

Abstract
Significance: Over-nutrition and sedentary lifestyle has led to a worldwide increase in obesity, insulin resistance, and type 2 diabetes (T2D) associated with an increased risk of development of cardiovascular disorders. Diabetic cardiomyopathy, independent of hypertension or coronary disease, is induced by a range of systemic changes and may through multiple processes result in functional and structural cardiac derangements. The pathogenesis of this cardiomyopathy is complex and multifactorial, and it will eventually lead to reduced cardiac working capacity and increased susceptibility to ischemic injury. Recent Advances: Metabolic disturbances such as altered lipid handling and substrate utilization, decreased mechanical efficiency, mitochondrial dysfunction, disturbances in nonoxidative glucose pathways, and increased oxidative stress are hallmarks of diabetic cardiomyopathy. Interestingly, several of these disturbances are found to precede the development of cardiac dysfunction. Critical Issues: Exercise training is effective in the prevention and treatment of obesity and T2D. In addition to its beneficial influence on diabetes/obesity-related systemic changes, it may also amend many of the metabolic disturbances characterizing the diabetic myocardium. These changes are due to both indirect effects, exercise-mediated systemic changes, and direct effects originating from the high contractile activity of the heart during physical training. Future Directions: Revealing the molecular mechanisms behind the beneficial effects of exercise training is of considerable scientific value to generate evidence-based therapy and in the development of new treatment strategies. Antioxid. Redox Signal. 22, 1587–1605.
Introduction
The world is facing an epidemic increase in obesity and insulin resistance due to over-nutrition and sedentary lifestyle. Obesity and insulin resistance are risk factors for type 2 diabetes (T2D), and cardiovascular disease is the leading cause of mortality in patients with T2D. In addition to increased incidence of hypertension and concomitant macro- and microvascular disorders, diabetic patients are also at risk of developing a specific diabetic cardiomyopathy (66). The development of diabetic cardiomyopathy is most likely induced by a range of systemic changes, which through multiple signaling pathways will lead to functional and structural derangements (Fig. 1).
FIG. 1.

Hallmarks of diabetic cardiomyopathy. A range of systemic changes contribute to the onset of molecular mechanisms leading to cardiac dysfunction and inefficiency in diabetes. NO, nitric oxide; AGE, advanced glycation-end product; RAGE, AGE receptors; ROS, reactive oxygen species; RAS, renin-angiotensin system; LV, left ventricle; O-GlcNAc, O-linked N-acteylglucosamine.
Hallmarks of the diabetic heart include oxidative stress, fibrosis, apoptosis, impaired autophagy, inflammation, and altered calcium handling. In addition, these hearts display metabolic-related changes, including altered substrate utilization, decreased mechanical efficiency, lipid accumulation, mitochondrial dysfunction, impaired insulin signaling, increased advanced glycation end-products (AGEs), and sustained O-linked N-acteylglucosamine (O-GlcNAc) modification of proteins. The pathogenesis of this cardiomyopathy is, by no doubt, multifactorial and complex, eventually leading to an energetically compromised heart with reduced working capacity and increased susceptibility to ischemia-reperfusion.
Aerobic fitness, measured as maximal oxygen uptake (VO2max), has been shown to be a strong and independent predictor for total and cardiovascular mortality both in healthy individuals and in patients with cardiovascular disease (109, 144). Exercise is a low-cost strategy for prevention and treatment of obesity and T2D (120, 207). It is well known that exercise can induce cardioprotection in normal hearts through a range of molecular mechanisms (79, 158). Beneficial cardiac effects after exercise training in diabetes/obesity have been reported in clinical (94, 167, 172, 173) and experimental animal studies (21, 61, 85, 111, 128, 176, 184). This review highlights existing knowledge of how metabolic derangements contribute to the development of diabetic cardiomyopathy, and of how exercise training may influence these changes and the progression of diabetic cardiomyopathy.
Myocardial Substrate Utilization in Diabetes
The requirement to continuously pump blood makes the myocardium the most metabolically active tissue in the body. The majority of its ATP production is derived from mitochondrial oxidative phosphorylation (OxPhos), and to meet the high energetic demand, a constant flow of substrate to the mitochondria is pivotal. The cardiac metabolic network has developed a versatile system, capable of metabolizing all carbon substrates for energy production, where glucose and fatty acids (FA) are the main substrates (Fig. 2). An important feature of the normal heart is metabolic flexibility and the ability to ensure appropriate ATP production rate under diverse physiologic and dietary conditions. Lack of this flexibility has been regarded as fundamental in the development of heart failure, including diabetic cardiomyopathy, but exactly how remains unclear.
FIG. 2.
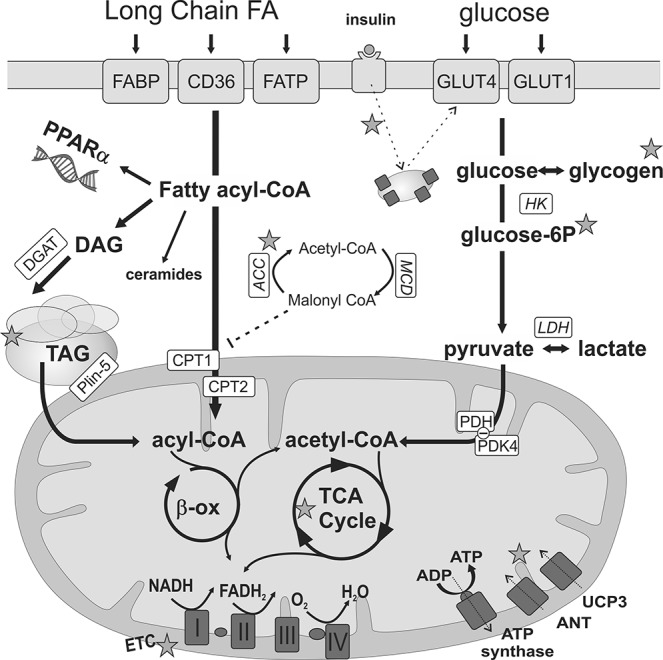
Glucose and FA uptake and oxidation in the diabetic cardiomycyte. A continuous elevation in FAs supply leads to increased long-chain FA (LCFA) utilization due to activation of the peroxisome proliferator-activated receptor (PPAR)-α and, consequently, transcriptional induction of enzymes involved in FA transport (FAs transport protein [FATP], FA binding protein [FABP] and FA translocase [FAT/CD36]) and β-oxidation (β-ox). The entry of Acyl-CoA into the mitochondria is mediated by the carnitine-palmitoyltransferase (CPT1 and 2) system. After β-oxidation, acetyl Co-A enters the tricarboxylic acid (TCA) cycle to form reducing equivalents (FADH2/NADH) that drive the electron transport chain (ETC) and generate the proton motive force that powers ATP synthesis. Glucose uptake (facilitated diffusion by the glucose transporters 1 (GLUT1) and the insulin-sensitive GLUT4) are decreased in diabetes due to impaired insulin signaling. Pyruvate formed from glycolysis is taken up by the mitochondria, and acetyl-CoA is generated by the pyruvate dehydrogenase complex (PDH). Myocardial glucose oxidation is reduced in diabetes due to increased pyruvate dehydrogenase kinase 4 (PDK4), which inhibits the PDH. Excess acyl-CoA is stored as triacylglycerol (TAG) in lipid droplets and may result in the accumulation of toxic intermediates such as diacylglycerol (DAG) and ceramides. Impaired mitochondrial respiration may be due to reduced respiration capacity and/or efficiency, increased uncoupling via uncoupling proteins (UCP), and/or the adenine nucleotide translocase (ANT). Exercise-induced changes reported in the literature are indicated by stars and described in detail in the text. See “Myocardial Substrate Utilization in Diabetes” and “Lipotoxicity in Diabetic Cardiomyopathy” sections for references. DGAT, diglyceride acyltransferase; MCD, malonyl-CoA decarboxylase; ACC, acetyl-CoA carboxylase; Plin-5, perilipin-5.
Despite the presence of hyperglycemia, the diabetic heart relies predominantly on FA oxidation, with a concomitant reduction in glucose oxidation (1, 17, 155). Obesity and diabetes-induced enhancement of myocardial FA uptake and oxidation is due to the activation of peroxisomal proliferator-activated receptor-α (PPAR-α) and, consequently, transcriptional induction of enzymes involved in FA transport and β-oxidation. This also leads to a concomitant suppression of glucose oxidation, due to increased expression of pyruvate dehydrogenase kinase 4 (PDK4), which inhibits the pyruvate dehydrogenase complex (PDH) (129). Recent studies in skeletal muscle have also suggested carnitine acetyltransferase (CrAT) to be a key regulator in metabolic flexibility, and that deficits in CrAT activity in diabetes might contribute to a more rigid and inflexible metabolism (140).
Altered cardiac substrate metabolism has been shown in experimental and clinical studies to precede the development of ventricular dysfunction (1, 155). Studies using genetically modified mice in which cardiac FA uptake and/or oxidation resemble that of the diabetic metabolic phenotype also describe development of cardiac dysfunction (48, 49, 67), and interventions that reduce the substrate switch have been shown to ameliorate the development of ventricular dysfunction (17, 81, 96).
Taken together, these studies support the notion that altered myocardial lipid metabolism has a causative role in diabetic cardiomyopathy and that targeting myocardial metabolism is a therapeutic intervention for the maintenance of cardiovascular health in diabetes (129). It should be noted, however, that the presence of FAs or maintenance of FA oxidation has also been reported to have beneficial functional effects in diabetic hearts and cardiomyocytes (64, 88, 191, 203). These discrepancies may suggest that while preventing the switch in substrate utilization may ameliorate the development of diabetic cardiomyopathy, diabetic hearts in an untreated state may undergo adaptive changes that eventually make these hearts dependent on FAs, specifically as an energy substrate.
Endurance training improves myocardial metabolism
An acute exercise session will lead to an altered metabolic state with high levels of plasma lactate (from glycolysis in exercising muscle) and nonesterified FAs (β-adrenergic stimulation and lipolysis). Consequently, FA and lactate become the major substrates for the heart during exercise (76). The myocardial metabolic adaptation in response to endurance training is, however, less clarified. Several studies have reported altered expression of genes and proteins related to myocardial metabolism in normal (83, 126, 185) and diabetic hearts (85, 177) in response to chronic exercise. Studies directly addressing the effects of exercise training on metabolic fluxes in the hearts are, however, few and diverging.
In the normal heart, Broderick et al. (37) did not find treadmill running to alter myocardial glucose oxidation and glycolysis, whereas Burelle et al. (39) reported an increased rate of both glucose and FA oxidation while glycolysis was decreased. These discrepancies may be related to variations in the intensity of the exercise training protocol and in line with this, Hafstad et al. (83) recently reported that only high-intensity training (and not moderate intensity) altered myocardial substrate utilization in normal mice.
In type 1 diabetic (T1D) rats, induced by streptozotocin (STZ), exercise training was reported to increase rates of glucose oxidation and glycolysis (37) whereas FA oxidation was unaltered (153). Similarly, in a recent study using diet-induced insulin-resistant mice, treadmill running increased myocardial glucose oxidation while oxidation rates of FAs were less affected (85). An important finding from the latter study was that in contrast to that reported in healthy mice, both high- and moderate-intensity training induced a similar metabolic response in obese mice.
Evidence from studies on skeletal muscle has shown that exercise training can influence insulin pathways and improve insulin resistance. Insulin signaling seems to have a central role in the exercise-induced hypertrophic response in the healthy heart (164). Exercise has also been shown to increase the ability of insulin to phosphorylate the insulin receptor (IR), insulin receptor substrate-1 (IRS-1), IRS-2, Akt, and Foxo1, as well as to affect other pathways influencing the insulin signaling cascade in cardiac tissue from diet-induced obese rats (134, 157). The metabolic consequence (i.e., substrate utilization) of these changes has, however, not been investigated. It should be noted that compelling experimental evidence has linked excessive cardiac insulin signaling with adverse effects after chronic pressure overload in normal and T1D (180), implying that improved systemic insulin levels by exercise training may provide beneficial cardiac effects under certain types of stress, such as hypertension.
