a derivative of camptothecin acts against a number of hematological malignancies and has now become part of high-dose chemotherapy regimen [36]. In fact, topotecan has also been widely used in the treatment of lymphoblastic, myeloid acute leukemia and non-Hodgkin lymphomas [36]. Studies have demonstrated topotecan was an innovative and effective treatment in resistant and/or relapsed multiple myeloma cases [12]. HM910, a novel camptothecin derivate, displayed high efficacy and low toxicity in the inhibition of MM cell growth in vitro and in vivo. These suggest HM910 is promising to develop as a novel chemotherapeutical drug, encouraging further study in clinic. Up to date, HM910 is in phase I clinical trial in China.
Many cytostatic agents are known to influence the apoptosis and proliferation of cells. Cyclin-dependent kinase 4 (CDK4) and CDK6 associate with D-type cyclins to promote cell-cycle entry and progression through G1 by inactivating the retinoblastoma protein (Rb) in opposition to the INK4 family of CDK inhibitors (CKIs) [37]. CDK inhibitors (CDKIs) have attracted a lot of attention recently as an inhibitory factor against tumor cell proliferation. At present, pan-CDK inhibitors, specifically highly selective CDK inhibitors, are in clinical trials [14]. Reports have shown CDKIs were effective on the treatment of solid tumors. For example, CDK4 inhibitor PD0332991 was in Phase II/III clinical trials for the patients with CDK4-amplified well-differentiated or dedifferentiated liposarcoma [38].
Although germline mutations of CDK4 CDK6 in human cancers are rare, dysregulation of CDK4 and CDK6 by gain of function or loss of inhibition is common [39]. So halting unscheduled cell-cycle progression by inhibition of CDK4/CDK6 may significantly improve treatment efficacy. Multiple evidences further suggest that dysregulation of CDK4/CDK6 is pivotal for the loss of cell-cycle control in MM. In MM, malignant plasmacytoid cells retain the self-renewing potential as opposed to normal plasma cells, which are permanently arrested in early G1 because of inhibition of CDK4 and CDK6 by p18INK4c [40-42]. CDK4 and CDK6 thus appear to be promising targets for cell-cycle control of MM. PD0332991 is the only known selective and potent inhibitor for CDK4 and CDK6, and induces early G1 arrest in primary human myeloma cells in the presence of BM stromal cells (BMSCs) ex vivo [43]. In a pilot study, PD0332991 cooperated with bortezomibprolonged the survival of mice developing tumors in the immune-competent 5T myeloma model [44]. PD0332991 has now opened a new vista in the first single-agent phase I clinical study and is being actively investigated in MM (www.clinicaltrials.gov). Here, we demonstrate for the first time that the mechanism of down-regulation of CDK4 expression by HM910 had no influence on transcriptional regulation, but related to promoting its degradation. Importantly, HM910 could trigger MM cell cycle arrest in phase G1. These data suggest the inhibition of MM cell growth by HM910 is associated with cell cycle arrest related to the downregulation of CDK4/6, c-myc and cyclin D1 expression levels.
Apoptosis is a common mechanism for most of chemotherapeutic drugs that induce cancer cell death. Apoptosis is regulated in an orderly way by a series of signaling cascades and occurs by two connected pathways. The extrinsic pathway is initiated by cell surface death receptor stimulation and activation of caspase-8, while the intrinsic pathway involves cytochrome c release from mitochondria and subsequent caspase-9 and -3 activations.
The results showed that HM910 induced the generation of reactive oxygen species (ROS) and decreased mitochondrial membrane potential and led to the release of cytochrome c. Further investigation revealed that HM910 increased protein expression levels of Bad, Bim and activation of caspase-9 and -3; and decreased the expression levels of Bcl-2 and Bcl-XL to trigger mitochondrial-dependent apoptosis pathway.
