

Abstract
Introduction
The Tie2/angiopoietin (Tie2/Ang) and vascular endothelial growth factor receptor-ligand systems (VEGFR/VEGF) are recognized to play important roles in the regulation of microvascular endothelial function. Downregulation of these genes during sepsis has been implicated in the pathogenesis of sepsis-related microvascular leak and multiple organ dysfunction syndrome. Mechanisms responsible for dysregulation of angiogenic genes in sepsis are poorly defined.
Methods
Western blot, reverse transcription-polymerase chain reaction, and multiplex chromatin immunoprecipitation platform (Matrix ChIP) were used to investigate serum albumin leak, changes in gene expression, and associated epigenetic alterations in a murine model of acute lung injury-induced sepsis (ALI-sepsis).
Results
Experimental ALI-sepsis induced microvascular leak and downregulation of expression of Angpt1 (Ang1), Tek (Tie2), and Kdr (Vegfr2 or Flk-1) genes in the lung, kidney, and liver. These changes correlate with a decrease in RNA polymerase II density at these genes, and the greatest response was observed in the lung. ALI-sepsis reduced levels of transcription-permissive histone H3 lysine acetylation (H3KAc) at these loci in all examined tissues. Decreases in permissive H3K4m3 and H3Km2 marks were detected only in the lung. In contrast, only minimal alterations in transcription-repressive histone modifications (H3K27m3, H3K9m2, H3K9m3, and H4K20m3) were observed in all tissues.
Conclusions
Our results demonstrate that decreases in transcription-permissive, but not increases in transcription-repressive, histone modifications at Angpt1, Tek, and Kdr are a systemic, rather than a lung-restricted, response, involving key end-organs in experimental ALI-sepsis. Given that ventilator-associated pneumonia is a major cause of sepsis in critically ill patients, elucidation of mechanisms mediating epigenetic alterations during sepsis provides fundamental new insights into the pathogenesis of sepsis-induced microvascular leak and subsequent end-organ injury/dysfunction.
Electronic supplementary material
The online version of this article (doi:10.1186/s13054-015-0943-4) contains supplementary material, which is available to authorized users.
Introduction
Sepsis is a common and devastating complication in critically ill patients and remains a major cause of morbidity and mortality [1]. Pneumonia and ventilator-associated pneumonia (VAP) represent leading causes of sepsis in intensive care units (ICUs) [2–5]. In large part, rapid progression to major end-organ (that is, lung, kidney, and liver) injury/dysfunction is responsible for the high morbidity and mortality of clinical sepsis. The last few decades of intense research and clinical trials have failed to improve clinical outcomes in sepsis management, and there is a pressing need to develop therapies of greater efficacy [5–9]. Historically, severe systemic inflammation—that is, release of tumor necrosis factor (TNF), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), and other inflammatory mediators—was thought to play the major role in sepsis-induced multiple organ dysfunction syndrome (MODS) [10]. However, more recently, endothelial dysfunction and associated microvascular leak have emerged as critical pathogenic mechanisms in sepsis and MODS [9, 11, 12].
Tie receptors and their angiopoietin ligands (Tie2/Ang) and vascular endothelial growth factor receptors and their ligands (Vegfr/Vegf) are two endothelial receptor tyrosine kinase systems that play critical roles in the regulation of angiogenesis and endothelial function [13, 14]. Components of the Tie2/Ang and Vegfr/Vegf systems act, and are predominantly expressed, in the endothelium [13]. Although the role of abnormal Tie2/Ang signaling in mediating detrimental microvascular leak in sepsis and MODS is well established [15], recent studies have also implicated altered Vegfr/Vegf signaling in the pathogenesis of sepsis and MODS [16]. Sepsis-induced endothelial dysfunction driven by dysregulated Tie2/Ang and Vegfr/Vegf signaling reflects, in part, decreased levels of these angiogenic mediators and their receptors that, in turn, result from downregulation of these genes [15, 17]. Both Angpt1 and Angpt2 bind to the Tie2 receptor but often trigger opposite biological effects. Whereas Angpt1 can be protective, Angpt2 can worsen outcomes in sepsis, observations that are being used to develop biomarkers in sepsis [18–20]. Despite increasing appreciation of the role played by angiogenic genes in sepsis-induced MODS, the molecular mechanisms underlying their transcriptional regulation in this complex syndrome remain poorly understood.
Epigenetic processes play a critical role in transcriptional regulation by influencing genomic regulatory sequences in a cell type-specific and extracellular environment-dictated manner. Epigenetic language encompasses covalent histone modifications and DNA CpG methylation. Together these epigenetic modifications control chromatin accessibility to factors that regulate RNA polymerase II (Pol II) transcription (Additional file 1: Figure S1) [21, 22]. Although epigenetic regulation is one of the most intensely studied fields of biology today, relatively little is known about epigenetic regulation in sepsis and MODS. Identification of transcription factors and epigenetic modifiers engaged at injury-related genes could allow the development of small-molecule drugs to therapeutically target these factors in an effort to improve clinical outcomes in sepsis and MODS. In this regard, Nicodeme et al. showed that an epigenetic drug, bromodomain and extra terminal domain inhibitor (I-BET), decreased inflammatory responses and protected mice from lipopolysaccharide-induced sepsis and death [23].
Given the importance of pneumonia as a leading cause of sepsis in critically ill patients in the ICU, we set out to define transcriptional and epigenetic changes at genes encoding components of the Tie2/Ang and Vegfr/Vegf systems in the lung, kidney, and liver in an experimental mouse model of acute lung injury-induced sepsis (ALI-sepsis). We hypothesized that experimental ALI-sepsis (employing a previously validated murine model of VAP that combines Staphylococcus aureus lung infection and mechanical ventilation, resulting in lung injury and extra-pulmonary organ dysfunction [24]) would be associated with both decreased permissive and increased repressive epigenetic modifications at downregulated genes in the Tie2/Ang and Vegfr/Vegf systems in the lung. Contrary to our hypothesis, we found that ALI-sepsis was associated with only decreased permissive epigenetic modifications at these genetic loci in the lung. Moreover, these epigenetic changes were not confined to the lung but extended to the kidney and liver, two prominent extra-pulmonary end-organs affected in sepsis-related MODS [24, 25].
Methods
Reagents
Bovine serum albumin, phosphate-buffered saline (PBS), salmon sperm DNA, and protein A were from Sigma-Aldrich (St. Louis, MO, USA), and proteinase K was from Invitrogen (part of Thermo Fisher Scientific, Waltham, MA, USA). Ninety-six-well polypropylene plates for multiplex chromatin immunoprecipitation platform (Matrix ChIP) were from BioExpress (Kaysville, UT, USA). Formaldehyde, ethanol, NaCl, EDTA, Triton X-100, NP-40, Tris, leupeptin, PMSF, p-nitrophenyl phosphate, NaF, Na3VO4, Na2MoO4, and β-glycerophosphate were from Sigma-Aldrich. The antibodies were commercially available and are listed in Additional file 1: Table S2.