Diabetes-Induced Hyperglycemia Stimulates Nonoxidative Glucose Pathways in Diabetic Cardiomyopathy
Hyperglycemia is an important risk factor for developing cardiovascular disease. In addition to generating pyruvate for oxidation, elevated plasma levels of glucose may also affect nonoxidative glucose pathways (Fig. 3). Both intracellular and extracellular lipids and protein exposed to high levels of sugars may become irreversibly glycated and form AGEs. AGE modifications of the extracellular matrix may render the tissue less compliant and induce myocardial stiffness (43). AGEs can also bind to AGE receptors (RAGEs), which may activate redox-sensitive transcription factors, such as nuclear factor-κB (NF-κB), and, consequently, affect intracellular signaling pathways leading to remodeling of the extracellular matrix, cell growth, cytokine formation, increased production of reactive oxygen species (ROS), and reduced nitric oxide (NO) bioavailability (80).
FIG. 3.
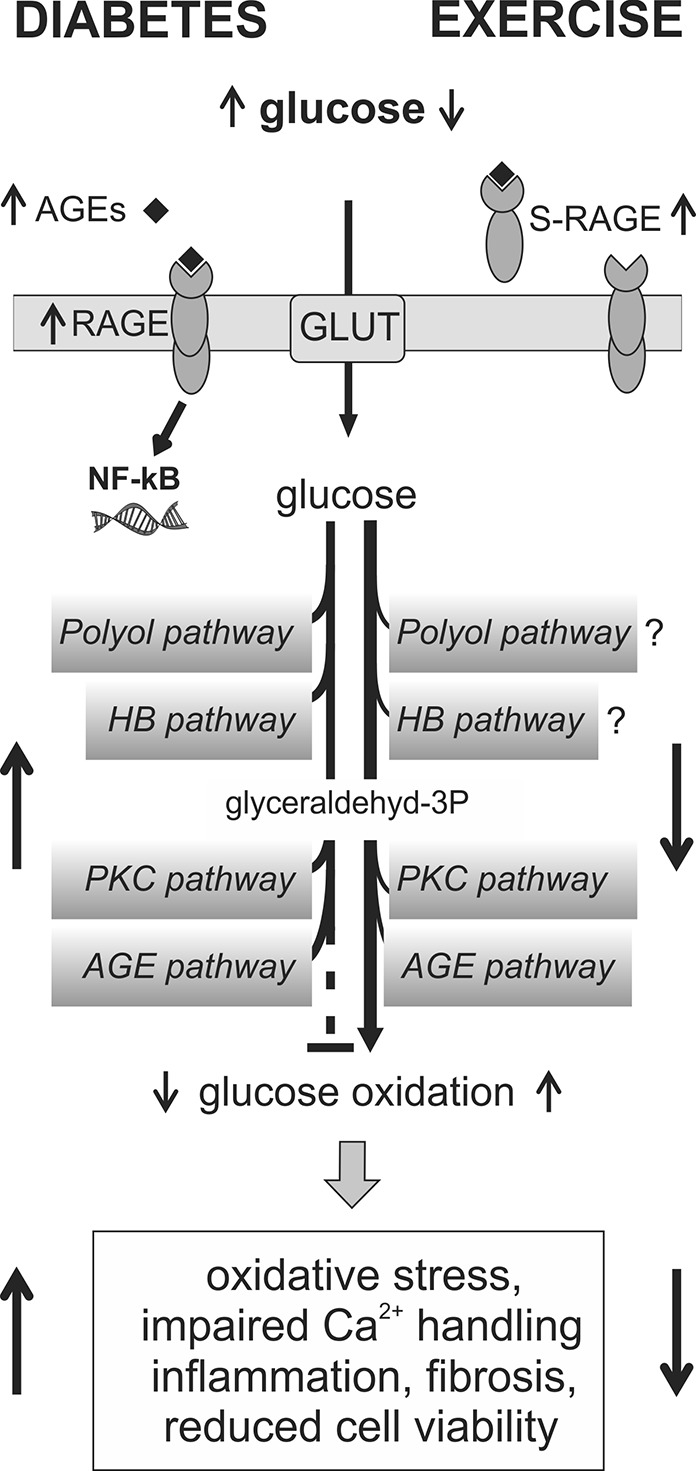
Nonoxidative pathways in diabetic cardiomyopathy and proposed effects of exercise training. Hyperglycemia and reduced shunting of carbons from glycolysis into glucose oxidation may lead to the induction of nonoxidative glucose pathways, including the polyol, hexosamine biosynthetic (HBP), protein kinase C (PKC), and advanced glycation end-products (AGE) pathway in the diabetic heart. Increased plasma levels of AGEs can also bind to AGE receptors (RAGEs), which together with PKC and oxidative stress are known activators of nuclear factor-κB (NF-κB). Increased NF-κB activity together with these pathways may mediate adverse effects in the diabetic heart due to impaired myocardial reductive capacity, formation of AGEs, and sustained O-GlcNAcylation of proteins. Exercise training may, through improved glucose homeostasis and increased myocardial glucose oxidation, reverse the induction of several nonoxidative glucose pathways in the diabetic heart. In addition, exercise can increase plasma levels of the soluble form of RAGE (sRAGE), which may work as a scavenger for AGEs and therefore reduce the intracellular effects of AGE/RAGE signaling. See “Diabetes-Induced Hyperglycemia Stimulates Nonoxidative Glucose Pathways in Diabetic Cardiomyopathy” section for references.
There is substantial evidence that AGEs are key mediators in the development of vascular injury in diabetic patients (80), and recent studies also support the role of AGEs and RAGEs in the development of diabetic cardiomyopathy (43, 114). In STZ-induced diabetes, treatment with the AGE cross-link breaker ALT-711 was shown to reduce cardiac AGEs, accompanied by a normalization of collagen III deposition, improvement of calcium handling, and attenuation of diabetes-associated myocardial structural changes (43, 114). The soluble form of RAGE (sRAGE), lacking an intracellular domain, is believed to work as a scavenger for AGE (205). In diabetic patients, an inverse correlation has been reported between plasma levels of sRAGE and HbA1c, insulin resistance index, and C-reactive protein (13).
The polyol pathway, which branches off the glycolytic pathway, is activated after increased concentrations of intracellular glucose. Aldose reductase (AR, the rate-limiting step of this pathway) oxidizes NADPH to NADP and can thus impair myocardial reductive/antioxidant capacity. Introducing hyperglycemia to isolated hearts has been shown to increase activation of the polyol pathway, accompanied by increased oxidative stress and impaired left ventricular diastolic function, while inhibition of AR attenuated these changes (188). Increased AR activity has been demonstrated in T1D hearts (105), while reports from T2D models are lacking.
Hyperglycemia can also increase the synthesis of diacylglycerol (DAG), an activator of the protein kinase C (PKC) signaling pathway in the diabetic heart (104). Increased activity of this pathway is associated with cardiac hypertrophy, fibrosis, adverse Ca2+ handling, and reduced cardiac performance (197). Treatment with a PKCβ inhibitor has been found to reduce collagen deposition, increase SERCA2a expression, and preserve diastolic function in T1D hearts (54).
Elevated intracellular glucose may also increase flux of carbons from glucose through the hexosamine biosynthetic pathway (HBP). This produces uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which is used as a substrate by O-GlcNAc transferase to modify serine/threonine residues in proteins (206). O-GlcNAcylation is reported to be upregulated in obese and diabetic hearts (19, 56, 97, 135). Although O-GlcNAcylation of proteins integrates glucose metabolism with posttranslational modifications, this process is not simply regulated by substrate availability, as multiple stressors have also been shown to induce O-GlcNAcylation in cardiac tissue (47, 198, 199).
Altered O-GlcNAcylation may influence many of the processes known to be affected in diabetic cardiomyopathy, including mitochondrial respiratory capacity (98), insulin signaling (89), FA utilization (118), Ca2+ handling (63, 97), autophagy (131), ER stress (147, 198), and fibrosis (3). Protein O-GlcNAcylation seems, however, to be paradoxical in nature; while a reduction in the overall O-GlcNAcylation in diabetic hearts is beneficial in terms of Ca2+ handling and ventricular function (97, 133), and O-GlcNAcylation has also been reported to be fundamental in cardiac protection (47, 125, 147, 199). This has led to the suggestion that acute elevation of cardiac protein O-GlcNAcylation is protective, while sustained protein O-GlcNAcylation is detrimental (103).
Exercise training reduces nonoxidative glucose pathways
Given the beneficial effects of exercise on glycemic control (85, 190, 200), one would expect that this should amend the adverse effects of the increased nonoxidative glucose pathways in the diabetic heart (Fig. 3). Interestingly, studies addressing these pathways often report exercise-induced effects that are independent of systemic effects (19, 29, 136). As exercise training also increases myocardial glucose oxidation (85), this may be a mechanism contributing to the shunting of carbons away from nonoxidative glucose pathways.
Although reports on cardiac effects of exercise on the RAGE/AGE system are sparse, exercise has been found to reduce fibrosis together with reduced AGEs in the ventricles of aged rats (202). In T2D patients, exercise training increased plasma levels of sRAGE, associated with reduction of several cardiometabolic risk factors (50). However, in obese Zucker rats, exercise lowered plasma AGEs without affecting circulating glucose, insulin, lipid profile, oxidative status, or inflammatory markers (29). Exercise training has also been shown to ameliorate the obesity-induced activation of both the NF-κB and JNK pathways in the heart (134), supporting an anti-inflammatory effect of exercise in cardiac tissue (30), which could potentially work through AGE pathways.
The role of the polyol pathway or PKC pathway in exercise-attenuated insulin resistance and cardiac dysfunction in diabetes remains unclear. The exercise-induced reduction in myocardial DAG levels has been associated with improved left ventricular function in T1D rats, although this was not found to be associated with altered PKCβII protein expression or activity (128).
Long-term intensive swim-training has been reported to reduce total cardiac protein O-GlcNAcylation in both lean mice (15) and STZ mice (19). In db/db mice, however, treadmill running was found to increase cardiac protein O-GlcNAcylation (56). A plausible explanation for these findings may be the use of different experimental models, training modes, and intensities. Recently, it has been demonstrated that acute exercise training altered cardiac protein O-GlcNAcylation in a very time- and spatial-restricted manner without any change in overall O-GlcNAc protein levels (136). This suggests a complex regulation of cardiac O-GlcNAcylation in response to exercise and may also partially clarify the discrepancies in the studies addressing cardiac protein O-GlcNAcylation after exercise interventions.
Lipotoxicity in Diabetic Cardiomyopathy
As a consequence of excess cardiac FA uptake relative to oxidation, long-chain FAs are incorporated into triacylglycerols (TAG), leading to cardiac steatosis in diabetes/obesity (85, 165). Elevated intramyocellular TAG content in skeletal muscle, in the absence of exercise, is a strong predictor of global insulin resistance (139), and intramyocardial TAG accumulation has been found to be an independent predictor of cardiac dysfunction in T2D patients (165).
Although TAG is not regarded as a toxic molecule in itself, shunting of FAs into nonoxidative pathways can lead to accumulation of intermediate metabolites, including DAG and ceramides at high concentrations (Fig. 2), and may increase fibrosis through the activation of protein kinases (PKCβ or PKA) (10), induce apoptosis (151), and disrupt insulin signaling (208). In addition, long-chain acylcarnitines and acyl-CoAs accumulate in skeletal muscle in diabetes due to incomplete β-oxidation and impaired CrAT activity (175). This has fostered support for a lipid-induced insulin resistance associated with obesity (113, 140).
Accumulation of mitochondrial long-chain acylcarnitine was also shown to impair glucose metabolism in the heart (130). Furthermore, shunting of lipid intermediates into TAG in lipid droplets has been shown to ameliorate ventricular dysfunction in models of lipotoxicity by maintaining a low concentration of lipotoxic intermediates (126, 127). In this regard, Perilipin-5 (Plin-5) is believed to play an important role in the stabilization and facilitation of lipolysis in the cardiomyocyte and in the direct transfer of FA between lipid droplets and mitochondria (Fig. 3) (115). Interestingly, although Plin-5 knockout mice showed a decline in cardiac function with age, they were resistant to STZ-induced cardiac dysfunction (116). These findings suggest that the diabetic heart may undergo adaptations, enabling them to cope with elevated lipid supply.