In conclusion, we found HM910, a novel CPT derivative, was a potent inhibitor on multiple myeloma cells growth in vitro and in vivo. The studies on the modes of action showed that HM910 induced cell cycle arrest in G1 phase via the downregulation of CDK4/6, cyclin D1 and c-myc expression levels. On the other hand, HM910 also induced MM cell apoptosis through the mitochondrial-dependent apoptotic pathway triggered by an increase of ROS (Figure 8). This discovery opens up new vistas for multiple myeloma treatment with HM910.
Figure 8.
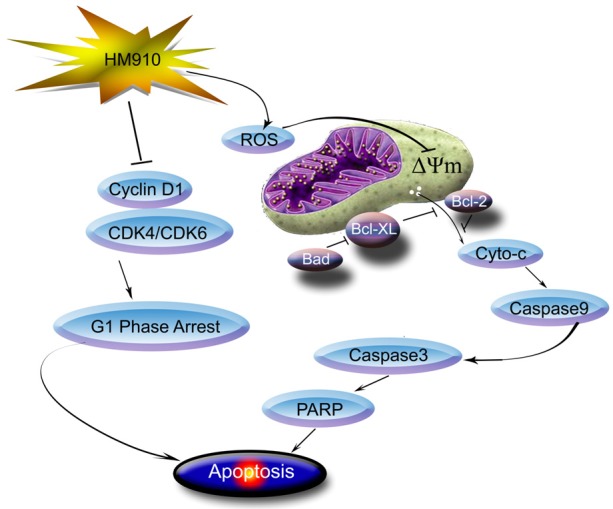
A schematic model illustrating the possible pathway targeting NCI-H929 cell by HM910. HM910 induced apoptosis via the activation of mitochondrial-dependent apoptotic pathway. More importantly, HM910 induced cell cycle arrest in G1 phase via downregulating the expressions of CDK4 and 6, resulting in down-regulation of cyclin D1.
Acknowledgements
This work was supported by Program of National Key Clinical Specialties and Natural Science Foundation of Guangdong Province, China (No. S2013010016838) and Scientific foundation of Guangzhou City, China (No. 12C32061587).
Disclosure of conflict of interest
None.
References
- 1.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, Dispenzieri A, Kumar S, Clark RJ, Baris D, Hoover R, Rajkumar SV. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113:5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyle RA, Kumar S. The significance of monoclonal gammopathy of undetermined significance. Haematologica. 2009;94:1641–1644. doi: 10.3324/haematol.2009.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 6.van de Donk NW, Lokhorst HM. New developments in the management and treatment of newly diagnosed and relapsed/refractory multiple myeloma patients. Expert Opin Pharmacother. 2013;14:1569–1573. doi: 10.1517/14656566.2013.805746. [DOI] [PubMed] [Google Scholar]
- 7.Sriram D, Yogeeswari P, Thirumurugan R, Bal TR. Camptothecin and its analogues: a review on their chemotherapeutic potential. Nat Prod Res. 2005;19:393–412. doi: 10.1080/14786410412331299005. [DOI] [PubMed] [Google Scholar]
- 8.Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Li X, Cao Z, Xu Y, Lin H, Zhao Y, Wei Y, Qian Z. Camptothecin nanocolloids based on N,N,N-trimethyl chitosan: efficient suppression of growth of multiple myeloma in a murine model. Oncol Rep. 2012;27:1035–1040. doi: 10.3892/or.2012.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kazmi SM, Saliba RM, Donato M, Wang M, Hosing C, Qureshi S, Anderlini P, Popat U, Champlin RE, Giralt SA, Qazilbash MH. Phase II trial of high-dose topotecan, melphalan and CY with autologous stem cell support for multiple myeloma. Bone Marrow Transplant. 2011;46:510–515. doi: 10.1038/bmt.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraut EH, Crowley JJ, Wade JL, Laufman LR, Alsina M, Taylor SA, Salmon SE. Evaluation of topotecan in resistant and relapsing multiple myeloma: a Southwest Oncology Group study. J. Clin. Oncol. 1998;16:589–592. doi: 10.1200/JCO.1998.16.2.589. [DOI] [PubMed] [Google Scholar]