Animal experiments
The specific protocol used in this study was approved by the Institutional Animal Care and Use Committee at the University of Washington. S. aureus was prepared and mice were inoculated and mechanically ventilated as previously described [25]. Briefly, for each experiment, a frozen aliquot of methicillin-sensitive S. aureus originally isolated from a patient with bacteremia was thawed and cultured on a sheep blood agar plate. The next morning, a single colony was selected and cultured overnight at 37 °C in tryptic soy broth. The morning after that, the bacteria were washed twice with saline and then resuspended in 2 mL of filtered, distilled water. Serial log dilutions were made and turbidity measured by OD540 (optical density at 540 nm). With a standard curve generated previously with OD540 versus bacterial concentration determined by quantitative culture, a working solution of approximately 2 × 108 ± 10% was prepared with filtered, distilled water.
For each experiment, four mice were used. Two mice were anesthetized with 5% isoflurane while suspended by the front teeth at a 60° angle. The tongue was extruded with forceps, and 50 μL of S. aureus (approximately 107 colony-forming units) was deposited in the oropharynx. After visual confirmation of aspiration, mice were placed on a nose cone with 5% isoflurane and intubated via tracheostomy with a 20-gauge blunt metal catheter. Intubated mice were connected to a MiniVent rodent ventilator (Harvard Biosciences, Hollison, MA, USA) and mechanically ventilated with a tidal volume of 10 mL/kg, a respiratory rate of 150 breaths per minute, a fraction of inspired oxygen (FiO2) of 0.21, and no end-expiratory pressure. Anesthesia was maintained with isoflurane (1.5%). Neuromuscular blockade was induced with pancuronium (0.02 mg in 0.2 mL subcutaneously). A second dose of pancuronium (0.01 mg in 0.1 mL) was given after 2 hours. Control mice were maintained in their cages and given an equivalent volume of PBS subcutaneously at time 0 and 2 hours.
After 6 hours, mice were deeply anesthetized with 5% isoflurane and euthanized by cardiac puncture and exsanguination. Lungs, kidneys, and livers were harvested, flash-frozen, and stored at −80 °C.
Microvascular albumin leak measurements
Microscular leak was assessed by tissue albumin levels. Briefly, frozen tissues were homogenized in low detergent buffer (150 mM NaCl, 50 mM Tris, 5 mM EDTA, 0.005% NP-40, 0.01% Triton X-100, pH 7.5) by using a Bioruptor (Diagenode, Seraing, Belgium) (15 minutes, high power 30 seconds ON 30 seconds OFF, 4 °C). Homogenate was centrifuged (10,000 rpm, 5 minutes, 4 °C), and supernatant was boiled in SDS Laemmli buffer. Albumin levels were assessed by SDS-PAGE and Western blot analysis (anti-albumin SC-46293; Santa Cruz Biotechnology, Inc., Dallas, TX, USA; 1:2,000 dilution). Anti-β-actin antibody (A5441; Sigma-Aldrich; 1:5,000 dilution) was used as loading control. Albumin and β-actin band intensities were measured from scanned membrane images by using ImageJ64 software.
RNA extraction and cDNA synthesis
RNA was extracted from frozen tissue fragments by using Trizol reagent in accordance with the protocol of the manufacturer. To synthesize cDNA, 400 ng of Trizol-extracted total RNA was reverse-transcribed with 200 units of MMLV reverse transcriptase (Invitrogen) and oligo-dT primers in 20-μL reactions. Reverse transcription (RT) reactions were diluted 100-fold with water prior to use in quantitative polymerase chain reaction (PCR) [26].
Chromatin preparation and multiplex Matrix ChIP assay
The same protocol was used for all organs. The multiplex microplate Matrix ChIP method was previously described [27–29]. Briefly, for ChIP assays, frozen tissue fragments (10 to 20 mg) were cross-linked with formaldehyde, and chromatin was sheared by using a Bioruptor [27]. ChIP assays were done by using protein A-coated 96-well polypropylene microplates as described before [27]. Eluted DNA (1 to 2 μL) was used in 2-μL real-time PCRs (ABI7900HT). All PCRs were run in quadruplicates. PCR calibration curves were generated for each primer pair from a dilution series of total mouse genomic DNA. The PCR primer efficiency curve was fit to cycle threshold (Ct) versus log [genomic DNA concentration] by using an r2 best fit. DNA concentration values for each ChIP and input DNA sample were calculated from their respective average Ct values. Final results are expressed as fraction of input DNA [28]. Matrix ChIP PCR primers are shown in Additional file 1: Table S1, and a list of antibodies is shown in Additional file 1: Table S2.
Statistical analysis and data display
GraphGrid Excel-based software tools were used to acquire, store, and analyze large data sets generated by the high-throughput Matrix ChIP platform [29]. Pair-wise t test was used to measure statistically significant differences, which are represented by the size of a circle for each comparison made: P <0.05 by a small circle, P <0.01 by a large circle, and non-significance by no circle [29].
Results
Serum albumin leak in the lung, kidney, and liver in an experimental ALI-sepsis model
We previously developed a clinically relevant experimental murine model of ALI-sepsis in which mice with S. aureus pneumonia progress to sepsis-induced MODS when mechanically ventilated [25]. This model simulates a common clinical scenario in the ICU in which mechanically ventilated critically ill patients develop sepsis-induced MODS from a pulmonary source of infection. In this model, profound systemic release of multiple inflammatory mediators occurs within 6 hours, and it is not associated with extra-pulmonary dissemination of S. aureus [25]. These mediators or systemic release of pathogen-associated molecular pattern molecules (PAMPs) or endogenous danger-associated molecular pattern molecules (DAMPs/alarmins) or a combination of these could affect multiple organs. This ALI-sepsis model is characterized by increased serum creatinine and transaminases, indicating renal and hepatic impairment, respectively [25]. Endothelial dysfunction is another hallmark of sepsis [30, 31]. Serum albumin leak is often used to assess integrity of the microvascular barrier. Western blot analysis of proteins extracted from fragments of the lung, kidney, and liver showed higher tissue albumin levels (normalized to β-actin) in ALI-sepsis mice compared with controls (Fig. 1). The magnitude of observed differences is comparable to those reported for albumin leak in other experimental models of sepsis [32]. Thus, these tissue albumin measurements provide evidence for lung, kidney, and liver endothelial dysfunction and associated microvascular leak in this ALI-sepsis model.
Fig. 1.
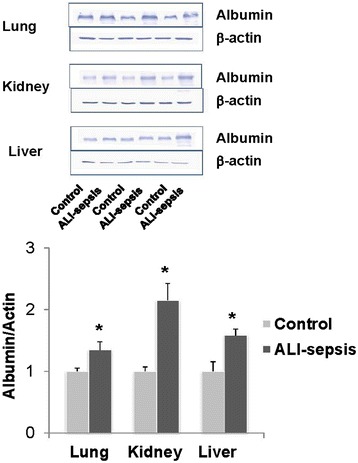
Increased albumin leak in the lung, kidney, and liver in an experimental acute lung injury-induced sepsis (ALI-sepsis) model. Combined Staphylococcus aureus pneumonia and mechanical ventilation in anesthetized mice was performed as previously described (Methods) [25]. Mice without any intervention served as controls. After 6 hours of treatment, the lungs, kidneys, and livers were harvested, flash-frozen, and stored at −80 °C. Western blot analysis of tissue albumin, top panel, was used to assess microvascular leak (Methods). Albumin and β-actin band intensities were measured from scanned membrane images by using ImageJ64 [54]. Albumin-to-β-actin ratios normalized to control tissues were calculated for each tissue; the graph depicts mean ± standard error of the mean (n = 3 mice in each group). *P <0.01 (t test) bottom panel
Experimental ALI-sepsis induced downregulation of angiogenic gene expression in the lung and extra-pulmonary organs
We examined changes in expression of several angiogenic genes in the lung, kidney, and liver. As shown in Fig. 2, mRNA levels of Tek receptor and its main cognate agonist/ligand, Angpt1, decreased in all three organs after 6 hours of experimental ALI-sepsis. These changes were most prominent in the lungs.