The effects of exercise on myocardial lipotoxicity
In healthy individuals, an acute bout of exercise can increase cardiac lipid content through exercise-induced elevation of FA concentrations in the blood (22). In normal mice, increased cardiac TAG storage after short-term intensive swim training was found to be accompanied by a marked upregulation of the TAG storage enzyme diacylglycerol transferase 1 (DGAT1), whereas the level of ceramide and DAG was reported to be reduced (126) (Fig. 2).
In obese subjects, endurance/strength training was shown to reduce myocardial TAG content in combination with improved ejection fraction (172). This finding, however, was not reproduced in obese T2D patients where myocardial TAG contents were found to be unaltered (107, 173). Exercise-induced reduction in myocardial TAG content has been reported in diet-induced obese mice (85), while the effects on myocardial DAG and ceramides levels in obese/diabetic models are not known. Although increased Plin-5 expression has been reported after acute exercise in human skeletal muscle (112), the effects of exercise on Plin-5 in the heart remain to be elucidated.
Mitochondrial Disturbances in Diabetic Cardiomyopathy
The heart is highly dependent on mitochondrial OxPhos, which must be closely linked to the rate of ATP hydrolysis. Mitochondrial dysfunction has been suggested to play a central role in the pathogenesis of diabetic cardiomyopathy, as an imbalance between energy availability and demand will lead to an energetically compromised heart with reduced working capacity. In human diabetic atria fibers, Anderson et al. (5) found that ADP-stimulated respiration (state 3 respiration) was decreased when using both palmitate and glutamate as substrates. In experimental studies from obese/diabetic models, decreased and unchanged mitochondrial respiratory capacity has been reported using carbohydrate-based substrates (2, 14, 32, 33, 53, 68, 85), while there is less agreement when FAs are used as substrates (2, 14, 31–33, 53, 68).
Mitochondrial uncoupling is regarded as a key mediator of mitochondrial dysfunction in the diabetic heart. The underlying mechanisms mediating diabetes-induced uncoupling are unclear, but both uncoupling proteins (UCPs) (31, 33, 142) and adenine nucleotide translocator (33) have been proposed as candidates. Although there are inconsistent findings regarding the gene and protein expression of myocardial UCPs in models of T2D (32, 142), UCP activity is known to be increased by FAs, ROS, and lipid peroxidation products (60). Recent studies have demonstrated that both inhibition (53, 141) and genetic deletion (31) of UCP3 improve P/O ratios in models of diet-induced obesity.
The coupling efficiency of diabetic mitochondria evaluated by respiratory control ratio (i.e., RCR or state 3/state 4) or P/O ratio (a measure independent of the oxygen consumption) is decreased or unchanged in diabetic conditions (31–33, 53, 85, 142). Finally, mitochondrial respiration after administration of oligomycin (an inhibitor of ATP synthase) has also in some cases been reported to be increased (32, 33). Although the role of this proton leak is unclear, it has been suggested to lower mitochondrial membrane potential and, subsequently, reduce mitochondrial ROS production. Further research is, however, required to better elucidate the precise physiological role and/or the pathophysiological consequence of mitochondrial uncoupling in the heart.
Increased mitochondrial mass, area, and number have been reported in diabetic hearts (178, 179), possibly as an adaptive mechanism to overcome impaired mitochondrial respiratory capacity. In line with this notion, Shen et al. (178) found in hearts from T1D mice that improving the antioxidant capacity amended mitochondrial respiration, which was accompanied by normalization of mitochondrial density and morphometry (178).
Calcium is an important activator of key metabolic enzymes in the mitochondria (168) and accelerates the production of NADH (34). Mitochondrial Ca2+ cycles via the uniporter entry and export system (12, 168) and will be influenced by changes in cytosolic Ca2+ homeostasis. Depression of mitochondrial Ca2+ uptake and handling has been reported in diabetic cardiomyocytes (65, 156), as a consequence of impaired Ca2+ handling (as will be discussed later), and is therefore a plausible contributing factor to impaired energy metabolism in diabetic cardiomyopathy. Mitochondria can also induce cell necrosis via opening of the Ca2+-sensitive permeability transition pore (mPTP). Several studies have shown that mitochondria from diabetic hearts are sensitized to mPTP opening (6, 182), which may be partly responsible for the increased susceptibility to ischemic injury described in diabetic hearts.
The effect of exercise on mitochondrial function
Increased palmitoyl-carnitine and glutamate supported respiration capacity has been demonstrated after exercise training in cardiac mitochondria from normal (83, 148) and obese/diabetic rodent models (68, 85). Exercise-mediated improvement in mitochondrial capacity may be linked to mitochondrial Ca2+ handling. Although this has not been specifically studied, there could be an obvious link between enhanced mitochondrial calcium handling in diabetes and the beneficial effects on mitochondrial respiratory capacity and efficiency observed in hearts from endurance trained diabetic models (68, 85, 148).
Hafstad et al. (85) did not find exercise training to improve RCR, but to normalize P/O ratios, which was accompanied by increased proton leak after inhibition with oligomyocin. This may suggest that the mitochondrial adaptations induced by endurance training compensate for mild mitochondrial proton leak and preserve the efficiency of ATP synthesis. Interestingly, similar mitochondrial changes were recently reported in skinned cardiac fibers from high-fat-fed UCP3KO mice (31).
The impact of exercise training on mPTP opening remains largely unknown. Targeted treatment to reduce mitochondrial ROS was shown to improve resistance to mPTP opening in diabetic hearts (182). One could therefore speculate that exercise-induced effects on mitochondrial redox environment (as will be discussed later) may potentially increase resistance to mPTP opening in the diabetic heart and, consequently, increase cell survival in response to insults such as ischemia or Ca2+ overload.
Although exercise training has been shown to induce mitochondrial biogenesis in skeletal muscle (83, 123), its effect on mitochondrial biogenesis in the heart is less clear. In a study using lean mice, it was demonstrated that exercise induced skeletal muscle but not cardiac mitochondrial biogenesis (123). In contrast, high-intensity interval training was reported to increase cardiac mitochondrial content both in lean (83) and in obese (85) animal models. Interestingly, the same studies reported that this was not found after moderate-intensity continuous training (83, 85), suggesting a role of intensity in exercise-induced cardiac mitochondrial biogenesis.
Sources of ROS and Their Roles in the Progression of Diabetic Cardiomyopathy
Apart from being the final electron acceptor in electron transport chain (ETC), oxygen plays a role in cellular redox signaling, and may become toxic through formation of high concentrations of ROS. Thus, redox modulation of cellular proteins comes in “different flavors”; while moderate reversible oxidations may increase protein activity, and a more sustained ROS may induce pathological modifications through stable interactions with lipids, proteins, and DNA (40).
Several systemic alterations (Fig. 4) are believed to contribute to increased ROS formation in diabetes and obesity. In particular, the cardiovascular system is characterized by increased mitochondrial and extra-mitochondrial ROS generation as well as by impaired antioxidant defense systems (5, 33, 77, 162). Clinical and experimental studies support increased oxidative damage in diabetes (5, 33, 191, 195), and oxidative stress is likely to be a key mediator in the pathogenesis of diabetic cardiomyopathy and to be causative for the increased ischemic susceptibility in the diabetic heart (42, 57).
FIG. 4.
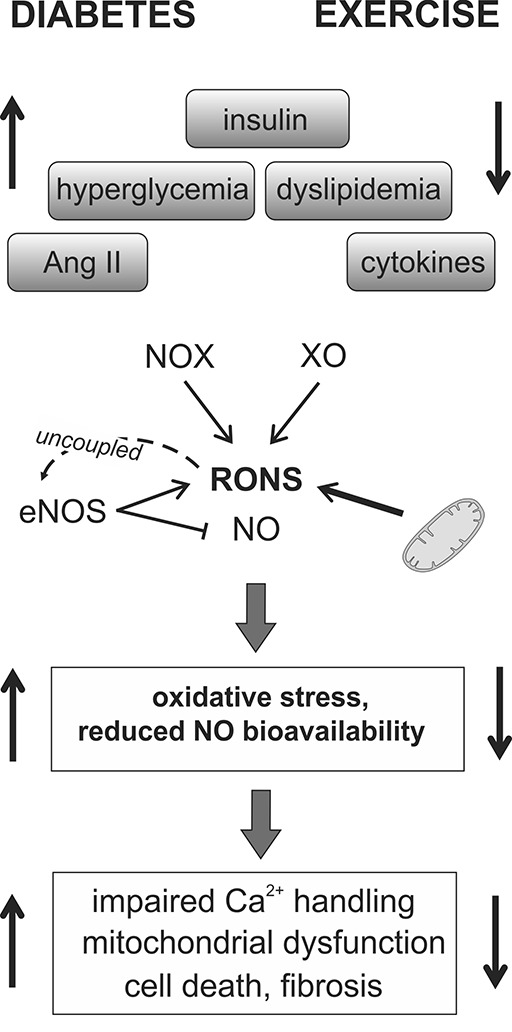
Myocardial ROS producing systems in diabetes and the effect of exercise training. Systemic changes in diabetes such as increased levels of angiotensin II (Ang II), insulin, cytokines, glucose, and dyslipidemia may induce the production of reactive oxygen and nitrogen species (RONS) from both mitochondrial and nonmitochondrial sources, including NADPH oxidases (NOX), xanthine oxidase (XO), and nitric oxide synthase (NOS). Reactive oxygen species may also lead to the uncoupling of endothelial NOS (eNOS), which will decrease NO production, limiting its bioavailability. These changes can lead to several adverse effects in the diabetic myocardium, which can be ameliorated through the systemic effects of exercise. Direct effects of exercise on ROS-producing enzymes are also reported and described in the text.
Despite substantial evidence from experimental studies showing beneficial cardiac effects of different pharmacological and genetic antioxidant therapies (124, 178, 189, 204), clinical trials have failed to show beneficial effects of conventional antioxidant therapies (24). A plausible explanation for these discrepancies is that general exogenous antioxidant therapies target all sources of ROS, not only pathophysiological ROS but also physiological ROS signaling in the heart.
In hearts from both humans (5) and animal models (33, 64, 191), a high rate of mitochondrial ROS generation is believed to contribute to diabetes-induced mitochondrial dysfunction. In accordance, Shen et al. (178) demonstrated that cardiac overexpression of the mitochondrial antioxidant manganese superoxide dismutase (MnSOD) was able to reduce ROS and normalize mitochondrial function.
Mitochondrial ROS production in diabetes is suggested to be due to an increased supply of reducing equivalents from myocardial FA oxidation to the ETC, without a parallel increase in the OxPhos capacity. The subsequent loss of electrons from the ETC leads to the generation of superoxide (O2•−) at complex I and II. This notion has been challenged by Fauconnier et al. (64), who found that although palmitate profoundly increased mitochondrial ROS production in wild-type cardiomyocytes, this was not observed in cardiomyocytes from ob/ob mice. In addition, Tocchetti et al. (191) recently found that palmitate restored redox balance and counteracted the exaggerated ROS emission induced by hyperglycemia and β-adrenergic stimulation in isolated cardiomyocytes from db/db mice. Again, this suggests a complex role for FAs in the diabetic heart and the possibility that an adaptive response in diabetic mitochondria allows them to maintain homeostasis in the presence of high FAs.
A major nonmitochondrial source of ROS in cardiomyocytes are NADPH oxidases (NOXs) that produce ROS by using reducing equivalents from NADPH to molecular oxygen. NOX2 and NOX4 are the main isoforms in the cardiomyocyte, and their activation may modulate multiple signaling pathways and redox-sensitive proteins (7, 8). NOX signaling has been shown to influence hypertrophic pathways, fibrosis, cell viability, calcium handling, ER stress, and antioxidant capacity (40, 86, 145, 170). Potent activators of NOXs are angiotensin II, hyperglycemia, cytokines, oxidized low-density lipoprotein, mechanical forces, and hypoxia (8, 145). Interestingly, the two isoforms differ in mode of activation, subcellular location and they mediate distinct redox responses to agonist stimulation (8).
