

Abstract
Gastric cancer (GC) is one of the most common malignant tumors and recent data demonstrates the tumor suppressor role of VGLL4 in GC, but the mechanisms for VGLL4 downregulation in GC remain to be elucidated. Here, we confirmed the suppressor role of VGLL4 on proliferation and invasion in GC cells with over-activated YAP-TEAD signal, and indicated the reverse correlation between expression patters of VGLL4 and miR-222. Bioinformatics analysis combined with experimental confirmation revealed VGLL4 is a direct target of miR-222 in GC cells. Functionally, miR-222 inhibitor significantly inhibited GC cells proliferation and invasion and VGLL4 knockdown abolished the effects of miR-222 inhibitor. Moreover, TEAD1 knockdown resulted in decrease of miR-222 expression and increase of VGLL4 expression, and also resulted in reduction of luciferase activity driven by miR-222 promoter in GC cells, suggesting over-activated TEAD1 positively feedback transcriptionally regulates miR-222 expression via physically binding to the miR-222 promoter indicated by ChIP assay. Collectively, our findings implied the important role of miR-222/VGLL4/YAP-TEAD1 regulatory loop maintaining over-activated YAP-TEAD1 signal in GC cells, and enriched the rationale of VGLL4 in GC based on which a promising therapeutic strategy will be developed.
Keywords: Gastric cancer, VGLL4, miR-222, TEAD1
Introduction
Gastric cancer (GC) is the fourth most common type of cancer worldwide, with 989 000 new cases, accounting for about 7.86% of total global cancers, and 738000 deaths, accounting for about 9.7% of total global cancer deaths, annually [1]. Surgical removal of early stage GC tumors is critical to effective treatment, but because of few symptoms accompanying early GC, most GCs are found at an advanced stage. GC recurrence rates are high among all populations, and surgery and combination chemotherapies have been shown to confer only modest survival benefits in advanced GC, resulting in an overall 5 year survival rate of 24% [2]. Many genetic and epigenetic transformations have been shown to contribute to the multistep process of GC development [3], such as HNF4α is a targetable oncoprotein in GC, which is regulated by AMPK signal and resides upstream of WNT signal [4], and GC patients with methylated DACT1 promoter is significantly associated with the poorer survival [5].
Recently, Jiao et al. demonstrated the elevated expression of YAP and its target genes in GC tissues than those in paired control tissues, and overexpression of YAP target genes have been suggested to correlate with GC progression [6]. YAP is the key component of Hippo pathway which controls organ size in diverse species from Drosophila to human. Hippo pathway features a central kinase cascade formed by Hippo (Hpo; MST1/2 in mammals) and Warts (Wts; LATS1/2 in mammals), whose activations lead to phosphorylation of the downstream transcriptional coactivator Yorkie (Yki; YAP/TAZ in mammals), thus preventing its interaction with and therefore transactivation of the DNA binding transcriptional factor Scalloped (Sd; TEADs/TEF in mammals), and inactivation of the Hippo signaling leads to pro-proliferation and anti-apoptosis associated with cancer development and progression [7-9]. Moreover, Jiao et al. found that VGLL4 expression frequently decreased and its decrease inversely correlated with 5 year survival rate of GC patients, and that among YAP-positive GC patients, those with VGLL4-positive expression have a better clinical outcome compared with VGLL4-negative expression. Mechanically, VGLL4 suppresses GC growth in vitro and in vivo via directly competing with YAP for binding TEADs to inhibit YAP-TEADs transcriptional activity [6].
VGLL (Vestigial-like; Vg in Drosophila) proteins are transcriptional cofactors and four VGLL proteins (VGLL1-4) have been found in mammals. Similar to YAP, the VGLL proteins do not contain DNA-binding domain and they also exert their transcriptional regulatory functions through pairing with TEADs. Most investigations have identified VGLL1-3 as TEADs-related transcriptional coactivators required for cancers growth [10-12], but VGLL4 has been identified as a transcriptional repressor that inhibits YAP induced overgrowth and tumorigenesis in Drosophila and human, in regard to cancer such as pancreatic adenocarcinoma, lung cancer and esophageal squamous cell carcinoma [6,13-17]. However, the mechanisms for VGLL4 downregulation remain unclear. Here, we showed that VGLL4 expression decrease was accompanied with miR-222 expression increase in GC tissues and that miR-222 directly targets VGLL4 and VGLL4 responsible for the role of miR-222 in GC cell lines. Furthermore, we identified TEAD1 physically binds to the miR-222 promoter and positively transcriptionally regulates miR-222 expression, suggesting the miR-222/VGLL4/TEAD1 regulatory loop in GC as valuable biomarker and potential therapeutic target.