Fig. 2.
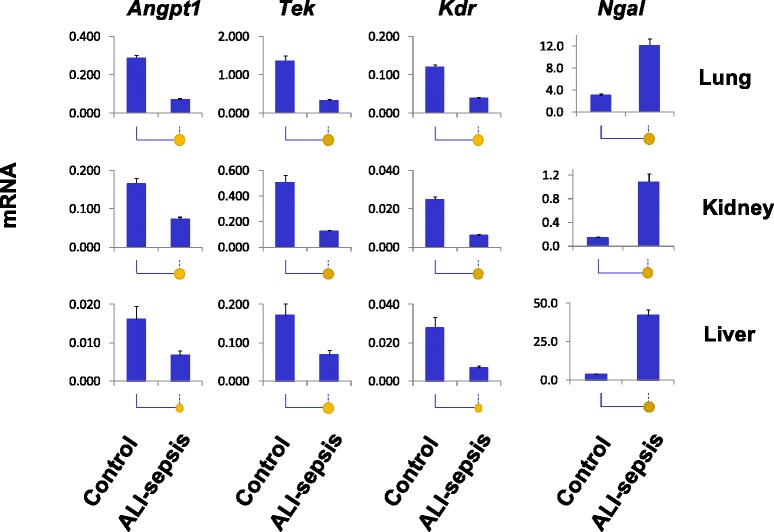
Analysis of changes in lung, kidney, and liver mRNA expression in experimental acute lung injury-induced sepsis (ALI-sepsis). Total RNA from ALI-sepsis mouse lungs, kidneys, and livers was used in reverse transcription reactions with oligo-dT primers. cDNA was used in real-time polymerase chain reaction with gene-specific primers (Additional file 1: Table S1). mRNA level of a given gene in each sample was normalized to β-actin transcript. Data are represented as mean ± standard error of the mean (n = 12 mice in each group). Statistical differences between two means (P value) are shown by the size of the solid circles: P <0.05 by a small circle, P <0.01 by a large circle, and non-significance by no circle
Vegfr2 is a homodimer receptor composed of Flk1 protein encoded by the Kdr gene [33]. Sepsis-induced downregulation of Kdr mRNA in the lung, kidney, and liver was similar in magnitude to the Tek mRNA changes (Fig. 2). In contrast to the marked downregulation of Angpt1, Tek, and Kdr expression in the lung, reductions of Angpt2, Vegfα, and Flt1 (Vegfr1) mRNAs were small (Additional file 1: Figure S2). These genes were not further pursued in this study.
Neutrophil gelatinase-associated lipocalin (Ngal) (also known as Lcn2) is expressed in neutrophils and also in solid organs, where it is thought to provide protection against bacterial infection by sequestration of iron, thereby limiting bacterial growth [34]. Ngal has also gained much interest as a biomarker of acute kidney injury [35]. Ngal transcript levels increased in the lung, kidney, and liver in septic mice. We used upregulated Ngal expression as a pathophysiologically relevant control for transcriptional and epigenetic changes at downregulated angiogenic genes.
Experimental ALI-sepsis decreased Pol II density at angiogenic genes
Sepsis-induced changes in mRNA levels (Fig. 2) can result from changes in transcription or from changes in mRNA stability/degradation [29, 36]. To examine this issue, we used a monoclonal antibody to the Pol II C-terminal domain (CTD) in Matrix ChIP assays [28] to assess density of Pol II at angiogenic genes in the lungs, kidneys, and livers of septic mice. Matrix ChIP measurements were done at 5′ and 3′ ends of genes (Fig. 3A). After 6 hours of ALI-sepsis, Pol II levels at both the 5′ and 3′ ends of downregulated Angpt1, Tek, and Kdr genes were decreased in all three organs compared with control animals (Fig. 3B). ALI-sepsis-induced decreases were most pronounced in the lung. In contrast to the angiogenic genes, Pol II levels at the upregulated Ngal locus were higher in ALI-sepsis animals compared with controls (Fig. 3B). The magnitude of sepsis-induced Pol II changes at all the loci matched cognate mRNA changes (Fig. 2), suggesting that, at least in part, changes in mRNA levels reflected altered transcription of Angpt1, Tek, Kdr, and Ngal genes.
Fig. 3.
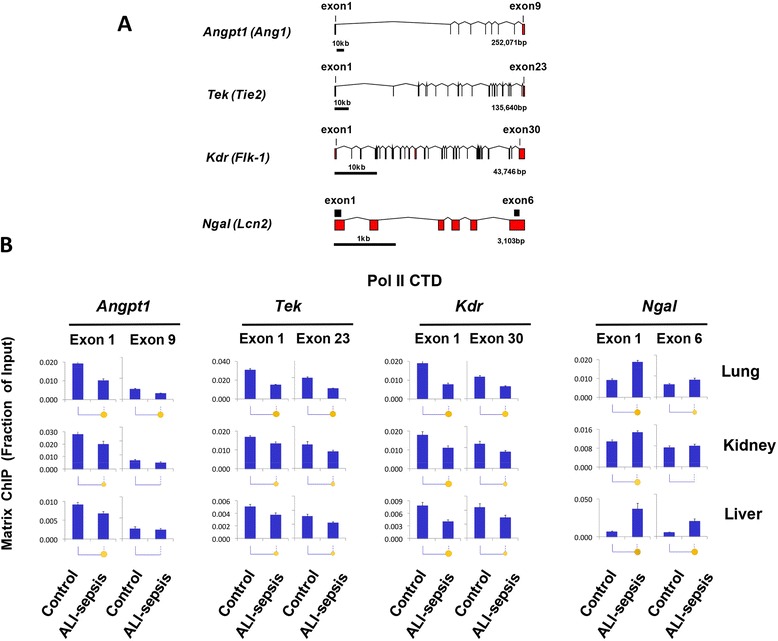
RNA polymerase II (Pol II) changes at Angpt1, Tek, Kdr, and Ngal genes in experimental acute lung injury-induced sepsis (ALI-sepsis). (A) Intron-exon organization of genes analyzed in these studies. Exons are shown as rectangles and introns as lines. Chromatin immunoprecipitation platform polymerase chain reaction (ChIP PCR) amplicons are shown as black boxes. (B) ALI was induced in anesthetized mice as in Fig. 1. Mice without any intervention served as controls. After 6 hours of treatment, the lungs, kidneys, and livers were harvested, flash-frozen, and stored at −80 °C. Sheared cross-linked chromatin was prepared from frozen lungs, kidneys, and livers. ChIP analysis was done by using an antibody to the Pol II C-terminal domain (CTD). ChIP DNA samples were analyzed by real-time quantitative PCR with primers to the first and last gene exons (A). Data represent mean ± standard error of the mean (n = 12 animals from each group), expressed as a fraction of input. Statistical differences are shown as in Fig. 2
Experimental ALI-sepsis decreased levels of transcription-permissive histone modifications at angiogenic genes
Compact chromatin structure is maintained, in part, by electrostatic interaction of positive histone amino acid residues and negatively charged DNA. Acetylation of histone lysine residues removes positive charges, reducing chromatin compaction and thereby permitting higher rates of Pol II transcription (Additional file 1: Figure S1) [37]. Histone acetyl-lysine modifications also serve as docking sites for factors that promote transcription via bromodomain interactions [38]. Histone lysine acetylation is higher at the 5′ compared with the 3′ ends of genes, suggesting their importance in transcription initiation and early steps of elongation [39]. Reduced transcription of angiogenic genes in organs of ALI-sepsis mice (Figs. 2 and 3) could conceivably be associated with deacetylation of histones. We used antibody against acetylated lysines 9 and 14 of histone H3 (H3KAc) in Matrix ChIP assay to assess changes in this transcriptionally permissive modification (Fig. 4). In the lungs of ALI-sepsis mice, H3KAc levels at 5′ ends of Angpt1, Tek, and Kdr were lower compared with control animals. Similar ALI-sepsis-induced histone H3 lysine deacetylation was observed in the kidney and the liver, but the decreases were smaller and at some sites not statistically significant. Total histone H3 levels at the genes remained unaltered or tended to be higher in organs of ALI-sepsis animals (Additional file 1: Figure S3), suggesting that reduced H3KAc levels did not reflect loss of nucleosomes. There was an increase in H3KAc at Ngal in all three organs, and the magnitude of change was greater in the lung and liver compared with the kidney. In summary, sepsis-induced H3KAc changes (Fig. 4) generally paralleled Pol II levels at the genes (Fig. 3), suggesting a functional link between ALI-sepsis-induced changes in histone lysine acetylation and rates of transcription.