NOXs have been shown to play key roles in the pathogenesis of vascular disease in diabetes (74), and in the myocardium increased expression of NOXs has been found in type 1 and T2D models (72, 195). Both genetic and pharmacological inhibition of NOX2 has been found in T1D mice to reduce myocardial fibrosis, improve ER stress markers and cardiac function (122). Furthermore, treatment of T2D rodents with an angiotensin receptor blocker was found to reduce the cardiac expression of NOX2 together with reduced cardiac ROS and fibrosis (72, 195).
NO produced by different NO synthase (NOS) isoforms, working in discrete subcellular domains to induce S-nitrosylation, may alter the activity of proteins involved in physiological processes such as excitation-contraction (EC) coupling (174), β-adrenergic inotropic response (78), and also the Frank–Starling mechanism (159). However, under conditions of increased ROS, endothelial NOS (eNOS) may become uncoupled (45) and produce O2•−, which can react with inducible NOS (iNOS)-produced NO to form highly oxidant reactive nitric species.
Hyperglycemia favors iNOS expression in normal hearts (46), and in the diabetic myocardium an increased monomer-to-dimer ratio of eNOS leads to production of O2•− (209). Reduced NO bioavailability, increased nitrosative stress, and peroxynitrite formation are believed to contribute to the pathogenesis of vascular disease in diabetes (4, 150, 209), and nitrosative stress has also been shown to be associated with the progression of diabetic cardiomyopathy and increase of myocardial cell death (70, 108).
Another extra-mitochondrial source of ROS in the diabetic myocardium is the enzyme xanthine oxidase (XO) (161). Treatment with an inhibitor of XO (allopurinol) was shown to reduce the development of T1D-induced cardiac dysfunction together with decreased oxidative/nitrosative stress and fibrosis (75, 161).
Acute and long-term effects of exercise on ROS-producing enzymes
An acute bout of exercise will induce a cardiac stress (acute exercise stress, [AES]) and lead to a transient increase in reactive oxygen and nitrogen species (RONS) (26, 143, 160) (Fig. 4). This burst of RONS seems to be of utmost importance for the cardiac response to exercise since antioxidant therapies have been shown to impair health-promoting exercise effects in healthy young humans (166) and to impair beneficial cardiac responses in rodents (169).
Although the source of this transient increase in RONS is not clear, rigorous cardiac contractions during intense exercise have been suggested to increase the flow of electrons through the ETC and favor increased O2•− generation. In line with this, Bo et al. reported an elevated ROS production in isolated mitochondria from healthy rat hearts after prolonged acute exercise. This was associated with a transient increase in mitochondrial membrane potential, a marked increase in the expression of UCP2, increased uncoupled respiration, and reduced P/O ratio (26). Interestingly, mitochondrial ROS production was decreased and returned to resting levels as the exercise continued. The mitochondrial response was therefore regarded as “a first line of defense against ROS” where (at the expense of reduced mitochondrial efficiency) alleviation of mitochondrial membrane potential reduced O2•− generation (26).
It should be noted, however, that in the recently introduced “Redox-Optimized ROS Balance” (R-ORB) hypothesis (9), where the redox environment is taken into account as an important intermediary between mitochondrial respiration and ROS production, increased mitochondrial respiration is not accompanied by increased ROS, but rather by reduced ROS (55). This questions whether the mitochondria could be the main source of ROS during an acute exercise bout. Accordingly, several reports have suggested nonmitochondrial ROS-producing enzymes to be activated after AES.
An acute bout of exercise was shown to induce recruitment of catalytic subunits (p47phox and rac1) to NOX2 and increase its activity in ventricular tissue from dogs (169) as well as to induce a robust expression of NOX4 in mouse hearts (143). Furthermore, exhaustive exercise was reported to acutely increase XO activity in the myocardium of aging rats (99). Hence, there is evidence to support that several ROS-producing systems are activated in the heart after an AES; however, whether these systems work together or in discrete subcellular locations to induce specific cellular redox signaling remains to be elucidated.
Long-term endurance exercise may influence ROS-producing enzymes through very different mechanisms. First of all, several of the exercise-mediated systemic changes (improved insulin signaling, reduced inflammatory status, alteration of plasma lipids, and reduced renin-angiotensin system activity) most likely participate in decreasing ROS in the diabetic cardiomyocyte by dampening the activators of ROS-producing enzymes (Fig. 4). Bo et al. reported that the exercise-induced elevation of mitochondrial ROS production was attenuated in cardiac mitochondria from endurance trained rats (26), suggesting an adaptive response to better tolerate AES in the myocardium.
In the diabetic myocardium, where chronic NOX2 activation is believed to be detrimental, long-term endurance training significantly reduced NOX2 activity in T2D rats (82). Although the acute effects of exercise on cardiac NOS enzymes remain largely unexplored, long-term exercise was shown to increase NOS expression, NO levels, and NOS activity in healthy rat hearts (100). Such adaptations would be of particular benefit in the diabetic myocardium where NO bioavailability is reduced. In accordance with this, low-intensity endurance training of the T2D Goto-Kakizaki rat increased myocardial eNOS protein expression and eNOS dimer/monomer ratio, which facilitates NO generation compared with ROS production (82).
Impaired Endogenous Antioxidant Capacity in the Diabetic Myocardium
ROS formed is usually efficiently removed by endogenous antioxidants in the cardiomyocyte. Diabetic hearts, however, exhibit reduced expression and/or activity of several endogenous antioxidant enzymes and systems, including heme oxygenase 1 (HO-1) (117), superoxide dismutase (SOD) (4), catalase (195), glutathione perioxidase (GPx) (4), and the thioredoxin (Trx) system (149). The diabetic myocardium is also associated with a reduced ratio between reduced and oxidized glutathione (GSH/GSSG) (5, 68, 191). Genetic approaches aimed at increasing the endogenous antioxidants have been shown to reduce oxidative stress and rescue cardiomyocyte contractility, cardiac morphology, and/or mitochondrial function in diabetic models (124, 178, 192).
An important regulator of intracellular defense against ROS is the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2), which regulates the antioxidant response elements (ARE) mediated expression of detoxifying and antioxidant enzymes (106). Nrf2 stability and activity is regulated through redox modifications of the Kelch-like ECH-associated protein 1 (Keap1), where elevated H2O2 maintains the activation of Nrf2 (36). While acute hyperglycemia and short-term diabetes is associated with increased Nrf2 activation (91), sustained ROS and long-term diabetes seem to impair Nrf2 activity, most likely through the ERK pathway (187).
Exercise increases myocardial antioxidant capacity
In the study of an AES, Bo et al. (26) demonstrated a time-dependent increase in myocardial MnSOD protein and activity, which is likely an adaptive response to minimize exercise-induced increase in cardiac oxidative stress. Mutushamy et al. (143) also demonstrated that AES induced antioxidant defense pathways in the myocardium through activation of Nrf2. These changes were lost in Nrf2-/- mice, where AES resulted in increased oxidative stress and blunted the antioxidant responses in the myocardium (Fig. 5). Although there is no absolute consensus as to how endurance exercise influences antioxidant systems in the healthy heart, most studies have reported increased or unchanged gene expressions of antioxidant enzymes and systems after both long and short-term endurance training (11).
FIG. 5.

Proposed effects of exercise on endogenous antioxidant responses in the diabetic myocardium. In long-term diabetes, sustained reactive oxygen species (ROS) and impaired insulin signaling may lead to impaired nuclear factor erythroid 2-related factor 2 (Nrf2) activity due to exaggerated phosphorylation of the extracellular signal-related kinase (ERK) and reduced activity of phosphoinositol 3-kinase (PI3K). This may lead to reduced endogenous antioxidant capacity and oxidative stress, here illustrated by the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG). Acute exercise leads to a transient increase in reactive oxygen and nitrogen species (RONS) that stabilize and activate Nrf2, leading to transcriptional increase in endogenous antioxidant systems. Endurance training with repetitive bouts of RONS and improved insulin signaling can increase Nrf2 activity, which increases the antioxidant capacity of the myocardium. In conditions of impaired Nrf2 signaling and blunted antioxidant responses, exercise stress may lead to increased oxidative stress. See “Impaired Endogenous Antioxidant Capacity in the Diabetic Myocardium” section for references.
As the diabetic myocardium is associated with impaired antioxidant capacity, one would expect that exercise training could be particularly beneficial to the diabetic heart through enhancement of the myocardial antioxidant systems. Despite few studies on this issue, increased gene and protein expression of myocardial antioxidant systems have been reported in endurance trained diabetic and obese models (68, 85). Increased exercise-induced insulin sensitivity could potentially increase Nrf2 activity, as insulin signaling has been shown to increase myocardial Nrf2 activity via the PI3K pathway (187) (Fig. 5).
Finally, the progression and the severity of diabetes may be an important determination in how the diabetic heart tolerates and adapts to an AES and endurance training. Short-term (2 weeks) intense exercise of old and severely diabetic db/db mice using motorized wheels failed to induce the same antioxidant responses as seen in the myocardium of their control littermates (119). These hearts also exhibited reduced GSH/GSSG ratio, increased 4HNE content, and elevated fibrosis compared with untrained mice (119). Although not addressed or discussed in this article, this observation could be related to an impaired Nrf2 induction after the long-term diabetes (187). It could also be speculated that the use of motorized running wheels induced a considerable greater stress on the animals as compared with the use of treadmills or nonmotorized wheels where the animals can engage in more normal running behavior.
Autophagy in Diabetic Cardiomyopathy and the Potential Effects of Exercise
Elevated levels of nitrosative and oxidative stress are also believed to contribute to the disruption of autophagy that has been reported in diabetic cardiomyopathy and in hearts from rodents fed obesogenic diets (42, 194). Autophagy is viewed as a housekeeping process to maintain cellular homeostasis and is essential for the removal of protein and lipid aggregates, damaged and unwanted organelles. As the diabetic heart is more prone to adverse molecular modifications of proteins and lipids, impaired autophagy may be particularly harmful and thus a potential contributor to the development of diabetic cardiomyopathy (194). Autophagy has been shown to be an important adaptive response that counteracts the development of insulin resistance in response to a high-fat diet in mice, and loss of autophagy was shown to impair insulin action in obese mice (59).
Acute exercise has been demonstrated to induce autophagy both in skeletal and in cardiac muscle (90). Interestingly, both short- and long-term exercise has been shown to improve insulin sensitivity through an autophagy-dependent mechanism (90). Whether endurance exercise can amend blunted autophagy in diabetic myocardium is yet to be explored.
Altered Calcium Handling in the Diabetic Cardiomyocyte
Changes in intracellular Ca2+ handling are believed to be an important factor leading to ventricular dysfunction in diabetes. At the cellular level, diabetic cardiomyocytes are characterized by Ca2+ transients with lower amplitude and slower rate of decay, which is linked to impaired Ca2+ handling by the sarcoplasmic reticulum (SR) (Fig. 6). A reduced SERCA2a activity (18, 176, 184) most likely contributes to both diastolic and systolic dysfunction in diabetes, as it may lead to impaired SR Ca2+ release and reuptake and, subsequently, reduced SR Ca2+ load. Although the exact mechanisms behind these alterations are not fully elucidated, studies have reported a decrease in SERCA2a expression and/or changes in its regulator phospholamban (PLN) (18, 61, 114, 121, 152).
FIG. 6.