Materials and methods
Clinical specimens and cell culture
Eight paired fresh GC tissues and adjacent morphologically normal gastric tissues, and 52 formalin-fixed paraffin-embedded tissues including 9 normal gastric tissues and 43 GC tissues were collected at the Affiliated Tumor Hospital of Guangzhou Medical University between 2005-09–2010-05. Fresh tissue samples were cut into two parts and immediately snap-frozen in liquid nitrogen. One section was used for mRNA and miRNA extraction, and the other section was used for protein extraction. The study was approved by the ethics committee of the Affiliated Tumor Hospital of Guangzhou Medical University. Human GC cell lines, MKN-45, BGC-823, MGC-803 and HGC-27 were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) in a humidified cell incubator with an atmosphere of 5% CO2 at 37°C.
Real-time PCR for mRNA and miRNAs
The mRNAs and miRNAs were extracted simultaneously were isolated and purified with miRNA isolation system (OMEGA Bio-Tek). For mRNA qRT-PCR, cDNAs from the mRNAs were synthesized with the first-strand synthesis system (Thermo Scientific, Glen Brunie, MA, USA). Real-time PCR was carried out according to standard protocols using an ABI 7500 with SYBR Green detection (Applied Biosystems, Foster City, CA, USA). GAPDH was used as an internal control and the qRT-PCR was repeated three times. The primers for GAPDH were: forward primer 5’-ATTCCATGGCACCGTCAAGGCTGA-3’, reverse primer 5’-TTCTCCATGGTGGTGAAGACGCCA-3’; primers for VGLL4 were: forward primer 5’-TTGTCCTAGGAAACGGGCTG-3’, reverse primer 5’-GGGCTTACTGGTAGACGGTG-3’; primers for CTGF were: forward primer 5’-GTTTGGCCCAGACCCAACTA-3’, reverse primer 5’- GGCTCTGCTTCTCTAGCCTG-3’; primers for CYR61 were: forward primer 5’-CAGGACTGTGAAGATGCGGT-3’, reverse primer 5’-GCCTGTAGAAGGGAAACGCT-3’. For miRNA qRT-PCR, cDNA was generated with the miScript II RT Kit (QIAGEN) and the quantitative real-time PCR (qRT-PCR) was done by using the miScript SYBR Green PCR Kit (QIAGEN) following the manufacturer’s instructions. The miRNA sequence-specific qRT-PCR primers for miR-222 and endogenous control RNU6 were purchased from QIAGEN, and the qRT-PCR analysis was carried out using 7500 Real-Time PCR System (Applied Biosystems). The gene expression threshold cycle (CT) values of miRNAs were calculated by normalizing with internal control RNU6 and relative quantization values were calculated.
Immunohistochemistry
The sections were dried at 55°C for 2 h and then deparaffinized in xylene and rehydrated using a series of graded alcohol washes. The tissue slides were then treated with 3% hydrogen peroxide in methanol for 15 min to quench endogenous peroxidase activity and antigen retrieval then performed by incubation in 0.01 M sodium citrate buffer (pH 6.0) and heating using a microwave oven. After a 1 h preincubation in 10% goat serum, the specimens were incubated with primary antibody overnight at 4°C. The tissue slides were treated with a non-biotin horseradish peroxidase detection system according to the manufacturer’s instruction (DAKO, Glostrup, Denmark). Two different pathologists evaluated the immunohistological samples.
Western blot
Total proteins were extracted from corresponding cells using the RIPA buffer (Pierce) in the presence of Protease Inhibitor Cocktail (Pierce). The protein concentration of the lysates was measured using a BCA Protein Assay Kit (Pierce). Equivalent amounts of protein were resolved and mixed with 5 × Lane Marker Reducing Sample Buffer (Pierce), electrophoresed in a 10% SDS–acrylamide gel and transferred onto Immobilon-P Transfer Membrane (Millipore). The membranes were blocked with 5% non-fat milk in Tris-buffered saline and then incubated with primary antibodies followed by secondary antibody. The signal was detected using an ECL detection system (Millipore). The VGLL4 antibody was from Novus Biologicals. YAP antibody, TEAD1 antibody and β-Actin antibody were from Cell Signaling Technology. HRP-conjugated secondary antibody was from Thermo.