- 13.Lamb R, Lehn S, Rogerson L, Clarke RB, Landberg G. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity. Cell Cycle. 2013;12:2384–2394. doi: 10.4161/cc.25403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs. 2013;22:723–738. doi: 10.1517/13543784.2013.789859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Sanz R, Gonzalez-Fraile MI, Mateo G, Hernandez JM, Lopez-Berges MC, de las Heras N, Fernandez-Calvo J, Ortega F, Portero JA, Barez A, Galende J, Orfao A, San Miguel JF. Proliferative activity of plasma cells is the most relevant prognostic factor in elderly multiple myeloma patients. Int J Cancer. 2004;112:884–889. doi: 10.1002/ijc.20491. [DOI] [PubMed] [Google Scholar]
- 16.Glassford J, Rabin N, Lam EW, Yong KL. Functional regulation of D-type cyclins by insulin-like growth factor-I and serum in multiple myeloma cells. Br J Haematol. 2007;139:243–254. doi: 10.1111/j.1365-2141.2007.06789.x. [DOI] [PubMed] [Google Scholar]
- 17.Quinn J, Glassford J, Percy L, Munson P, Marafioti T, Rodriguez-Justo M, Yong K. APRIL promotes cell-cycle progression in primary multiple myeloma cells: influence of D-type cyclin group and translocation status. Blood. 2011;117:890–901. doi: 10.1182/blood-2010-01-264424. [DOI] [PubMed] [Google Scholar]
- 18.Glassford J, Kassen D, Quinn J, Stengel C, Kallinikou K, Khwaja A, Yong KL. Inhibition of cell cycle progression by dual phosphatidylinositol-3-kinase and mTOR blockade in cyclin D2 positive multiple myeloma bearing IgH translocations. Blood Cancer J. 2012;2:e50. doi: 10.1038/bcj.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogalska A, Koceva-Chyla A, Jozwiak Z. Aclarubicin-induced ROS generation and collapse of mitochondrial membrane potential in human cancer cell lines. Chem Biol Interact. 2008;176:58–70. doi: 10.1016/j.cbi.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Akbas SH, Timur M, Ozben T. The effect of quercetin on topotecan cytotoxicity in MCF-7 and MDA-MB 231 human breast cancer cells. J Surg Res. 2005;125:49–55. doi: 10.1016/j.jss.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 21.LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 22.Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, Shi L, Zangari M, Miles R, Bearss D, Tricot G, Zhan F. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23:48–62. doi: 10.1016/j.ccr.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu LW, He LR, Liang YJ, Chen LM, Xiong HY, Yang XP, Pan QC. Experimental chemotherapy against xenografts derived from multidrug resistant KBv200 cells and parental drug-sensitive KB cells in nude mice by annonaceous acetogenin 89-2. Yao Xue Xue Bao. 2003;38:565–570. [PubMed] [Google Scholar]
- 24.Yan Y, Su X, Liang Y, Zhang J, Shi C, Lu Y, Gu L, Fu L. Emodin azide methyl anthraquinone derivative triggers mitochondrial-dependent cell apoptosis involving in caspase-8-mediated Bid cleavage. Mol Cancer Ther. 2008;7:1688–1697. doi: 10.1158/1535-7163.MCT-07-2362. [DOI] [PubMed] [Google Scholar]
- 25.Shi Z, Liang YJ, Chen ZS, Wang XW, Wang XH, Ding Y, Chen LM, Yang XP, Fu LW. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther. 2006;5:39–47. doi: 10.4161/cbt.5.1.2236. [DOI] [PubMed] [Google Scholar]
- 26.Wan Q, Wang H, Lin Y, Gu L, Han M, Yang Z, Zhang Y, Ma R, Wang L, Wang Z. Effects of quercetin on CDK4 mRNA and protein expression in A549 cells infected by H1N1. Biomed Rep. 2013;1:766–770. doi: 10.3892/br.2013.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Ye Q, Ye L, Xu X, Huang B, Zhang X, Zhu Y, Chen X. Epigallocatechin-3-gallate suppresses 1-methyl-4-phenyl-pyridine-induced oxidative stress in PC12 cells via the SIRT1/PGC-1alpha signaling pathway. BMC Complement Altern Med. 2012;12:82. doi: 10.1186/1472-6882-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ou Y, Dong X, Liu XY, Cheng XC, Cheng YN, Yu LG, Guo XL. Mechanism of tetramethylpyrazine analogue CXC195 inhibition of hydrogen peroxide-induced apoptosis in human endothelial cells. Biol Pharm Bull. 2010;33:432–438. doi: 10.1248/bpb.33.432. [DOI] [PubMed] [Google Scholar]