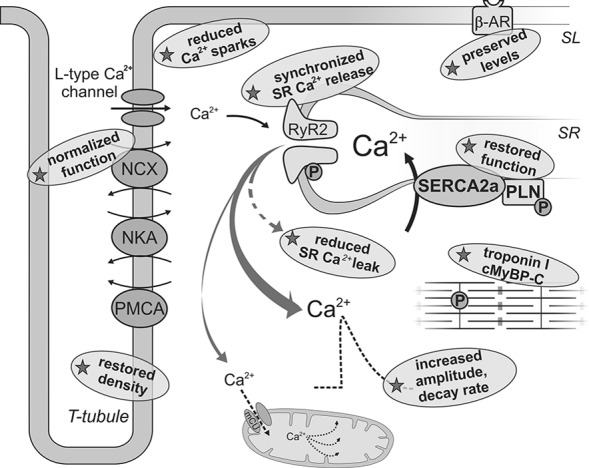
Impaired calcium homeostasis in diabetic cardiomyocyte is improved by exercise training. In diabetes, Ca2+ transients are altered due to impaired SERCA2a function and increased RyR2 Ca2+ leak. These cardiomyocytes are also more dependent on SL Ca2+ transport, have increased spontaneous Ca2+ spark frequency, dyssynchronous Ca2+ release, and reduced T-tubule density, as well as reduced β-adrenergic response. Mitochondrial Ca2+ cycling via the uniporter entry system (mCU) is essential in the activation of key metabolic enzymes. Indicated by stars are exercise-induced changes after endurance training in models of obesity/diabetes that may contribute to restoring myocardial Ca2+ homeostasis (see text for details and references). NKA, Na+/K+ ATPase; PLN, phospholamban: β-AR, β-adrenergic receptor; TCA, tricarboxylic acid; PDH, pyruvate dehydrogenase complex: cMyBp, cardiac myosin binding protein C. SL, sarcolemmal; SR, sarcoplasmatic reticulum; RyR2, ryanodine receptor 2 SERCA2a, SR Ca2+ ATPase; NCX, Na+/Ca2+ exchange; PMCA, SL Ca2+ ATPase.
Due to its slow turnover rate, SERCA2a is susceptible to post-translational modifications. An extensive SERCA2a glycation has been reported in the myocardium of STZ-induced diabetic rats (20), and treatment with the AGE cross-link breaker was found to partially normalize SR Ca2+ handling and cardiac function in an STZ model (114). Increased O-GlcNAcylation of PLN, SERCA2a and its transcription factor Sp1 have also been shown to be involved in myocardial SERCA2 downregulation and dysfunction after exposure to high glucose and in diabetes (52, 69). Although not extensively studied in diabetes, redox regulation of SERCA2a may also play a role, as ROS has been shown to reduce SERCA2a pump activity in heart failure (51).
It has also been shown that the decreased expression and function of SERCA2a is accompanied by an increased NCX expression and function in the diabetic myocardium (121, 184), which most likely serve as a compensatory mechanism to lower cytoplasmic Ca2+ levels. Furthermore, diabetic cardiomyocytes display increased SR Ca2+ leak through ryanodine receptors (RyR2) (16, 184), which may contribute toward reducing SR Ca2+ content. This does not seem to be associated with increased RyR2 protein expression (61, 152, 163, 184), but rather to altered RyR phosphorylation status due to changes in the activity of upstream kinases (149, 163, 176). O-GlcNAc-modified CaMKII was recently demonstrated in the heart of diabetic humans and rats by Erickson et al. (63), linking diabetic hyperglycemia to increased SR Ca2+ release events. In accordance with this, SR Ca2+ leak was normalized after inhibition of CaMKII in db/db cardiomyocytes (184).
Another plausible mediator of this SR Ca2+ leak is ROS, as it can increase the activity of upstream kinases such as PKA and CaMKII (35, 62). Oxidation of RyR2 can also promote increased opening probability of the channel and, consequently, promote Ca2+ leak (58). Increased spontaneous Ca2+ spark frequency and dyssynchronous Ca2+ release, in addition to reduced transverse (T)-tubule density, has also been reported in cardiomyocytes from db/db mice (184).
The diabetic myocardium is associated with an altered adrenergic contractile response due to reduced density of β-adrenoceptors (β-AR) (93), impaired β-adrenergic G-protein-adenylat cyclase system, and PLN phosphorylation (21, 73). Recent studies also suggest a cross-talk between IR activation and the β-AR2, where the contractile response to cardiac β-adrenergic stimuli was blunted through a β2AR-dependent mechanism (71).
It should be stressed that a common limitation in studies addressing calcium transport in cardiomyocytes is the lack of FAs as energy substrates. The diabetic cardiomyocyte most likely is adapted to and potentially dependent on FAs as an energy substrate. This notion is supported by studies showing that the addition of FA as a substrate significantly increases Ca2+ transients in diabetic cardiomyocytes (64, 191).
Exercise improves calcium homeostasis
Exercise training has been shown to improve cardiomyocyte contractility in obese/diabetic models, changes that have been associated with improved Ca2+ homeostasis (61, 137, 184). Exercise effectively restored myocardial expression and activity of SERCA2a (19, 61, 176, 184), associated with an increase in PLN phosphorylation at the Serine 16 (61, 184). In addition, exercise normalized the diabetic-induced Ca2+ SR leak in cardiomyocytes from db/db mice (184) and STZ rats (176). The reduced leak does not seem to be linked to altered RyR2 expression (61, 152, 176, 184) but rather to its phosphorylation status due to a normalization of CaMKII activity (176, 184). Stabilization of the RyR was also found to restore the synchronicity of SR Ca2+ release (184). In addition, Stolen et al. (184) reported that high-intensity interval training restored T-tubule density, which by improving the spacing between L-type Ca2+ channels and RyR2 may improve the efficiency of EC coupling.
Clinical studies have demonstrated that exercise interventions after the onset of diabetes can improve the responsiveness of the heart to β-adrenergic stimulation (196). In experimental models, this has been shown to be associated with preserved levels of β-adrenoceptors (21, 137). Finally, exercise-induced improvement of function in diabetic hearts may also be related to PKA-induced phosphorylation of the myofibrillar proteins troponin I and myosin-binding protein-C (111).
Cardiac Inefficiency and Oxygen Wasting in Diabetes
Despite its high metabolic activity, the heart has a relatively low content of high energy compounds. Consequently, the heart depends on optimal substrate metabolism and efficient ATP hydrolysis at energy consuming sites. Any imbalance between energy demand and availability can ultimately lead to an energetically compromised heart with reduced working capacity. In support of this notion, decreased myocardial energetics, measured as a reduced PCr:ATP ratio, has been shown to correlate with the severity of heart failure in patients with idiopathic dilated cardiomyopathy (146). Decreased PCr:ATP ratio has also been reported in hearts from obese and/or T2D subjects (154, 171), and impaired cardiac energetics due to cardiac inefficiency has been suggested to be an important mediator in the progression of obesity/diabetes-induced cardiac dysfunction.
Reduced cardiac mechanical efficiency, defined as the ratio between external work (stroke work) and MVO2 (23), has become a hallmark of the obese/diabetic heart, as it has been reported in a range of experimental studies (1, 32, 38, 53, 68, 85, 201) as well as in obese young women (155). Impaired mechanical efficiency also precedes the development of obesity/diabetes-related ventricular dysfunction (85, 155, 201).
A range of factors are known to determine MVO2; apart from heart rate, this includes factors related to the energy used for generating force/tension by the contractile machinery (i.e., pre- and after-load) and factors related to nonmechanical processes. It should be noted that mechanical efficiency is a load-dependent parameter and that afterload-dependent peak efficiency was not found to be affected in STZ-induced diabetes (87).
The underlying mechanisms leading to decreased efficiency in diabetic cardiomyopathy are far from clear. It is acknowledged that an acute elevation of FAs in the normal heart induces oxygen wasting (28, 53, 95, 138), and as FA is a less efficient substrate when compared with glucose (lower P/O ratio), increased FA oxidation is generally suggested to be causative for this increase in MVO2. Likewise, elevated FA oxidation in the diabetic heart is also regarded to be an important factor leading to decreased mechanical efficiency. It should be noted, however, that the heart should only use 11–12% more MVO2 when shifted from exclusively glucose to FA oxidation, which is too low to explain the reported increase in MVO2 under both increased FA load and diabetes where the change in substrate utilization is far less (95, 138).
Supporting the notion that additional oxygen consuming processes are activated are experimental data indicating that it is not the high myocardial FA oxidation rate (accompanying elevated FA concentration) that leads to the increased MVO2 in normal hearts (28, 102), but that FAs increase the nonwork-related oxygen demand for processes associated with EC coupling (28, 41). In contrast to what is observed in normal hearts, exposing hearts from T2D mice to high FA levels does not further increase MVO2 (despite elevation of FA oxidation rates) (28, 53, 95). Taken together, it is clear that the oxygen wasting effects of acute and a more long-term elevation of FAs levels are not necessarily via the same mechanisms and, importantly, FA oxidation rates per se do not seem to be the sole mechanism for the increased MVO2.
Assessment of left ventricular mechanoenergetic properties [by using the PVA-MVO2 framework (186)] has demonstrated that the diabetic heart uses more oxygen for nonmechanical processes (84, 85, 95), including processes associated with basal metabolism and EC coupling (27, 28, 85). The energy cost for EC coupling is primarily related to calcium transport, and changes in myocardial Ca2+ handling in the diabetic heart may therefore contribute to the observed increase in the oxygen cost of EC coupling. Oxygen wasting processes may include SR Ca2+ leak (16, 184) and subsequent futile cycling of Ca2+, increased use of the less energetically favorable NCX instead of SERCA2a for cytoplasmic Ca2+ clearance, as well as dyssynchronous SR Ca2+ release (181).
In addition, increased ventricular stiffness (and thus wall stress) can occur due to diabetes-induced fibrosis (101) and excessive accumulation of AGEs (114) may also contribute to a decreased mechanical efficiency in the diabetic heart. Myocardial oxygen consumption has been found to be correlated with levels of ROS in cardiac tissue (85), which suggests that oxidative stress may play a role in diabetes-induced oxygen wasting. In support of this, although not addressed in the diabetic myocardium, allopurinol (a XO inhibitor) infusion to heart failure patients significantly increased cardiac mechanical efficiency (44).
Exercise restores cardiac efficiency through oxygen sparing effects
Improved cardiac efficiency after endurance training is supported by studies done on patients with dilated cardiomyopathy (183, 193) and in a swine model of heart failure with a preserved ejection fraction (132). Recently, exercise training was also shown to improve mechanical efficiency due to reduced oxygen consumption for both basal metabolism and EC coupling in hearts from diet-induced obese mice (85). The exercise-induced reduction in oxygen wasting associated with EC coupling can be related to reduced RyR2 Ca2+ leak, improved SERCA2a function and normalization of SL Ca2+ transport, and synchronization of SR Ca2+ release. It is known that calcium-handling proteins are sensitive to the intracellular redox environment (35, 51, 58, 62), and it is, therefore, tempting to suggest that the reported positive correlation between myocardial ROS and MVO2 (85) reflects ROS-mediated changes in Ca2+ handling.
In addition, as exercise has been found to counteract obesity-induced left ventricular remodeling and to reduce ventricular fibrosis (85, 119, 128, 167); a reduced wall stress can also positively affect cardiac efficiency. Finally, as decreased efficiency will be particularly disadvantageous under conditions of reduced oxygen availability, one could also speculate that exercise-induced amendments of cardiac efficiency could also reduce ischemic susceptibility in the diabetic heart.
Concluding Remarks
Cardiovascular disease is the leading cause of mortality in people with diabetes, and there is no doubt that exercise training is efficient in the prevention and treatment of obesity and T2D. Exercise influences several of the systemic changes associated with diabetes/obesity such as insulin resistance, glycemic control, plasma lipid levels, and inflammatory status, and beneficial cardiac effects after exercise training in this patient group are reported.
Due to the complex nature of the development of T2D, the diversity of exercise training regimens, and wide range of physiological adaptations to exercise, it is not unexpected that reports regarding the mechanisms that may lead to the cardiac response to exercise training are diverging. In addition, studying molecular adaptions to exercise in the diabetic heart advocates the use of animal models where the severity and the progression of diabetes may vary, which may influence the tolerance and the adaptive response to acute and chronic exercise. It is essential that the models used in experimental studies of T2D closely resemble those of the human disease, and that changes in STZ-induced T1D models do not necessarily reflect the changes in obese T2D models. It is also likely that variations in animal diets can influence the adaptive response to exercise and the restitution between exercise sessions.