Cells transfection
MiR-222 inhibitor and relative control were purchased from Ambion. Cells were trypsinized, counted and seeded onto 6-well plates the day before transfection to ensure 70% cell confluence on the day of transfection. The transfection of inhibitor and related controls was carried out using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer’s procedure. The inhibitor and controls were used at a final concentration of 100 nM. At 48 h post-transfection, follow-up experiments were performed.
Luciferase reporter assay
For miRNA luciferase reporter assay: Two single strands of the wild type 3’UTR with miR-222 binding site and two single strands of the mutant type with 7 bases deleted in the miR-222 binding site (as mutant control), of VGLL4 were synthesized with restriction sites for SpeI and HindIII located at both ends of the oligonucleotides for further cloning. The single strands DNA sequences were following: the wild type 3’UTR of VGLL4 (sense: 5’-CTAGT TGAAGAACATTAATTTGTTAATGATATGTAGCTATTTAATTTTTCCCTTTCCT A-3’; antisense: 5’-AGCTT AGGAAAGGGAAAAATTAAATAGCTACATATCATTAACAAATTAATGTTCTTCA A-3’) and the mutated type 3’UTR of FOXO1 (sense: 5’-CTAGT TGAAGAACATTAATTTGTTAATGAT-------TATTTAATTTTTCCCTTTCCT A-3’; antisense: 5’-AGCTT AGGAAAGGGAAAAATTAAATA-------ATCATTAACAAATTAATGTTCTTCA A-3’). The corresponding sense and antisense strands were annealed and subsequently cloned into pMir-Report plasmid downstream of firefly luciferase reporter gene. Cells were seeded in 96 well-plates and co-transfected with pMir-Report luciferase vector, pRL-TK Renilla luciferase vector and miR-222 inhibitor or control. For promoter activity assay: To determine whether TEAD1 regulates the promoter activity of miR-222, a two kilobase region upstream of the miR-222 precursor starting site was cloned into the pGL4-reporter vector upstream of the luciferase gene. Cells were seeded in 96-well plates and co-transfected with the pGL4-reporter vector and the pRL-TK Renilla luciferase vector with or without the TEAD1 specific siRNAs. 48h after cotransfection, the luciferase activities were determined using a Dual-Luciferase Reporter Assay System (Promega) where the Renilla luciferase activity was used as internal control and the firefly luciferase activity was calculated as the mean ± SD after being normalized by Renilla luciferase activity.
Cell proliferation assay
Cell proliferation was monitored by the MTS assay using the CellTiter96®AQueous One Solution Cell Proliferation Assay kit (Promega) according to the manufacturer’s instructions. Related treated cells were seeded into 96-well plates at 2000 cells/well (0.20 ml/well). The cell proliferation assay was performed on days 0, 1, 2, 3, 4 and 5 by incubation with MTS (0.02 ml/well). After 2 h further incubation, the absorbance at 490 nm of each well was recorded on the BioTek Synergy 2 and the absorbance represented the cell number.
Cell invasion assay
Invasion of OS cells was assessed using the Cell Invasion Assay Kit (BD Biosciences) according to the manufacturer’s instructions. Briefly, at 36 h post-transfection, 3 × 104 cells in 300μl serum-free medium were added to the upper chamber precoated with ECMatrix™ gel. Then, 0.5 ml of 10% FBS-containing medium was added to the lower chamber as a chemoattractant. Cells were incubated for 24 h at 37°C, and then non-invading cells were removed with cotton swabs. Cells that migrated to the bottom of the membrane were fixed with pre-cold methanol and stained with 2% Giemsa solution. Stained cells were visualized under a microscope. To minimize the bias, at least three randomly selected fields with 100 × magnification were counted, and the average number was taken.