Fig. 4.
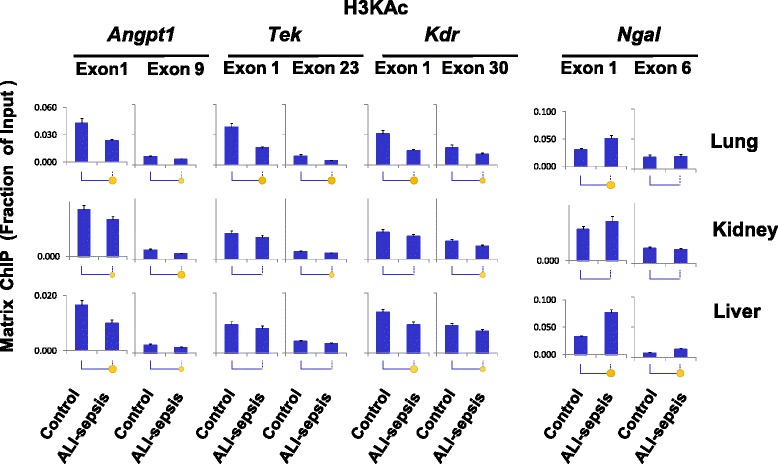
Matrix ChIP analysis of changes in histone H3 lysine acetylation (H3KAc) levels at Angpt1, Tek, Kdr, and Ngal genes in experimental acute lung injury-induced sepsis (ALI-sepsis). Sheared cross-linked lungs, kidneys, and livers were assayed by using an antibody to acetylated histone H3 lysine 9 and 14, H3KAc. ChIP analysis was done as in Fig. 3. Data represent mean ± standard error of the mean (n = 12 animals from each group), expressed as a fraction of input. Statistical differences between two means (P value) are shown as in Fig. 2. ChIP, chromatin immunoprecipitation platform
Di- and tri-methylation of histone H3 lysine 4, H3K4m2 and H3K4m3, are other well-studied permissive chromatin modifications [21]. Histone lysine methylation provides docking sites for chromatin modifiers but, unlike acetylation, does not alter histone charge [37]. It has been suggested that H3K4m2 and H3K4m3 modifications open chromatin by recruitment of ATP-dependent chromatin remodelers [40]. These modifications are enriched at the 5′ ends of genes [41]. Consistently, for all of the angiogenic genes examined, H3K4m2 and H3K4m3 were higher at the 5′ compared with the 3′ ends in all organs (Figs. 5 and 6). Pol II decreases (Fig. 3) at Tek and Kdr genes in mice with ALI-sepsis were accompanied by decreases in H3K4m2 (Fig. 5) and H3K4m3 (Fig. 6) signals in the lung. In contrast, H3K4m2 and HK4m3 levels were largely unaltered by ALI-sepsis in the kidney and liver. Thus, ALI-sepsis-induced decreases in H3K4m2 and HK4m3 may account for the greater sepsis-induced repression of Angp1, Tek, and Kdr transcription in the lung compared with extra-pulmonary organs (Fig. 3).
Fig. 5.
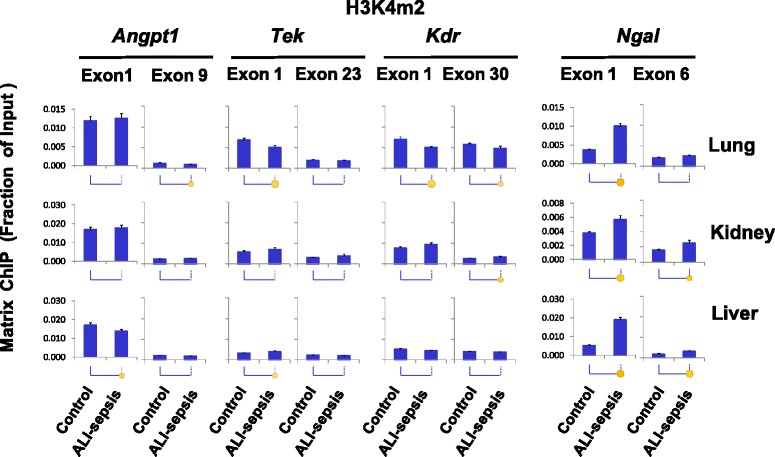
Changes in histone H3 lysine 4 di-methylation (H3K4m2) in experimental acute lung injury-induced sepsis (ALI-sepsis). Sheared cross-linked lung, kidney, and liver chromatin from mice were assayed by using an antibody to di-methylated histone H3 lysine 4, H3K4m2. Chromatin immunoprecipitation platform (ChIP) analysis was done as in Fig. 3. Data represent mean ± standard error of the mean (n = 12 animals from each group), expressed as a fraction of input. Statistical differences are shown as in Fig. 2
Fig. 6.