In many exercise studies, it is challenging to discriminate between the indirect effects linked to exercise-mediated systemic changes and the direct cardiac effect originating from the high contractile activity of the heart during bouts of physical training. In addition, there are clearly different molecular pathways involved in the cardiac response to an AES as opposed to the adaptive changes after repetitive bouts of high contractile activity during physical training. Moreover, there are most likely transient time-dependent changes during the cardiac adaptive responses to exercise.
The different training regimens with respect to exercise mode, duration, and intensity between studies may also cause discrepancies between findings. For instance, experimental and clinical studies have suggested that exercise interventions with higher intensities produce greater effects in terms of improving glycemic control and insulin sensitivity (85, 190, 200). High-intensity training has also been shown to produce greater aerobic capacity (92) and to produce greater cardiac adaptations than isocaloric low-moderate intensity training (83, 110).
Few experimental studies actually report how fitness and aerobic capacity are affected by the exercise protocols, and the role of exercise intensity in the adaptive responses in the diabetic heart is not well elucidated. As low aerobic capacity is a strong predictor of cardiovascular disease (25, 109), it would be a great advantage if the effect of the exercise regime used on VO2max was reported in experimental studies.
The beneficial cardiac effects of exercise in diabetic cardiomyopathy is supported by studies reporting exercise training to amend many of the metabolic disturbances characterizing the diabetic myocardium, including mitochondrial capacity and efficiency, carbohydrate oxidation, lipotoxicity, and disturbances in nonoxidative glucose pathways. In addition, direct and systemic effects of exercise training may influence ROS production and enhance endogenous antioxidant capacity, making exercise intervention a potent tool in amending the pro-oxidant milieu of the diabetic heart. Exercise-induced improvements of myocardial calcium handling are also of great therapeutic potential in the diabetic heart, as they may improve ventricular function, cardiac efficiency, and potentially also mitochondrial function. Revealing the molecular mechanisms behind the beneficial effects of exercise training is of considerable scientific value to generate evidence-based therapy and in the development of new therapeutic strategies.
Abbreviations Used
- β-AR
β-adrenoceptors
- 4HNE
4 hydroxynonenal
- ACC
acetyl-CoA carboxylase
- AES
acute exercise stress
- AGEs
advanced glycation end-products
- ANT
adenine nucleotide translocator
- AR
aldose reductase
- ARE
antioxidant response elements
- CaMKII
Ca2+-calmodulin-dependent protein kinase II
- cMyBp
cardiac myosin binding protein C
- CPT1 and 2
carnitine-palmitoyltransferases system 1 and 2
- CrAT
carnitine acetyltransferase
- DAG
diacylglycerol
- DGAT1
diacylglycerol transferase 1
- EC
excitation-contraction
- eNOS
endothelial NOS
- ERK
extracellular signal regulated kinase
- ETC
electron transport chain
- FA
fatty acids
- FABP
fatty acid binding protein
- FAT/CD36
fatty acid translocase
- FATP
fatty acid transport protein
- Foxo1
Forkhead box protein O1
- GLUT1
glucose transporter1
- GLUT4
glucose transporter4
- GPx
glutathione perioxidase
- GSSG
oxidized glutathione
- HbA1c
glycated hemoglobin
- HBP
hexosamine biosynthetic pathway
- HK
hexokinase
- HO-1
includes heme oxygenase 1
- iNOS
inducible NOS
- IR
insulin receptor
- IRS-1
insulin receptor substrate-1
- IRS-2
insulin receptor substrate-2
- JNK
c-Jun N-terminal kinase
- LCFA
long chain fatty acids
- LDH
lactate dehydrogenase
- LV
left ventricle
- MCD
malonyl-CoA decarboxylase
- mCU
mitochondrial Ca2+ uniporter
- MnSOD
manganese superoxide dismutase 0
- mPTP
mitochondrial permeability transition pore
- MVO2
myocardial oxygen consumption
- NADP
nicotinamide adenine dinucleotide phosphate
- NCX
sodium-calcium exchange
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- NOS
NO synthase
- NOX
NADPH oxidase
- Nrf2
nuclear factor erythroid 2-related factor 2
- O-GlcNAc
O-linked N-acteylglucosamine
- OxPhos
oxidative phosphorylation
- PCr:ATP
phosphocreatinine:adenosine triphosphate
- PDH
pyruvate dehydrogenase complex
- PDK4
pyruvate dehydrogenase kinase 4
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PKC
protein kinase C
- Plin-5
perilipin-5
- PLN
phospholamban
- PMCA
sarcolemmal Ca2+ ATPase
- PPAR-α
peroxisomal proliferator activated receptor- α
- PVA
pressure volume area
- RAGEs
AGE receptors
- RAS
renin-angiotensin system
- RCR
respiratory control ratio
- RONS
reactive oxygen and nitrogen species
- ROS
reactive oxygen species
- RyR2
ryanodine receptors
- Serca
sarcoplasmic reticulum Ca2+ ATPase
- SERCA2a
sarcoplasmatic reticulum Ca2+ ATPase
- SL
sarcolemmal
- SOD
superoxide dismutase
- SR
sarcoplasmic reticulum
- sRAGE
soluble form of RAGE
- STZ
streptozytocin
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TAG
triglyceride
- UCP
uncoupling proteins
- UDP-GlcNAc
uridine diphosphate N-acetylglucosamine
- VO2max
maximal oxygen uptake
- XO
xanthine oxidase
Acknowledgments
ADH is supported by a fellowship funded by the Norwegian Health Association. NTB is supported by a fellowship funded by UNIKARD (Norwegian Research Council and the Northern Norway Regional Health Authority).
References
- 1.Aasum E, Hafstad AD, Severson DL, and Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 52: 434–441, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Abdurrachim D, Ciapaite J, Wessels B, Nabben M, Luiken JJ, Nicolay K, and Prompers JJ. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta 1841: 1525–1537, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Aguilar H, Fricovsky E, Ihm S, Schimke M, Maya-Ramos L, Aroonsakool N, Ceballos G, Dillmann W, Villarreal F, and Ramirez-Sanchez I. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am J Physiol Cell Physiol 306: C794–C804, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aliciguzel Y, Ozen I, Aslan M, and Karayalcin U. Activities of xanthine oxidoreductase and antioxidant enzymes in different tissues of diabetic rats. J Lab Clin Med 142: 172–177, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, and Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 54: 1891–1898, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, and Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol 300: H118–H124, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anilkumar N, Sirker A, and Shah AM. Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front Biosci (Landmark Ed) 14: 3168–3187, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Anilkumar N, Weber R, Zhang M, Brewer A, and Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol 28: 1347–1354, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Aon MA, Cortassa S, and O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797: 865–877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asbun J. and Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol 47: 693–700, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Ascensao A, Ferreira R, and Magalhaes J. Exercise-induced cardioprotection—biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol 117: 16–30, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 34: 1259–1271, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Basta G, Sironi AM, Lazzerini G, Del TS, Buzzigoli E, Casolaro A, Natali A, Ferrannini E, and Gastaldelli A. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J Clin Endocrinol Metab 91: 4628–4634, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Beaudoin MS, Perry CG, Arkell AM, Chabowski A, Simpson JA, Wright DC, and Holloway GP. Impairments in mitochondrial palmitoyl-CoA respiratory kinetics that precede development of diabetic cardiomyopathy are prevented by resveratrol in ZDF rats. J Physiol 592: 2519–2533, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol (1985) 111: 157–162, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Belke DD. and Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 6: 424–429, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Belke DD, Larsen TS, Gibbs EM, and Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab 279: E1104–E1113, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Belke DD, Swanson EA, and Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 53: 3201–3208, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Bennett CE, Johnsen VL, Shearer J, and Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci 92: 657–663, 2013 [DOI] [PubMed] [Google Scholar]
- 20.Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, and Besch HR., Jr Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 53: 463–473, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Bidasee KR, Zheng H, Shao CH, Parbhu SK, Rozanski GJ, and Patel KP. Exercise training initiated after the onset of diabetes preserves myocardial function: effects on expression of beta-adrenoceptors. J Appl Physiol (1985) 105: 907–914, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bilet L, van de Weijer T, Hesselink MK, Glatz JF, Lamb HJ, Wildberger J, Kooi ME, Schrauwen P, and Schrauwen-Hinderling VB. Exercise-induced modulation of cardiac lipid content in healthy lean young men. Basic Res Cardiol 106: 307–315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bing RJ, Hammond , Handelsman JC, Powers SR, Spencer FC, Eckenhff JE, Goodal MD, Hafkenschiel JH, and Kety SS. The measurement of coronary blood flow, oxygen consumption, and efficiency of the left ventricle in man. Am Heart J 38: 1–24, 1949 [DOI] [PubMed] [Google Scholar]
- 24.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, and Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev 3: CD007176, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Blair SN. and Brodney S. Effects of physical inactivity and obesity on morbidity and mortality: current evidence and research issues. Med Sci Sports Exerc 31: S646–S662, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Bo H, Jiang N, Ma G, Qu J, Zhang G, Cao D, Wen L, Liu S, Ji LL, and Zhang Y. Regulation of mitochondrial uncoupling respiration during exercise in rat heart: role of reactive oxygen species (ROS) and uncoupling protein 2. Free Radic Biol Med 44: 1373–1381, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Boardman N, Hafstad AD, Larsen TS, Severson DL, and Aasum E. Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am J Physiol Heart Circ Physiol 296: H1373–H1379, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Boardman NT, Larsen TS, Severson DL, Essop MF, and Aasum E. Chronic and acute exposure of mouse hearts to fatty acids increases oxygen cost of excitation-contraction coupling. Am J Physiol Heart Circ Physiol 300: H1631–H1636, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Boor P, Celec P, Behuliak M, Grancic P, Kebis A, Kukan M, Pronayova N, Liptaj T, Ostendorf T, and Sebekova K. Regular moderate exercise reduces advanced glycation and ameliorates early diabetic nephropathy in obese Zucker rats. Metabolism 58: 1669–1677, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Botta A, Laher I, Beam J, Decoffe D, Brown K, Halder S, Devlin A, Gibson DL, and Ghosh S. Short term exercise induces PGC-1alpha, ameliorates inflammation and increases mitochondrial membrane proteins but fails to increase respiratory enzymes in aging diabetic hearts. PLoS One 8: e70248, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boudina S, Han YH, Pei S, Tidwell TJ, Henrie B, Tuinei J, Olsen C, Sena S, and Abel ED. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat-fed mice but not in leptin-deficient mice. Diabetes 61: 3260–3269, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, and Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112: 2686–2695, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, and Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Brandes R. and Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res 80: 82–87, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, and Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 281: 21827–21836, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Brewer AC, Murray TV, Arno M, Zhang M, Anilkumar NP, Mann GE, and Shah AM. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med 51: 205–215, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broderick TL, Poirier P, and Gillis M. Exercise training restores abnormal myocardial glucose utilization and cardiac function in diabetes. Diabetes Metab Res Rev 21: 44–50, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, and Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146: 5341–5349, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, and Allard MF. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol 287: H1055–H1063, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Burgoyne JR, Mongue-Din H, Eaton P, and Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res 111: 1091–1106, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, and Gerstenblith G. Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Physiol Heart Circ Physiol 261: H741–H750, 1991 [DOI] [PubMed] [Google Scholar]