ChIP assay
The ChIP assay was performed using the EZ-CHIPTM chromatin immunoprecipitation kit (Merck Millipore). Briefly: Chromatin proteins were cross-linked to DNA by addition of formaldehyde to the culture medium to a final concentration of 1%. After a 10 min incubation at room temperature, the cells were washed and scraped off in ice-cold phosphate-buffered saline (PBS) containing Protease Inhibitor Cocktail II. Cells were pelleted and then resuspended in lysis buffer containing Protease Inhibitor Cocktail II. The resulting lysate was subjected to sonication to reduce the size of DNA to approximately 200–1000 base pairs in length. The sample was centrifuged to remove cell debris and diluted ten-fold in ChIP dilution buffer containing Protease Inhibitor Cocktail II. A 5 μl sample of the supernatant was retained as “Input” and stored at 4°C. Then 5 µg of anti-RNA Polymerase antibody (positive control, included with the kit), or anti-TEAD1 antibody (cell signal technology) were added to the chromatin solution and incubated overnight at 4°C with rotation. After antibody incubation, protein G agarose was added and the sample incubated at 4°C with rotation for an additional 2 h. The protein/DNA complexes were washed with Wash Buffers four times and eluted with ChIP Elution Buffer. Cross-links were then reversed to free DNA by the addition of 5M NaCl and incubation at 65°C for 4 h. The DNA was purified according to the manufacturer’s instructions. 50 μl of DNA was obtained for each treatment. 0.2 μl of DNA from each group was used as a template for PCR. Primers for the miR-222 promoter containing putative TEAD1 binding sites were as follows, sense: 5’-ACCCCTGCTTCACCTTGTAAATTCC-3’, antisense: 5’-GGGAATGATTTAACTATTTAATCACAG-3’ (for site A); sense: 5’-GTACATGATTCTTCTCCTCCTAC-3’, antisense: 5’-CCTCCATAGATATGGACGGTC-3’ (for site B. Primers for the human GAPDH gene: sense, 5’-TACTAGCGGTTTTACGGGCG-3’, antisense, 5’-TCGAACAGGAGGAGCAGAGAGCGA-3’. The PCR conditions were as follows: 1 cycle of 95°C for 5 min; 32 cycles of 95°C for 20 s, 59°C for 30 s, and 72°C 30 s; and 1 cycle of 72°C for 10 min. PCR samples were resolved by electrophoresis in a 2% agarose gel and stained with ethidium bromide.
Statistical analysis
All data are expressed as means ± standard deviation from three independent experiments. Statistical analyses were performed using SPSS16.0 software (SPSS, Chicago, IL). The differences between groups were analyzed using Student’s t-test with only two groups or one-way analysis of variance (ANOVA) when more than two groups were compared. Pearson’s correlation analyses was used to determine the correlation between miR-222 expression and VGLL4 mRNA level in the 52 tissues. P values less than 0.05 were considered statistically significant.
Results
VGLL4 expression decrease accompanied with miR-222 expression increase in GC tissues
To confirm the VGLL4 expression pattern in GC, we firstly detected VGLL4 expression in freshly collected eight pairs of human GC and adjacent non-cancer tissues. As shown in Figure 1A, VGLL4 expression was significantly lower in GC tissues than in paired non-cancer tissues in mRNA and protein levels. VGLL4 has been reported to directly compete with YAP for binding TEADs to inhibit their transcriptionally activity, consistent with which, here we observed and elevated expression of YAP target genes CTGF and CYR61 in GC tissues than those in paired normal control tissues (Figure 1B). To investigate the mechanisms responsible for VGLL4 downregulation, we focused on the miRNAs mediated gene expression regulation. Potential miRNAs that will target VGLL4 were predicted using the public database-TargetScan (http://www.targetscan.org) and miR-222 with critically conserved binding site was selected for further expression and function confirmation (Figure 1C). Expectedly, the detection of miR-222 expression pattern in above tissues showed miR-222 expression in GC tissues were much higher than in paired non-cancer tissues, which is reverse with VGLL4 expression pattern (Figure 1D). In addition, we detected VGLL4 expression with qRT-PCR and IHC in formalin-fixed paraffin-embedded tissues from 9 normal gastric tissues and 43 GC patients. Results showed VGLL4 mRNA and protein levels were remarkably downregulated in 29 out of 43 GC tissues (Figure 1E and 1F). The results of miR-222 expression detection showed among the 29 VGLL4 negative tissues, 28 tissues represented with overexpressed miR-222 (Figure 1G). Moreover, we found miR-222 levels were inversely correlated with those of VGLL4 expression in GC tissues. In view of these findings, we confirm the decreased expression of VGLL4 in GC, which we propose closely due to miR-222 overexpression.
Figure 1.