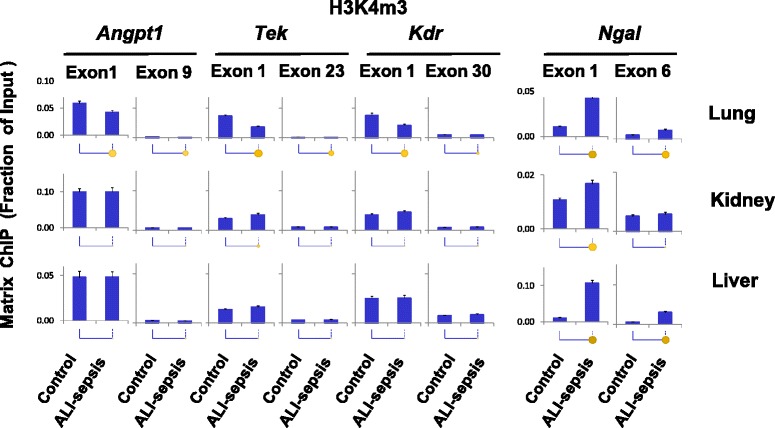
Changes in histone H3 lysine 4 tri-methylation (H3K4m3) in experimental acute lung injury-induced sepsis (ALI-sepsis). Sheared cross-linked lung, kidney, and liver chromatin from mice were assayed by using an antibody to H3K4m3. Chromatin immunoprecipitation platform (ChIP) analysis was done as in Fig. 3. Data represent mean ± standard error of the mean (n = 12 animals from each group), expressed as a fraction of input. Statistical differences are shown as in Fig. 2
These results suggest organ-specific differences in epigenetic responses to ALI-sepsis. Some of the differences may reflect the magnitude of local lung versus remote responses to ALI-sepsis. Consistent with elevated transcription, ALI-sepsis-induced increases in H3K4m2 and H3K4m3 levels were observed at Ngal in the lung, kidney, and liver. The magnitude of H3K4m2 (Fig. 5) and H3K4m3 (Fig. 6) changes at Ngal correlated well with the observed ALI-sepsis-induced increase in Pol II levels at this locus in the different organs (Fig. 3). Furthermore, the magnitude of ALI-sepsis-induced H3K4m2 and H3Km3 increases at Ngal in the different organs correlated with changes in H3 acetylation levels at this locus. These observations imply that these two permissive epigenetic modifications act in concert to generate robust Ngal transcriptional responses during ALI-sepsis.
Results of the above experiments imply that ALI-sepsis-induced decreases in both permissive histone lysine acetylation and methylation play a role in aberrant transcription of angiogenic genes in the lung. In contrast, alterations in histone lysine acetylation appear more important than alterations in histone methylation in aberrant transcription of angiogenic genes in extra-pulmonary organs.
Experimental ALI-sepsis induced minimal transcription-repressive histone modifications at angiogenic genes
We examined several histone modifications (H3K27m3, H3K9m2, H3K9m3, and H4K20m3) associated with repressed genes (Fig. 7 and Additional file 1: Figures S4-S6). These modifications downregulate gene expression by increasing chromatin compaction, thereby decreasing access of transcription factors and impeding Pol II recruitment [21, 37, 42]. In the ALI lung, there was an increase in H3K27m3 levels at the 5′ end of Kdr but not at other angiogenic loci. In the kidney, ALI-sepsis was associated with small increases in H3K27m3 levels at Angpt1, Tek, and Kdr, but in the liver the increase was detected at the Angpt1 gene. At the upregulated Ngal locus, H3K27m3 loss was detected only in the lung.
Fig. 7.
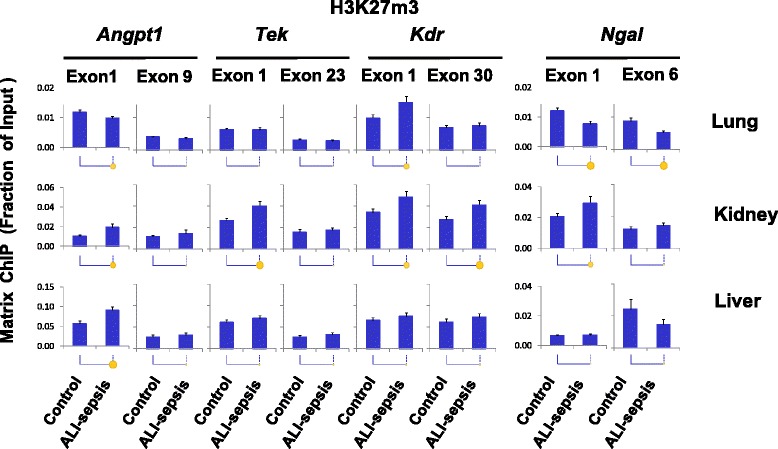
Changes in histone H3 lysine 27 tri-methylation (H3K27m3) in experimental acute lung injury-induced sepsis (ALI-sepsis). Sheared cross-linked lung, kidney, and liver chromatin from mice were assayed by using an antibody to H3K27m3. Chromatin immunoprecipitation platform (ChIP) analysis was done as in Fig. 3. Data represent mean ± standard error of the mean (n = 12 animals from each group), expressed as a fraction of input. Statistical differences between are shown as in Fig. 2
H3K9m2 and H3K9m3 are well-studied repressive marks that are enriched in compacted heterochromatin [42, 43]. There were no consistent sepsis-induced changes in H3K9m2/3 levels at the angiogenic genes (Additional file 1: Figures S4 and S5), suggesting that these epigenetic marks did not contribute to Angp1, Tie2, and Kdr repression in the organs examined. ALI-sepsis caused H3K9m2 and H3K9m3 de-methylation, albeit at low levels, at Ngal in the lung and kidney, and this could potentially contribute to increased Ngal transcription in these organs.
Histone H4 lysine 20 tri-methylation, H4K20m3, is another repressive mark enriched in compacted chromatin [21, 44]. Matrix ChIP analysis showed no consistent sepsis-induced changes in H4K20m3 at angiogenic and Ngal genes in any of the organs (Additional file 1: Figure S6).
Taken together, these results suggest that changes in repressive epigenetic modifications play an insignificant role in ALI-sepsis-induced downregulation of pulmonary and extra-pulmonary Angp1, Tie2, and Kdr. However, decreased repressive histone modifications may contribute to sepsis-induced Ngal expression in some organs (Fig. 7 and Additional file 1: Figures S4-S6).
Discussion
Sepsis is characterized by downregulation of Angpt1, Tek, Kdr, and other angiogenic genes. These alterations have been implicated in the pathogenesis of sepsis-induced microvascular leak and end-organ injury/dysfunction [17, 45, 46]. Here, we present evidence that dysregulation of Angpt1, Tek, and Kdr caused by experimental ALI-sepsis is associated with a reduction in levels of Pol II at these loci in the lung, kidney, and liver, thereby providing, for the first time, evidence for the systemic transcriptional repression of these angiogenic genes in sepsis. This finding prompted our hypothesis that ALI-sepsis would be associated with both decreased permissive and increased repressive epigenetic modifications at Angpt1, Tek, and Kdr.
To explore the epigenetic basis of sepsis-induced transcriptional dysregulation of angiogenic genes, we compared several important permissive and repressive histone modifications with Pol II levels along these genes. There was positive correlation between ALI-sepsis-induced reduction of Pol II levels (Fig. 3) at Angpt1, Tek, and Kdr and deacetylation of H3KAc at these loci in the lung, kidney, and liver (Fig. 4). There was also concordance between ALI-sepsis-induced increases in Pol II and H3KAc levels at the upregulated Ngal locus in these organs. Given that histone lysine acetylation at gene promoters is a well-established transcription-permissive epigenetic modification [37], this analysis suggests that H3KAc changes play a mechanistic role in sepsis-induced dysregulation of angiogenic genes in the lung, kidney, and liver. The observed ALI-sepsis-induced Pol II decline (Fig. 3) also correlated with loss of H3K4m2 (Fig. 5) and H3K4m3 (Fig. 6) at angiogenic genes in the lung. Changes in these modifications at angiogenic genes in the other organs were minimal. Thus, although loss of these permissive modifications in the lung could play a role in sepsis-induced downregulation of pulmonary angionenic genes, their relative contribution to transcriptional dysregulation in extra-pulmonary end-organs in this model of ALI-sepsis appears minimal.