- 42.Cai L. and Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol 3: 219–228, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, and Burrell LM. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res 92: 785–792, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, and Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104: 2407–2411, 2001 [DOI] [PubMed] [Google Scholar]
- 45.Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, and Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal 18: 1078–1099, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceriello A, Quagliaro L, D'Amico M, Di FC, Marfella R, Nappo F, Berrino L, Rossi F, and Giugliano D. Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes 51: 1076–1082, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Champattanachai V, Marchase RB, and Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol 294: C1509–C1520, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, and Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 96: 225–233, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, and Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107: 813–822, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi KM, Han KA, Ahn HJ, Hwang SY, Hong HC, Choi HY, Yang SJ, Yoo HJ, Baik SH, Choi DS, and Min KW. Effects of exercise on sRAGE levels and cardiometabolic risk factors in patients with type 2 diabetes: a randomized controlled trial. J Clin Endocrinol Metab 97: 3751–3758, 2012 [DOI] [PubMed] [Google Scholar]
- 51.Choudhary G. and Dudley SC., Jr Heart failure, oxidative stress, and ion channel modulation. Congest Heart Fail 8: 148–155, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, and Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 278: 44230–44237, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N, and Clarke K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 106: 447–457, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, and Gilbert RE. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail 2: 129–137, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Cortassa S, O'Rourke B, and Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta 1837: 287–295, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cox EJ. and Marsh SA. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol 12: 101, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di FC, Cuzzocrea S, Rossi F, Marfella R, and D'Amico M. Oxidative stress as the leading cause of acute myocardial infarction in diabetics. Cardiovasc Drug Rev 24: 77–87, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Eager KR. and Dulhunty AF. Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg2+ and ATP. J Membr Biol 163: 9–18, 1998 [DOI] [PubMed] [Google Scholar]
- 59.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, Kawamori R, Fujitani Y, and Watada H. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8: 325–332, 2008 [DOI] [PubMed] [Google Scholar]
- 60.Echtay KS, Murphy MP, Smith RA, Talbot DA, and Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem 277: 47129–47135, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Epp RA, Susser SE, Morissette MP, Kehler DS, Jassal DS, and Duhamel TA. Exercise training prevents the development of cardiac dysfunction in the low-dose streptozotocin diabetic rats fed a high-fat diet. Can J Physiol Pharmacol 91: 80–89, 2013 [DOI] [PubMed] [Google Scholar]
- 62.Erickson JR, He BJ, Grumbach IM, and Anderson ME. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev 91: 889–915, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, and Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502: 372–376, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R, Katz A, Bruton JD, and Westerblad H. Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes 56: 1136–1142, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, and Westerblad H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes 54: 2375–2381, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Fein FS. and Sonnenblick EH. Diabetic cardiomyopathy. Cardiovasc Drugs Ther 8: 65–73, 1994 [DOI] [PubMed] [Google Scholar]
- 67.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, and Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–130, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fisher-Wellman KH, Mattox TA, Thayne K, Katunga LA, La Favor JD, Neufer PD, Hickner RC, Wingard CJ, and Anderson EJ. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J Physiol 591: 3471–3486, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, Torres-Gonzalez M, Wang H, Ellrott I, Maya-Ramos L, Villarreal F, and Dillmann WH. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol 303: R689–R699, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, and Anversa P. Myocardial cell death in human diabetes. Circ Res 87: 1123–1132, 2000 [DOI] [PubMed] [Google Scholar]
- 71.Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, Zhang Y, Riehle C, Zhu Y, Rawlings T, Shi Q, Clark RB, Chen X, Abel ED, and Xiang YK. Insulin inhibits cardiac contractility by inducing a Gi-biased beta2-adrenergic signaling in hearts. Diabetes 63: 2676–2689, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, Ogawa H, and Kim-Mitsuyama S. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens 28: 340–352, 2010 [DOI] [PubMed] [Google Scholar]
- 73.Gando S, Hattori Y, Akaishi Y, Nishihira J, and Kanno M. Impaired contractile response to beta adrenoceptor stimulation in diabetic rat hearts: alterations in beta adrenoceptors-G protein-adenylate cyclase system and phospholamban phosphorylation. J Pharmacol Exp Ther 282: 475–484, 1997 [PubMed] [Google Scholar]
- 74.Gao L. and Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res 82: 9–20, 2009 [DOI] [PubMed] [Google Scholar]
- 75.Gao X, Xu Y, Xu B, Liu Y, Cai J, Liu HM, Lei S, Zhong YQ, Irwin MG, and Xia Z. Allopurinol attenuates left ventricular dysfunction in rats with early stages of streptozotocin-induced diabetes. Diabetes Metab Res Rev 28: 409–417, 2012 [DOI] [PubMed] [Google Scholar]
- 76.Gertz EW, Wisneski JA, Stanley WC, and Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest 82: 2017–2025, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giacco F. and Brownlee M. Oxidative stress and diabetic complications. Circ Res 107: 1058–1070, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Godecke A, Heinicke T, Kamkin A, Kiseleva I, Strasser RH, Decking UK, Stumpe T, Isenberg G, and Schrader J. Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J Physiol 532: 195–204, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Golbidi S. and Laher I. Molecular mechanisms in exercise-induced cardioprotection. Cardiol Res Pract 2011: 972807, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goldin A, Beckman JA, Schmidt AM, and Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114: 597–605, 2006 [DOI] [PubMed] [Google Scholar]
- 81.Golfman L, Wilson CR, Sharma S, Burgmaier M, Young ME, Guthrie PH, Van Varsdall M, Adrogue J, Brown KK, and Taegtmeyer H. Activation of PPAR enhances myocardial glucose oxidation and improves contractile function in isolated working hearts of ZDF rats. Am J Physiol Endocrinol Metab 289: E328–E336, 2005 [DOI] [PubMed] [Google Scholar]
- 82.Grijalva J, Hicks S, Zhao X, Medikayala S, Kaminski PM, Wolin MS, and Edwards JG. Exercise training enhanced myocardial endothelial nitric oxide synthase (eNOS) function in diabetic Goto-Kakizaki (GK) rats. Cardiovasc Diabetol 7: 34, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hafstad AD, Boardman NT, Lund J, Hagve M, Khalid AM, Wisloff U, Larsen TS, and Aasum E. High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J Appl Physiol (1985) 111: 1235–1241, 2011 [DOI] [PubMed] [Google Scholar]
- 84.Hafstad AD, Khalid AM, How OJ, Larsen TS, and Aasum E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab 292: E1288–E1294, 2007 [DOI] [PubMed] [Google Scholar]
- 85.Hafstad AD, Lund J, Hadler-Olsen E, Hoper AC, Larsen TS, and Aasum E. High- and moderate-intensity training normalizes ventricular function and mechanoenergetics in mice with diet-induced obesity. Diabetes 62: 2287–2294, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hafstad AD, Nabeebaccus AA, and Shah AM. Novel aspects of ROS signalling in heart failure. Basic Res Cardiol 108: 359, 2013 [DOI] [PubMed] [Google Scholar]
- 87.Han JC, Goo S, Barrett CJ, Mellor KM, Taberner AJ, and Loiselle DS. The afterload-dependent peak efficiency of the isolated working rat heart is unaffected by streptozotocin-induced diabetes. Cardiovasc Diabetol 13: 4, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harmancey R, Vasquez HG, Guthrie PH, and Taegtmeyer H. Decreased long-chain fatty acid oxidation impairs postischemic recovery of the insulin-resistant rat heart. FASEB J 27: 3966–3978, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hawkins M, Barzilai N, Liu R, Hu M, Chen W, and Rossetti L. Role of the glucosamine pathway in fat-induced insulin resistance. J Clin Invest 99: 2173–2182, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, and Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He X, Kan H, Cai L, and Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol 46: 47–58, 2009 [DOI] [PubMed] [Google Scholar]
- 92.Helgerud J, Hoydal K, Wang E, Karlsen T, Berg P, Bjerkaas M, Simonsen T, Helgesen C, Hjorth N, Bach R, and Hoff J. Aerobic high-intensity intervals improve VO2max more than moderate training. Med Sci Sports Exerc 39: 665–671, 2007 [DOI] [PubMed] [Google Scholar]
- 93.Heyliger CE, Pierce GN, Singal PK, Beamish RE, and Dhalla NS. Cardiac alpha- and beta-adrenergic receptor alterations in diabetic cardiomyopathy. Basic Res Cardiol 77: 610–618, 1982 [DOI] [PubMed] [Google Scholar]
- 94.Hollekim-Strand SM, Bjorgaas MR, Albrektsen G, Tjonna AE, Wisloff U, and Ingul CB. High-intensity interval exercise effectively improves cardiac function in patients with type 2 diabetes mellitus and diastolic dysfunction: a randomized controlled trial. J Am Coll Cardiol 64: 1758–1760, 2014 [DOI] [PubMed] [Google Scholar]
- 95.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, and Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 55: 466–473, 2006 [DOI] [PubMed] [Google Scholar]
- 96.How OJ, Larsen TS, Hafstad AD, Khalid A, Myhre ES, Murray AJ, Boardman NT, Cole M, Clarke K, Severson DL, and Aasum E. Rosiglitazone treatment improves cardiac efficiency in hearts from diabetic mice. Arch Physiol Biochem 113: 211–220, 2007 [DOI] [PubMed] [Google Scholar]
- 97.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, and Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res 96: 1006–1013, 2005 [DOI] [PubMed] [Google Scholar]
- 98.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, and Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 284: 547–555, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang CC, Tsai SC, and Lin WT. Potential ergogenic effects of L-arginine against oxidative and inflammatory stress induced by acute exercise in aging rats. Exp Gerontol 43: 571–577, 2008 [DOI] [PubMed] [Google Scholar]
- 100.Husain K. and Hazelrigg SR. Oxidative injury due to chronic nitric oxide synthase inhibition in rat: effect of regular exercise on the heart. Biochim Biophys Acta 1587: 75–82, 2002 [DOI] [PubMed] [Google Scholar]
- 101.Hutchinson KR, Lord CK, West TA, and Stewart JA., Jr Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS One 8: e72080, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hutter JF, Piper HM, and Spieckerman PG. Effect of fatty acid oxidation on efficiency of energy production in rat heart. Am J Physiol 249: H723–H728, 1985 [DOI] [PubMed] [Google Scholar]
- 103.Huynh K, Bernardo BC, McMullen JR, and Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther 142: 375–415, 2014 [DOI] [PubMed] [Google Scholar]
- 104.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, and King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 89: 11059–11063, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iwata K, Nishinaka T, Matsuno K, Kakehi T, Katsuyama M, Ibi M, and Yabe-Nishimura C. The activity of aldose reductase is elevated in diabetic mouse heart. J Pharmacol Sci 103: 408–416, 2007 [DOI] [PubMed] [Google Scholar]
- 106.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36: 1199–1207, 2004 [DOI] [PubMed] [Google Scholar]
- 107.Jonker JT, de MP, de Vries ST, Widya RL, Hammer S, van Schinkel LD, van der Meer RW, Gans RO, Webb AG, Kan HE, de Koning EJ, Bilo HJ, and Lamb HJ. Exercise and type 2 diabetes mellitus: changes in tissue-specific fat distribution and cardiac function. Radiology 269: 434–442, 2013 [DOI] [PubMed] [Google Scholar]
- 108.Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, and Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 50: 1414–1424, 2001 [DOI] [PubMed] [Google Scholar]
- 109.Kavanagh T, Mertens DJ, Hamm LF, Beyene J, Kennedy J, Corey P, and Shephard RJ. Prediction of long-term prognosis in 12 169 men referred for cardiac rehabilitation. Circulation 106: 666–671, 2002 [DOI] [PubMed] [Google Scholar]
- 110.Kemi OJ, Haram PM, Loennechen JP, Osnes JB, Skomedal T, Wisloff U, and Ellingsen O. Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function. Cardiovasc Res 67: 161–172, 2005 [DOI] [PubMed] [Google Scholar]