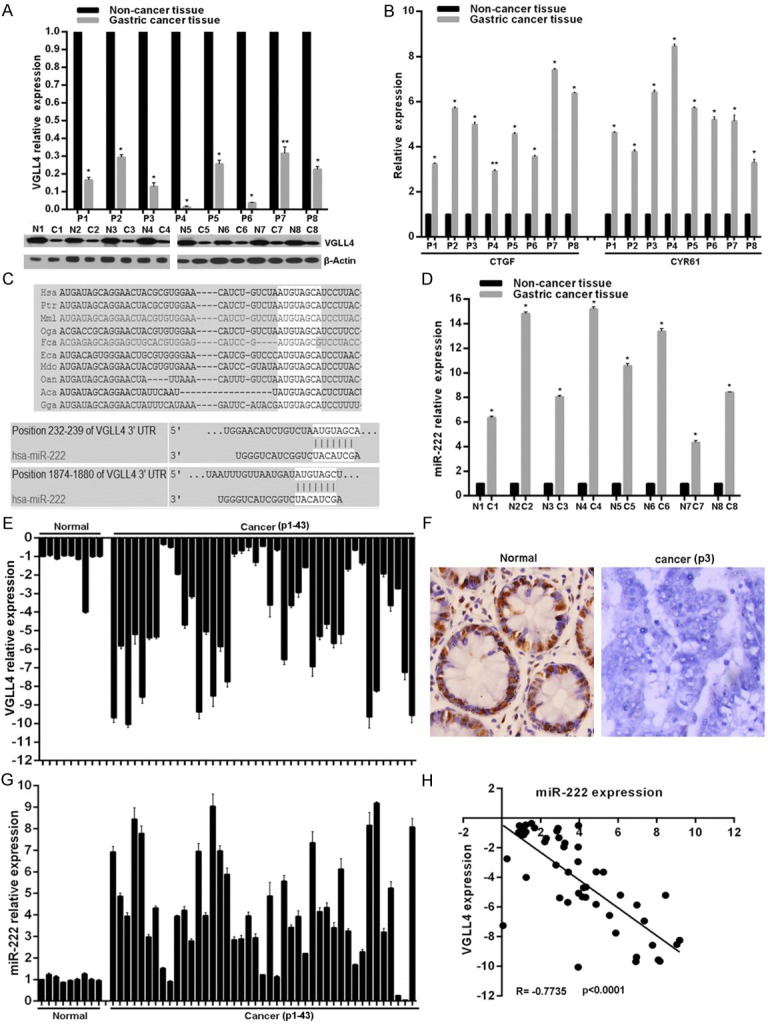
The expression patterns of VGLL4 and miR-222 in GC. A. VGLL4 expression in mRNA and protein levels was determined in eight GC and adjacent non-cancer tissues using qRT-PCR and western blot. B. Relative mRNA expression of YAP-TEADs target genes CTGF and CYR61 was determined by qRT-PCR. C. Schematic of the putative binding sites of miR-222 in 3′-UTR of VGLL4 is presented, which is broadly conserved among vertebrates. D. Relative miR-222 expression was determined by qRT-PCR. E and G. Relative VGLL4 and miR-222 expression in 52 formalin-fixed paraffin-embedded tissues were examined by qRT-PCT. F. Reprehensive images of VGLL4 protein level detected by immunohistochemical staining in 52 formalin-fixed paraffin-embedded tissues. H. Pearson’s correlation analyses between relative miR-222 expression and VGLL4 mRNA level in the 52 tissues. vs related normal control, **p < 0.05, *p < 0.01.