In contrast to the permissive epigenetic changes described above, experimental ALI-sepsis induced relatively few changes in repressive epigenetic modifications at Angpt1, Tek, and Kdr in the lung and extra-pulmonary end-organs. Except for changes in H3K27m3 levels in septic organs (Fig. 7), analysis of repressive post-translational modifications H3K9m2, H3K9m3, and H4K20m3 (Additional file 1: Figures S4-S6) revealed no significant ALI-sepsis-induced changes at the angiogenic genes in the lung, kidney, and liver. ALI-sepsis-induced H3K27m3 alterations were generally minimal and present primarily in the kidney as opposed to the other organs. Thus, alteration in repressive histone modifications may not play a major role in dysregulation of angiogenic genes in this model of ALI-sepsis and does not appear to be an epigenetic hallmark of sepsis, at least at the examined time point, contrary to our initial hypothesis.
There are several limitations in the present study. Given the remaining pre-analytical technical challenges in preparing a large number of samples for epigenetic analysis, we assessed only one time point of ALI-induced sepsis and have focused on Angpt1, Tek, and Kdr, which exhibited the most pronounced mRNA downregulation. At the time point examined, changes in Angpt2, Vagfα, and Flt1 expression were small (Additional file 1: Figure S2). Sepsis-induced alteration in angiogenic gene expression can be time-dependent and therefore it is possible that either downregulation [45] or upregulation [46] of Angpt2, Vagfα, and Flt1 was more robust at other time points. To evaluate whether mechanical ventilation has an independent effect on genes of interest, we analyzed microarray data from our previously published study that included normal mice and mice subjected to mechanical ventilation for 6 hours with the same protocol used in the present study, including anesthesia and paralysis [47]. Importantly, mechanical ventilation alone had a very modest effect on the expression of Tek, Kdr, Angpt1, Angpt2, or Ngal compared with control, unventilated mice (data not shown). Given the small changes in mRNA expression, we did not carry out epigenetic experiments with a mechanical ventilation-only group in the present study. It is anticipated that novel pre-analytical approaches and further advancement in epigenetic platforms as well as single-cell analysis will enable transcriptional and epigenetic studies of genes whose expression is not as markedly altered in whole tissue samples. Sepsis-induced reductions of angiogenic protein levels have previously been reported in the lung [46, 48, 49], kidney [50, 51], and liver [46]. Similar observations were made for sepsis downregulation of Kdr protein in the lung [45, 52]. Translation or protein degradation or both could be other important layers controlling expression of angiogenic genes in sepsis.
The level of any given histone modification reflects the balance between epigenetic “writers” and “erasers” [21]. Tek and Kdr are expressed predominantly in endothelial cells, whereas Angpt1 is expressed primarily in pericytes [17]. Considering the analysis performed in this study, we propose a model of sepsis-induced epigenetic modifications of angiogenic genetic loci at major target end-organs as depicted in Fig. 8. In contrast to ALI-sepsis-induced multi-organ increase in Ngal (Fig. 2) as well as nuclear factor-kappa-B (NF-κB)-responsive genes, including Tnf, Mcp-1, IL-6, and Cox2 (Additional file 1: Figure S7), the angiogenic gene expression decreased in the lung, kidney, and liver. Selective downregulation of these angiogenic genes may reflect ALI-sepsis effects on endothelial cell-specific transcription factors that control Pol II recruitment and epigenetic modifiers. Constitutively, the epigenetic balance is shifted toward higher levels of permissive epigenetic marks at angiogenic loci in both endothelial cells and pericytes by the activity of cell type-specific and general transcription factors. During sepsis, systemic release of PAMPs [42] or DAMPs [21, 29] or both initiates signal pathways that reduce levels of activating transcription factors at these angiogenic loci (Fig. 8). As a result, there is a reduction of permissive histone modifications at Tek and Kdr in endothelial cells and Angpt1 in pericytes, resulting in repression of Pol II transcription in both cell types (Fig. 8). Among the histone modifications examined, lysine acetylation was the most consistent sepsis-responsive epigenetic modification (Fig. 4). It is conceivable that transcription factors that control histone lysine acetyltransferases (HATs) and deacetylases (HDACs) are more sensitive to mediators of sepsis but that repressive histone lysine modifiers are relatively resistant. With increasing availability of small molecules that target different classes of chromatin modifiers, identification of epigenetic enzymes responsive to septic signals will be an important translational goal to elucidate key molecular mechanisms underlying the pathogenesis of clinical sepsis.
Fig. 8.
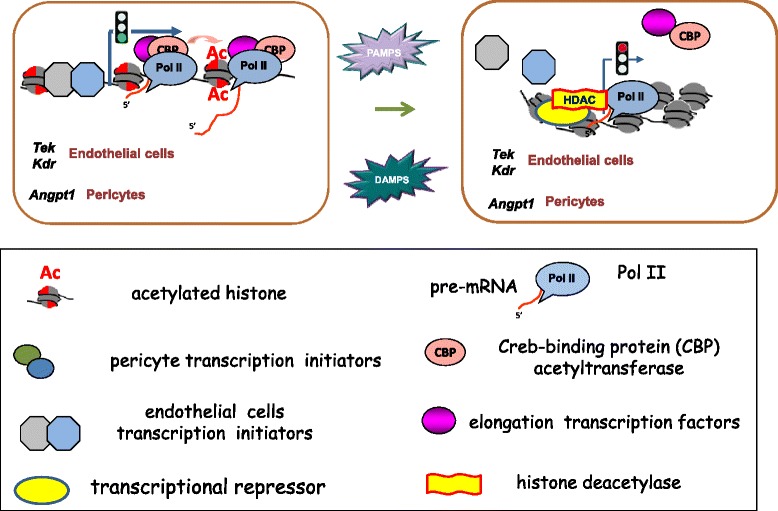
Proposed model for sepsis-induced downregulated transcription of angiogenic genes in endothelial cells and pericytes. Constitutively, epigenetic modifiers, including those that maintain permissive histone acetylation, are bound to genes by transcription factors and polymerase II (Pol II) machinery. During sepsis, systemic release of pathogen-associated molecular pattern molecules (PAMPs) or endogenous danger-associated molecular pattern molecules (DAMPs) or both initiates signal pathways that reduce activity of gene-bound transcription initiation or elongation factors or both. These transcription factor changes alter the balance of modifiers, shifting open chromatin toward more compact structures and contributing to downregulation of Pol II transcription of Tek and Kdr in endothelial cells and Angpt1 in pericytes
Conclusions
In summary, sepsis remains a major cause of morbidity and mortality in critically ill patients, and VAP is an important cause of sepsis in the ICU. Specific therapeutic options beyond supportive care and antimicrobial agents for pathogen eradication currently do not exist for patients with ALI-induced sepsis. With the increasing awareness that the endothelium plays a major role in end-organ responses to sepsis, we applied the multiplex Matrix ChIP platform to identify systemic transcriptional and epigenetic changes associated with sepsis-mediated downregulation of angiogenic genes in target organs. This analysis revealed important changes in permissive epigenetic histone modifications at Angpt1, Tek, and Kdr in the lung, kidney, and liver. However, contrary to our initial hypothesis, ALI-induced changes in repressive epigenetic histone modifications at these genetic loci were relatively minor. This approach and our findings open up a fundamentally new venue to define the epigenetic basis of sepsis-induced microvascular leak and end-organ injury/dysfunction while also identifying the epigenetic modifiers underlying these alterations. Our data suggest that transcriptionally mediated alterations in Angpt1, Tek, and Kdr during sepsis may potentially be modulated through targeting histone acetylases/deacetylases. Several drugs that target this pathway are currently in clinical testing for other diseases [53] but have not been evaluated in sepsis. Mapping epigenetic modifiers tethered to angiogenic loci during sepsis is a promising approach to discover novel therapeutic targets for innovative drug development to mitigate pulmonary injury and extra-pulmonary organ injury in ALI and sepsis.