- 30.Rubio S, Quintana J, Lopez M, Eiroa JL, Triana J, Estevez F. Phenylbenzopyrones structure-activity studies identify betuletol derivatives as potential antitumoral agents. Eur J Pharmacol. 2006;548:9–20. doi: 10.1016/j.ejphar.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 31.Marchal JA, Carrasco E, Ramirez A, Jimenez G, Olmedo C, Peran M, Agil A, Conejo-Garcia A, Cruz-Lopez O, Campos JM, Garcia MA. Bozepinib, a novel small antitumor agent, induces PKR-mediated apoptosis and synergizes with IFNalpha triggering apoptosis, autophagy and senescence. Drug Des Devel Ther. 2013;7:1301–1313. doi: 10.2147/DDDT.S51354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu YC, Chen MJ, Huang TY. Inducement of mitosis delay by cucurbitacin E, a novel tetracyclic triterpene from climbing stem of Cucumis melo L. , through GADD45gamma in human brain malignant glioma (GBM) 8401 cells. Cell Death Dis. 2014;5:e1087. doi: 10.1038/cddis.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 34.Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol. 2003;195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- 35.Podar K, Tai YT, Hideshima T, Vallet S, Richardson PG, Anderson KC. Emerging therapies for multiple myeloma. Expert Opin Emerg Drugs. 2009;14:99–127. doi: 10.1517/14728210802676278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrari S, Danova M. [Topotecan: a new field of use] . Tumori. 1999;85:S23–28. [PubMed] [Google Scholar]
- 37.Liu S, Bolger JK, Kirkland LO, Premnath PN, McInnes C. Structural and functional analysis of cyclin D1 reveals p27 and substrate inhibitor binding requirements. ACS Chem Biol. 2010;5:1169–1182. doi: 10.1021/cb1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, Landa J, Qin LX, Rathbone DD, Condy MM, Ustoyev Y, Crago AM, Singer S, Schwartz GK. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013;31:2024–2028. doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007;17:60–65. doi: 10.1016/j.gde.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, Shaffer AL 3rd, Louie T, Chen I, Randolph S, Hahn WC, Staudt LM, Niesvizky R, Moore MA. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120:1095–1106. doi: 10.1182/blood-2012-03-415984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morse L, Chen D, Franklin D, Xiong Y, Chen-Kiang S. Induction of cell cycle arrest and B cell terminal differentiation by CDK inhibitor p18(INK4c) and IL-6. Immunity. 1997;6:47–56. doi: 10.1016/s1074-7613(00)80241-1. [DOI] [PubMed] [Google Scholar]
- 42.Tourigny MR, Ursini-Siegel J, Lee H, Toellner KM, Cunningham AF, Franklin DS, Ely S, Chen M, Qin XF, Xiong Y, MacLennan IC, Chen-Kiang S. CDK inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity. 2002;17:179–189. doi: 10.1016/s1074-7613(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 43.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, Chen-Kiang S. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 44.Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008;68:5519–5523. doi: 10.1158/0008-5472.CAN-07-6404. [DOI] [PubMed] [Google Scholar]