MiR-222 directly targets VGLL4 in GC cells
To confirm the biological role of miR-222 on VGLL4, we first checked the VGLL4 and miR-222 expression patterns in four GC cell lines including MKN-45, BGC-823, MGC-803 and HGC-27. As shown, all the four cell lines possess comparative YAP and TEAD1 protein level, but MKN-45 cells has relative high VGLL4 expression in mRNA and protein levels compared to other three GC cell lines (Figure 2A), but adversely, MKN-45 possesses relative low miR-222 expression (Figure 2B). To determine whether VGLL4 is regulated by miR-222, BGC-823, MGC-803 and HGC-27 cell lines were selected for further experiments and were transfected with miR-222 inhibitor. As shown, miR-222 inhibitor significantly increased VGLL4 mRNA and protein levels in BGC-823, MGC-803 and HGC-27 cell lines (Figure 2C), but led to YAP target genes CTGF and CYR61 mRNA decrease (Figure 2D). To assess whether VGLL4 is a direct target of miR-222 via the 3’-UTR binding site, the luciferase reporter vectors with the putative VGLL4 3’-UTR target site for miR-222 downstream of the luciferase gene (pMir-VGLL4-Wt, set as wild-type) and mutant version with a deletion of 7bp in the seed region (pMir-VGLL4-Mut) were constructed. As shown in Figure 2E, miR-222 inhibitor in BGC-823, MGC-803 and HGC-27 cell lines significantly enhanced luciferase activity of the vector with the wild-type VGLL4 3’-UTR, but the mutant version abrogated the suppressive ability of miR-222. These results strongly suggest miR-222 negatively regulates VGLL4 expression via direct binding to putative binding site in the VGLL4 3’-UTR region.
Figure 2.

miR-222 targets VGLL4 in GC cells. A and B. The expression pattern of VGLL4, YAP, TEAD1 and miR-222 were examined using qRT-PCR or western blot in GC cell lines. C. The expression change of VGLL4 in mRNA and protein levels, and YAP target genes after miR-222 inhibitor transfection in GC cell lines were determined by qRT-PCR or western blot. D. Relative mRNA expression of YAP-TEADs target genes CTGF and CYR61 was determined by qRT-PCR. E. Luciferase reporter assay in GC cell lines cotransfected with miR-222 inhibitor, a luciferase reporter containing wild-type VGLL4 3’-UTR or a mutant version, and a renilla luciferase reporter for normalization. The mean of the results from cells transfected with pMir- VGLL4-Wt and inhibitor-control was set as 1. vs related control, *p < 0.01.
MiR-222 promotes GC cells proliferation and invasion via downregulating VGLL4 expression
As shown above, miR-222 was up-regulated in GC tissues and cell lines, which is negatively associated with VGLL4 expression. To explore the role of miR-222 and whether miR-222 exerts its effect via regulating VGLL4 expression, we firstly screened the VGLL4 specific siRNAs and found si-2# which was selected for followed experiments significantly attenuated VGLL4 protein level in MKN-45 cells (Figure 3A). The effects of miR-222 inhibitor transfection combined with/no VGLL4 siRNA on proliferation and invasion was measured using MTS assay and Transwell assay. We found miR-222 inhibition significantly inhibited the proliferation and invasion potential of BGC-823, MGC-803 and HGC-27 GC cell lines, but VGLL4 knockdown impaired the effects of miR-222 inhibitor on proliferation and invasion in GC cell lines (Figure 3B-D).
Figure 3.

VGLL4 knockdown reveres the effect of miR-222 inhibitor in GC cell lines on proliferation and in vasion. A. Selection of effective siRNAs targeting VGLL4 in GC cells detected by western blot. B. Knockdown of VGLL4 reserved the proliferation inhibition mediated by miR-222 inhibitor in GC cell lines measured with MTS assay. C and D. Knockdown of VGLL4 reserved the invasion inhibition mediated by miR-222 inhibitor in GC cell lines. *p < 0.01.
TEAD1 transcriptionally activates miR-222 expression in GC cells
VGLL4 has been reported to compete with YAP for binding TEADs to inhibit their transcriptionally activity, which also has been confirmed here. We also found VGLL4 downregulation in GC cells and tissues was due to miR-222 overexpression. To verify the presumption whether YAP-TEAD1 feedback transcriptionally regulates miR-222 expression to form positive regulatory loop to maintain constitutive activation of YAP-TEAD1, we firstly selected effective siRNAs targeted TEAD1 and found si-2# transfection obviously led to TEAD1 knockdown in BGC-823, MGC-803 and HGC-27 GC cell lines (Figure 4A), which was used in the followed experiments. As shown, TEAD1 knockdown significantly downregulated miR-222 expression in BGC-823, MGC-803 and HGC-27 GC cell lines (Figure 4B), accompanied with VGLL4 upregulation (Figure 4C). TEAD1 acts as a transcription factor, so we analyzed the response elements of a cohort of transcription factors within a two kilobase region upstream of the miR-222 precursor start site using the online software “The JASPAR database” and found there are two putative TEAD1 binding sites within it (Figure 4D). To confirm the direct association of TEAD1 with the miR-222 promoter, we performed a ChIP assay in BGC-823 cells for the two putative TEAD1 binding sites within the two kilobase region. ChIP results revealed that TEAD1 factually binds to Site A and Site B within the potential miR-222 promoter (Figure 4D). To investigate further the effects of TEAD1 on miR-222 expression, the putative two kilobase miR-222 promoter was cloned into a luciferase reporter vector and luciferase activity assays subsequently performed. As expected, TEAD1 knockdown remarkably suppresses the luciferase activity driven by miR-222 promoter in BGC-823, MGC-803 and HGC-27 GC cell lines (Figure 4E). These results factually imply TEAD1 could physically bind to the promoter region of miR-222 to promote its transcription in GC cells.