Key messages
Experimental ALI downregulates angiogenic gene expression not only in the lung but also in extra-pulmonary organs.
Experimental ALI-induced downregulation of angiogenic genes in all examined organs is transcriptionally mediated.
Experimental ALI-induced downregulation of angiogenic genes is associated with decreased permissive but minimal changes in repressive histone modifications.
Identification of histone deacetylation at downregulated angiogenic genes as a systemic response to sepsis opens up new possibilities of using rational-design epigenetic therapy (for example, histone deacetylase inhibitor) to mitigate sepsis-associated microvascular dysfunction and endothelial leak.
Acknowledgments
This work was supported by NIH R01 DK083310, R21 GM111439, R21 DK09881, R33 CA191135, and R01 HL086883 research funding from the University of Washington Department of Medicine and an anonymous private donation to UW Medicine Research.
Abbreviations
- ALI-sepsis
acute lung injury-induced sepsis
- Ang
angiopoietin
- ChIP
chromatin immunoprecipitation platform
- Ct
cycle threshold
- DAMP
danger-associated molecular pattern molecule
- ICU
intensive care unit
- IL-6
interleukin-6
- Matrix ChIP
multiplex chromatin immunoprecipitation platform
- MCP-1
monocyte chemoattractant protein-1
- MODS
multiple organ dysfunction syndrome
- OD540
optical density at 540 nm
- PAMP
pathogen-associated molecular pattern molecule
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- Pol II
polymerase II
- TNF
tumor necrosis factor
- VAP
ventilator-associated pneumonia
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
Additional file
Polymerase chain reaction (PCR) primers. Table S2. Chromatin immunoprecipitation platform (ChIP) antibodies. Figure S1. Example of epigenetic regulation of chromatin structure and transcription. Figure S2. Analysis of changes in lung Angpt2, Vegfα, and Flt1 mRNA expression in experimental acute lung injury-induced sepsis (ALI-sepsis). Figure S3. Multiplex chromatin immunoprecipitation platform (Matrix ChIP) analysis of histone H3 density at Angpt1, Tek, Kdr, and Ngal in experimental acute lung injury-induced sepsis (ALI-sepsis). Figure S4. Multiplex chromatin immunoprecipitation platform (Matrix ChIP) analysis of changes in repressive histone H3 lysine 9 di-methylation (H3K9m2) at Angpt1, Tek, Kdr, and Ngal genes in experimental acute lung injury-induced sepsis (ALI-sepsis). Figure S5. Multiplex chromatin immunoprecipitation platform (Matrix ChIP) analysis of changes in repressive histone H3 lysine 9 tri-methylation (H3K9m3) in experimental acute lung injury-induced sepsis (ALI-sepsis). Figure S6. Multiplex chromatin immunoprecipitation platform (Matrix ChIP) analysis of changes in histone H4 lysine 20 tri-methylation (H4K20m3) in experimental acute lung injury-induced sepsis (ALI-sepsis). Figure S7. Analysis of changes in the lung, kidney, and liver of Tnf, Mpc-1, IL-6, and Cox2 mRNA expression in experimental acute lung injury-induced sepsis (ALI-sepsis).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KB conceived and designed the study, performed analysis, and drafted the manuscript. DM performed Matrix ChIP and data analysis. DA performed animal experiments. RS performed quantitative PCR. MM contributed to data interpretation and analysis. SAG contributed to data analysis and editing of the manuscript. WAA conceived and designed the study, supervised animal experiments, and edited the manuscript. WCL conceived and designed the study and edited the manuscript. OD conceived and designed the study, supervised assays, performed analysis, and edited the manuscript. All authors read and approved the final manuscript.
Contributor Information
Karol Bomsztyk, Email: karolb@u.washington.edu.
Daniel Mar, Email: danmar@uw.edu.
Dowon An, Email: dowon@u.washington.edu.
Roya Sharifian, Email: roya_sharifian@yahoo.com.
Michal Mikula, Email: mikula.michal@gmail.com.
Sina A Gharib, Email: sagharib@u.washington.edu.
William A Altemeier, Email: billa@u.washington.edu.
W Conrad Liles, Email: wcliles@uw.edu.
Oleg Denisenko, Email: odenis@u.washington.edu.