Figure 4.

miR-222 is upregulated by TEAD1 in GC cells. A. Effective siRNA targeting TEAD1 was selected. B and C. The expression changes of miR-222 and VGLL4 after TEAD1 knockdown were determined by qRT-PCR. D. A schematic representation of TEAD1 binding sites in the 2kb putative miR-222 promoter upstream of the first base of the miR-222 precursor start site and the first base of the 2kb set as 1. ChIP assays identified TEAD1 binding sites within the putative miR-222 promoter. Primers specific for sites A and B yielded PCR reaction products from TEAD1–DNA immunoprecipitates. The input represents DNA directly after lysis. The PCR reaction product for immunoprecipitates obtained using the RNA Polymerase antibody represents the positive control. E. Luciferase reporter assays revealed changes in luciferase activity after TEAD1 knockdown in GC cells. *p < 0.01.
Discussion
Recently, the roles of VGLL4 in the cancer have attracted much attention. VGLL4 has been considered as a tumor suppressor in several cancer types [13-17]. Importantly, the recent study has indicated that VGLL4 directly competed with YAP for binding TEADs and a peptide mimicking this function of VGLL4 potently suppressed tumor growth of GC in vitro and in vivo [6], and here we confirmed the tumor suppressor role of VGLL4 in GC with inhibitive effects on proliferation and invasion. However, the detailed mechanisms responsible for VGLL4 expression decrease remained to be elucidated. In our present study, we focus on epigenetic roles on expression regulation of VGLL4 and found miR-222 was frequently upregulated in GC tissues and cells accompanied with YAP-TEADs activation, which was adverse with VGLL4 expression pattern. Bioinformatics analysis combined experimental verification demonstrated VGLL4 is a direct target of miR-222 and miR-222 functions as tumor promoter on proliferation and invasion via suppressing VGLL4 expression in GC cells.
In recent times, miRNAs have emerged as an established class of well conserved, short noncoding RNAs (about 19-25 nucleotide long) that play major roles in various biological processes including cell-cycle regulation, cell differentiation, development, apoptosis, angiogenesis and metabolism, by controlling stability and translation of mRNAs in a sequence-specific manner [18,19]. Growing evidence has indicated the important roles of miRNAs the multistep carcinogenesis process through the dysregulation of oncogenes and tumor suppressor genes [20]. MiR-222, known as an oncomiR, has been reported to induce cell growth and cell cycle progression via targeting p27 [21]. Overexpression of miR-222 has been observed in many types of cancers. MiR-22 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing Akt signaling [22]. In glioblastoma, miR-222 overexpression increases the invasive potential by targeting the protein phosphate PTPμ [23]. Upregulation of miR-222 in the clinically more aggressive basal-like subtype compared to luminal subtype of breast cancer promotes epithelial-mesenchymal transition by targeting ADIPOR1 [24]. MiR-222 also has been reported to be involved in GC, but the mechanisms for the biological role, especial for the expression regulation of miR-222 itself still remains to be clarified. Here, we revealed miR-222 promotes GC cells proliferation and invasion via downregulating VGLL4 expression to promote YAP-TEAD1 activation, and importantly, over-activated TEAD1 positively transcriptionally regulates miR-222 overexpression in GC. The precise detailed mechanisms behind the TEAD1 on miR-222 will be explored in our further studies.
Collectively, we confirmed the important role of VGLL4 in GC, and showed the inverse expression association between VGLL4 and miR-222. MiR-222 directly targets VGLL4 and exerts its biological role in GC cells via suppressing VGLL4 expression resulting in YAP-TEAD1 activation. TEAD1 transcriptionally enhances miR-222 expression to maintain the miR-222/VGLL4/YAP-TEAD1 regulatory loop contributing to proliferation and invasion of GC cells.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (8140101483) and General Financial Grant from the China Postdoctoral Science Foundation (2014M552185)
Disclosure of conflict of interest
The authors disclose no potential conflicts of interest.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 3.Jang BG, Kim WH. Molecular pathology of gastric carcinoma. Pathobiology. 2011;78:302–10. doi: 10.1159/000321703. [DOI] [PubMed] [Google Scholar]
- 4.Chang HR, Nam S, Kook MC, Kim KT, Liu X, Yao H, Jung HR, Lemos R Jr, Seo HH, Park HS, Gim Y, Hong D, Huh I, Kim YW, Tan D, Liu CG, Powis G, Park T, Liang H, Kim YH. HNF4α is a therapeutic target that links AMPK to WNT signalling in early-stage gastric cancer. Gut. 2014 doi: 10.1136/gutjnl-2014-307918. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng J, Liang H, Zhang R, Ying G, Xie X, Yu J, Fan D, Hao X. Methylated CpG site count of dapper homolog 1 (DACT1) promoter prediction the poor survival of gastric cancer. Am J Cancer Res. 2014;4:518–27. [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, Wang X, Guo T, Li P, Zhao Y, Ji H, Zhang L, Zhou Z. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25:166–80. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–34. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–74. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faucheux C, Naye F, Tréguer K, Fédou S, Thiébaud P, Théze N. Vestigial like gene family expression in Xenopus: common and divergent features with other vertebrates. Int J Dev Biol. 2010;54:1375–82. doi: 10.1387/ijdb.103080cf. [DOI] [PubMed] [Google Scholar]
- 11.Pobbati AV, Chan SW, Lee I, Song H, Hong W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure. 2012;20:1135–40. doi: 10.1016/j.str.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Hélias-Rodzewicz Z, Pérot G, Chibon F, Ferreira C, Lagarde P, Terrier P, Coindre JM, Aurias A. YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes Chromosomes Cancer. 2010;49:1161–71. doi: 10.1002/gcc.20825. [DOI] [PubMed] [Google Scholar]
- 13.Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev Cell. 2013;25:388–401. doi: 10.1016/j.devcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo T, Lu Y, Li P, Yin MX, Lv D, Zhang W, Wang H, Zhou Z, Ji H, Zhao Y, Zhang L. A novel partner of Scalloped regulates Hippo signaling via antagonizing Scalloped-Yorkie activity. Cell Res. 2013;23:1201–14. doi: 10.1038/cr.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ Australian Pancreatic Cancer Genome Initiative. Chang DK, Biankin AV, Waddell N, Kassahn KS, Grimmond SM, Rust AG, Adams DJ, Jenkins NA, Copeland NG. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109:5934–41. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Gao Y, Li P, Shi Z, Guo T, Li F, Han X, Feng Y, Zheng C, Wang Z, Li F, Chen H, Zhou Z, Zhang L, Ji H. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014;24:331–43. doi: 10.1038/cr.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang W, Yao F, He J, Lv B, Fang W, Zhu W, He G, Chen J, He J. Downregulation of VGLL4 in the progression of esophageal squamous cell carcinoma. Tumour Biol. 2015;36:1289–1297. doi: 10.1007/s13277-014-2701-7. [DOI] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–87. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–9. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 21.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong QW, Ching AK, Chan AW, Choy KW, To KF, Lai PB, Wong N. MiR-222 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing AKT signaling. Clin Cancer Res. 2010;16:867–75. doi: 10.1158/1078-0432.CCR-09-1840. [DOI] [PubMed] [Google Scholar]
- 23.Quintavalle C, Garofalo M, Zanca C, Romano G, Iaboni M, del Basso De Caro M, Martinez-Montero JC, Incoronato M, Nuovo G, Croce CM, Condorelli G. miR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate PTPμ. Oncogene. 2012;31:858–68. doi: 10.1038/onc.2011.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang MS, Yu N, Stinson SY, Yue P, Newman RJ, Allan BB, Dornan D. miR-221/222 targets adiponectin receptor 1 to promote the epithelial-to-mesenchymal transition in breast cancer. PLoS One. 2013;8:e66502. doi: 10.1371/journal.pone.0066502. [DOI] [PMC free article] [PubMed] [Google Scholar]