References
- 1.Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014;5:4–11. doi: 10.4161/viru.27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leligdowicz A, Dodek PM, Norena M, Wong H, Kumar A. Association between source of infection and hospital mortality in patients who have septic shock. Am J Respir Crit Care Med. 2014;189:1204–13. doi: 10.1164/rccm.201310-1875OC. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–40. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siempos II, Vardakas KZ, Kyriakopoulos CE, Ntaidou TK, Falagas ME. Predictors of mortality in adult patients with ventilator-associated pneumonia: a meta-analysis. Shock. 2010;33:590–601. doi: 10.1097/SHK.0b013e3181cc0418. [DOI] [PubMed] [Google Scholar]
- 5.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–51. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 6.Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 7.Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N, et al. High versus low blood-pressure target in patients with septic shock. N Engl J Med. 2014;370:1583–93. doi: 10.1056/NEJMoa1312173. [DOI] [PubMed] [Google Scholar]
- 8.Caironi P, Tognoni G, Masson S, Fumagalli R, Pesenti A, Romero M, et al. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med. 2014;370:1412–21. doi: 10.1056/NEJMoa1305727. [DOI] [PubMed] [Google Scholar]
- 9.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med. 2011;3:88ps25. doi: 10.1126/scitranslmed.3002011. [DOI] [PubMed] [Google Scholar]
- 10.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329–42. doi: 10.1189/jlb.0912437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 12.Lee WL, Liles WC. Endothelial activation, dysfunction and permeability during severe infections. Curr Opin Hematol. 2011;18:191–6. doi: 10.1097/MOH.0b013e328345a3d1. [DOI] [PubMed] [Google Scholar]
- 13.Jeltsch M, Leppanen VM, Saharinen P, Alitalo K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb Perspect Biol. 2013;5:a009183. doi: 10.1101/cshperspect.a009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saharinen P, Bry M, Alitalo K. How do angiopoietins Tie in with vascular endothelial growth factors? Curr Opin Hematol. 2010;17:198–205. doi: 10.1097/MOH.0b013e3283386673. [DOI] [PubMed] [Google Scholar]
- 15.David S, Kumpers P, van Slyke P, Parikh SM. Mending leaky blood vessels: the angiopoietin-Tie2 pathway in sepsis. J Pharmacol Exp Ther. 2013;345:2–6. doi: 10.1124/jpet.112.201061. [DOI] [PubMed] [Google Scholar]
- 16.Xu C, Chang A, Hack BK, Eadon MT, Alper SL, Cunningham PN. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014;85:72–81. doi: 10.1038/ki.2013.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parikh SM. Dysregulation of the angiopoietin-Tie2 axis in sepsis and ARDS. Virulence. 2013;4:517–24. doi: 10.4161/viru.24906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med. 2014;42:e654–62. doi: 10.1097/CCM.0000000000000524. [DOI] [PubMed] [Google Scholar]
- 19.van der Heijden M, van Nieuw Amerongen GP, Chedamni S, van Hinsbergh VW, Johan Groeneveld AB. The angiopoietin-Tie2 system as a therapeutic target in sepsis and acute lung injury. Expert Opin Ther Targets. 2009;13:39–53. doi: 10.1517/14728220802626256. [DOI] [PubMed] [Google Scholar]
- 20.David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis. Crit Care Med. 2012;40:3034–41. doi: 10.1097/CCM.0b013e31825fdc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bomsztyk K, Denisenko O. Epigenetic alterations in acute kidney injury. Semin Nephrol. 2013;33:327–40. doi: 10.1016/j.semnephrol.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 23.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Mahony DS, Liles WC, Altemeier WA, Dhanireddy S, Frevert CW, Liggitt D, et al. Mechanical ventilation interacts with endotoxemia to induce extrapulmonary organ dysfunction. Crit Care. 2006;10:R136. doi: 10.1186/cc5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhanireddy S, Altemeier WA, Matute-Bello G, O’Mahony DS, Glenny RW, Martin TR, et al. Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab Invest. 2006;86:790–9. doi: 10.1038/labinvest.3700440. [DOI] [PubMed] [Google Scholar]
- 26.Nelson JD, Denisenko O, Sova P, Bomsztyk K. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 2006;34 doi: 10.1093/nar/gnj004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Feng Q, Ruan Y, Komers R, Kiviat N, Bomsztyk K. Microplate-based platform for combined chromatin and DNA methylation immunoprecipitation assays. BMC Mol Biol. 2011;12:49. doi: 10.1186/1471-2199-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagin S, Nelson JD, Castner DG, Denisenko O, Bomsztyk K. Microplate-based chromatin immunoprecipitation method: Matrix ChIP: a platform to study signaling of complex genomic events. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bomsztyk K, Flanagin S, Mar D, Mikula M, Johnson A, Zager R, et al. Synchronous recruitment of epigenetic modifiers to endotoxin synergistically activated Tnf-alpha gene in acute kidney injury. PLoS One. 2013;8 doi: 10.1371/journal.pone.0070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Backer D, Orbegozo Cortes D, Donadello K, Vincent JL. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence. 2013;5:1. doi: 10.4161/viru.26482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawiger J, Musser JM. How to approach genome wars in sepsis? Crit Care. 2011;15:1007. doi: 10.1186/cc10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill SE, Taneja R, Rohan M, Wang L, Mehta S. Pulmonary microvascular albumin leak is associated with endothelial cell death in murine sepsis-induced lung injury in vivo. PLoS One. 2014;9 doi: 10.1371/journal.pone.0088501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinn TP, Peters KG, De Vries C, Ferrara N, Williams LT. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A. 1993;90:7533–7. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 1826;2012:129–69. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srisawat N, Murugan R, Lee M, Kong L, Carter M, Angus DC, et al. Plasma neutrophil gelatinase-associated lipocalin predicts recovery from acute kidney injury following community-acquired pneumonia. Kidney Int. 2011;80:545–52. doi: 10.1038/ki.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naito M, Bomsztyk K, Zager RA. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. 2008;19:1321–30. doi: 10.1681/ASN.2007121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–66. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 38.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–31. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–36. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 40.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Nelson JD, Flanagin S, Kawata Y, Denisenko O, Bomsztyk K. Transcription of laminin γ1 chain gene in rat mesangial cells: constitutive and inducible RNA polymerase II recruitment and chromatin states. Am J Physiol Renal Physiol. 2008;294:F525–33. doi: 10.1152/ajprenal.00299.2007. [DOI] [PubMed] [Google Scholar]
- 42.Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012;2:a010272. doi: 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 44.Bierhoff H, Dammert MA, Brocks D, Dambacher S, Schotta G, Grummt I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol Cell. 2014;54:675–82. doi: 10.1016/j.molcel.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 45.Jesmin S, Zaedi S, Islam AM, Sultana SN, Iwashima Y, Wada T, et al. Time-dependent alterations of VEGF and its signaling molecules in acute lung injury in a rat model of sepsis. Inflammation. 2012;35:484–500. doi: 10.1007/s10753-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 46.Mofarrahi M, Nouh T, Qureshi S, Guillot L, Mayaki D, Hussain SN. Regulation of angiopoietin expression by bacterial lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2008;294:L955–63. doi: 10.1152/ajplung.00449.2007. [DOI] [PubMed] [Google Scholar]
- 47.Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol. 2005;175:3369–76. doi: 10.4049/jimmunol.175.5.3369. [DOI] [PubMed] [Google Scholar]
- 48.David S, Ghosh CC, Kumpers P, Shushakova N, Van Slyke P, Khankin EV, et al. Effects of a synthetic PEG-ylated Tie2 agonist peptide on endotoxemic lung injury and mortality. Am J Physiol Lung Cell Mol Physiol. 2011;300:L851–62. doi: 10.1152/ajplung.00459.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karmpaliotis D, Kosmidou I, Ingenito EP, Hong K, Malhotra A, Sunday ME, et al. Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L585–95. doi: 10.1152/ajplung.00048.2002. [DOI] [PubMed] [Google Scholar]
- 50.van Meurs M, Kurniati NF, Wulfert FM, Asgeirsdottir SA, de Graaf IA, Satchell SC, et al. Shock-induced stress induces loss of microvascular endothelial Tie2 in the kidney which is not associated with reduced glomerular barrier function. Am J Physiol Renal Physiol. 2009;297:F272–81. doi: 10.1152/ajprenal.00137.2009. [DOI] [PubMed] [Google Scholar]
- 51.David S, Park JK, Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, et al. Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine. 2011;55:251–9. doi: 10.1016/j.cyto.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Tsokos M, Pufe T, Paulsen F, Anders S, Mentlein R. Pulmonary expression of vascular endothelial growth factor in sepsis. Arch Pathol Lab Med. 2003;127:331–5. doi: 10.5858/2003-127-0331-PEOVEG. [DOI] [PubMed] [Google Scholar]
- 53.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–8. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 54.ImageJ64 software. http://rsb.info.nih.gov/ij/download.html. Accessed date 03-15-2015