- 111.Korte FS, Mokelke EA, Sturek M, and McDonald KS. Exercise improves impaired ventricular function and alterations of cardiac myofibrillar proteins in diabetic dyslipidemic pigs. J Appl Physiol (1985) 98: 461–467, 2005 [DOI] [PubMed] [Google Scholar]
- 112.Koves TR, Sparks LM, Kovalik JP, Mosedale M, Arumugam R, DeBalsi KL, Everingham K, Thorne L, Phielix E, Meex RC, Kien CL, Hesselink MK, Schrauwen P, and Muoio DM. PPARgamma coactivator-1alpha contributes to exercise-induced regulation of intramuscular lipid droplet programming in mice and humans. J Lipid Res 54: 522–534, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, and Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008 [DOI] [PubMed] [Google Scholar]
- 114.Kranstuber AL, Del RC, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, and Lacombe VA. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front Physiol 3: 292, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kuramoto K, Okamura T, Yamaguchi T, Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K, Usuda N, Matsumura S, Inoue K, Fushiki T, Kojima Y, Hashimoto T, Sakai F, Hirose F, and Osumi T. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J Biol Chem 287: 23852–23863, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kuramoto K, Sakai F, Yoshinori N, Nakamura TY, Wakabayashi S, Kojidani T, Haraguchi T, Hirose F, and Osumi T. Deficiency of a lipid droplet protein, perilipin 5, suppresses myocardial lipid accumulation, thereby preventing type 1 diabetes-induced heart malfunction. Mol Cell Biol 34: 2721–2731, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.L'Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, Barsacchi R, Paolicchi A, Masiello P, Drummond GS, McClung JA, and Abraham NG. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol 293: H3532–H3541, 2007 [DOI] [PubMed] [Google Scholar]
- 118.Laczy B, Fulop N, Onay-Besikci A, Des RC, and Chatham JC. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS One 6: e18417, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Laher I, Beam J, Botta A, Barendregt R, Sulistyoningrum D, Devlin A, Rheault M, and Ghosh S. Short-term exercise worsens cardiac oxidative stress and fibrosis in 8-month-old db/db mice by depleting cardiac glutathione. Free Radic Res 47: 44–54, 2013 [DOI] [PubMed] [Google Scholar]
- 120.Lancaster GI. and Febbraio MA. The immunomodulating role of exercise in metabolic disease. Trends Immunol 35: 262–269, 2014 [DOI] [PubMed] [Google Scholar]
- 121.Larocca TJ, Fabris F, Chen J, Benhayon D, Zhang S, McCollum L, Schecter AD, Cheung JY, Sobie EA, Hajjar RJ, and Lebeche D. Na+/Ca2+ exchanger-1 protects against systolic failure in the Akitains2 model of diabetic cardiomyopathy via a CXCR4/NF-kappaB pathway. Am J Physiol Heart Circ Physiol 303: H353–H367, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J, Zhu H, Shen E, Wan L, Arnold JM, and Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59: 2033–2042, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li L, Muhlfeld C, Niemann B, Pan R, Li R, Hilfiker-Kleiner D, Chen Y, and Rohrbach S. Mitochondrial biogenesis and PGC-1alpha deacetylation by chronic treadmill exercise: differential response in cardiac and skeletal muscle. Basic Res Cardiol 106: 1221–1234, 2011 [DOI] [PubMed] [Google Scholar]
- 124.Liang Q, Carlson EC, Donthi RV, Kralik PM, Shen X, and Epstein PN. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes 51: 174–181, 2002 [DOI] [PubMed] [Google Scholar]
- 125.Liu J, Marchase RB, and Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol 42: 177–185, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, and Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284: 36312–36323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu L, Yu S, Khan RS, Homma S, Schulze PC, Blaner WS, Yin Y, and Goldberg IJ. Diacylglycerol acyl transferase 1 overexpression detoxifies cardiac lipids in PPARgamma transgenic mice. J Lipid Res 53: 1482–1492, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loganathan R, Novikova L, Boulatnikov IG, and Smirnova IV. Exercise-induced cardiac performance in autoimmune (type 1) diabetes is associated with a decrease in myocardial diacylglycerol. J Appl Physiol (1985) 113: 817–826, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, and Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010 [DOI] [PubMed] [Google Scholar]
- 130.Makrecka M, Kuka J, Volska K, Antone U, Sevostjanovs E, Cirule H, Grinberga S, Pugovics O, Dambrova M, and Liepinsh E. Long-chain acylcarnitine content determines the pattern of energy metabolism in cardiac mitochondria. Mol Cell Biochem 395: 1–10, 2014 [DOI] [PubMed] [Google Scholar]
- 131.Marsh SA, Powell PC, Dell'Italia LJ, and Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92: 648–656, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marshall KD, Muller BN, Krenz M, Hanft LM, McDonald KS, Dellsperger KC, and Emter CA. Heart failure with preserved ejection fraction: chronic low-intensity interval exercise training preserves myocardial O2 balance and diastolic function. J Appl Physiol (1985) 114: 131–147, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McLarty JL, Marsh SA, and Chatham JC. Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sci 92: 621–627, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Medeiros C, Frederico MJ, da LG, Pauli JR, Silva AS, Pinho RA, Velloso LA, Ropelle ER, and De Souza CT. Exercise training reduces insulin resistance and upregulates the mTOR/p70S6k pathway in cardiac muscle of diet-induced obesity rats. J Cell Physiol 226: 666–674, 2011 [DOI] [PubMed] [Google Scholar]
- 135.Medford HM, Chatham JC, and Marsh SA. Chronic ingestion of a Western diet increases O-linked-beta-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci 90: 883–888, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Medford HM, Porter K, and Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol 305: H114–H123, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mishra PK, Awe O, Metreveli N, Qipshidze N, Joshua IG, and Tyagi SC. Exercise mitigates homocysteine - beta2-adrenergic receptor interactions to ameliorate contractile dysfunction in diabetes. Int J Physiol Pathophysiol Pharmacol 3: 97–106, 2011 [PMC free article] [PubMed] [Google Scholar]
- 138.Mjøs OD. Effect of fatty acids on myocardial function and oxygen consumtion in intact dogs. J Clin Invest 50: 1386–1389, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Muoio DM. and Neufer PD. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab 15: 595–605, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, Covington JD, Bajpeyi S, Ravussin E, Kraus W, Koves TR, and Mynatt RL. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab 15: 764–777, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Murray AJ, Knight NS, Little SE, Cochlin LE, Clements M, and Clarke K. Dietary long-chain, but not medium-chain, triglycerides impair exercise performance and uncouple cardiac mitochondria in rats. Nutr Metab (Lond) 8: 55, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Murray AJ, Panagia M, Hauton D, Gibbons GF, and Clarke K. Plasma Free Fatty Acids and Peroxisome Proliferator-Activated Receptor {alpha} in the Control of Myocardial Uncoupling Protein Levels. Diabetes 54: 3496–3502, 2005 [DOI] [PubMed] [Google Scholar]
- 143.Muthusamy VR, Kannan S, Sadhaasivam K, Gounder SS, Davidson CJ, Boeheme C, Hoidal JR, Wang L, and Rajasekaran NS. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med 52: 366–376, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Myers J, Prakash M, Froelicher V, Do D, Partington S, and Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 346: 793–801, 2002 [DOI] [PubMed] [Google Scholar]
- 145.Nabeebaccus A, Zhang M, and Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev 16: 5–12, 2011 [DOI] [PubMed] [Google Scholar]
- 146.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, and Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 96: 2190–2196, 1997 [DOI] [PubMed] [Google Scholar]
- 147.Ngoh GA, Hamid T, Prabhu SD, and Jones SP. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am J Physiol Heart Circ Physiol 297: H1711–H1719, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, and Abel ED. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab 6: 294–306, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Okatan EN, Tuncay E, and Turan B. Cardioprotective effect of selenium via modulation of cardiac ryanodine receptor calcium release channels in diabetic rat cardiomyocytes through thioredoxin system. J Nutr Biochem 24: 2110–2118, 2013 [DOI] [PubMed] [Google Scholar]
- 150.Pacher P, Obrosova IG, Mabley JG, and Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem 12: 267–275, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, and Lavandero S. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res 77: 387–397, 2008 [DOI] [PubMed] [Google Scholar]
- 152.Paulino EC, Ferreira JC, Bechara LR, Tsutsui JM, Mathias W, Jr., Lima FB, Casarini DE, Cicogna AC, Brum PC, and Negrao CE. Exercise training and caloric restriction prevent reduction in cardiac Ca2+-handling protein profile in obese rats. Hypertension 56: 629–635, 2010 [DOI] [PubMed] [Google Scholar]
- 153.Paulson DJ, Mathews R, Bowman J, and Zhao J. Metabolic effects of treadmill exercise training on the diabetic heart. J Appl Physiol (1985) 73: 265–271, 1992 [DOI] [PubMed] [Google Scholar]
- 154.Perseghin G, Ntali G, De CF, Lattuada G, Esposito A, Belloni E, Canu T, Costantino F, Ragogna F, Scifo P, Del MA, and Luzi L. Abnormal left ventricular energy metabolism in obese men with preserved systolic and diastolic functions is associated with insulin resistance. Diabetes Care 30: 1520–1526, 2007 [DOI] [PubMed] [Google Scholar]
- 155.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, and Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 109: 2191–2196, 2004 [DOI] [PubMed] [Google Scholar]
- 156.Pierce GN. and Dhalla NS. Heart mitochondrial function in chronic experimental diabetes in rats. Can J Cardiol 1: 48–54, 1985 [PubMed] [Google Scholar]
- 157.Pieri BL, Souza DR, Luciano TF, Marques SO, Pauli JR, Silva AS, Ropelle ER, Pinho RA, Lira FS, and De Souza CT. Effects of Physical Exercise on the P38MAPK/REDD1/14–3-3 Pathways in the Myocardium of Diet-Induced Obesity Rats. Horm Metab Res 46: 621–627, 2014 [DOI] [PubMed] [Google Scholar]
- 158.Powers SK, Quindry JC, and Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med 44: 193–201, 2008 [DOI] [PubMed] [Google Scholar]
- 159.Prendergast BD, Sagach VF, and Shah AM. Basal release of nitric oxide augments the Frank-Starling response in the isolated heart. Circulation 96: 1320–1329, 1997 [DOI] [PubMed] [Google Scholar]
- 160.Radak Z, Taylor AW, Ohno H, and Goto S. Adaptation to exercise-induced oxidative stress: from muscle to brain. Exerc Immunol Rev 7: 90–107, 2001 [PubMed] [Google Scholar]
- 161.Rajesh M, Mukhopadhyay P, Batkai S, Mukhopadhyay B, Patel V, Hasko G, Szabo C, Mabley JG, Liaudet L, and Pacher P. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med 13: 2330–2341, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ramakrishna V. and Jailkhani R. Oxidative stress in non-insulin-dependent diabetes mellitus (NIDDM) patients. Acta Diabetol 45: 41–46, 2008 [DOI] [PubMed] [Google Scholar]
- 163.Reuter H, Gronke S, Adam C, Ribati M, Brabender J, Zobel C, Frank KF, Wippermann J, Schwinger RH, Brixius K, and Muller-Ehmsen J. Sarcoplasmic Ca2+ release is prolonged in nonfailing myocardium of diabetic patients. Mol Cell Biochem 308: 141–149, 2008 [DOI] [PubMed] [Google Scholar]
- 164.Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, and Abel ED. Insulin Receptor Substrates (IRS) are Essential for the Bioenergetic and Hypertrophic Response of the Heart to Exercise Training. Mol Cell Biol 34: 3450–3460, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de RA, and Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52: 1793–1799, 2008 [DOI] [PubMed] [Google Scholar]
- 166.Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, and Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 106: 8665–8670, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sacre JW, Jellis CL, Jenkins C, Haluska BA, Baumert M, Coombes JS, and Marwick TH. A six-month exercise intervention in subclinical diabetic heart disease: Effects on exercise capacity, autonomic and myocardial function. Metabolism 63: 1104–1114, 2014 [DOI] [PubMed] [Google Scholar]
- 168.Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, and Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol 571: 253–273, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sanchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Hartel S, Hidalgo C, and Donoso P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res 77: 380–386, 2008 [DOI] [PubMed] [Google Scholar]
- 170.Santos CX, Nabeebaccus AA, Shah AM, Camargo LL, Filho SV, and Lopes LR. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 20: 121–134, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, and Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 107: 3040–3046, 2003 [DOI] [PubMed] [Google Scholar]
- 172.Schrauwen-Hinderling VB, Hesselink MK, Meex R, van der Made S, Schar M, Lamb H, Wildberger JE, Glatz J, Snoep G, Kooi ME, and Schrauwen P. Improved ejection fraction after exercise training in obesity is accompanied by reduced cardiac lipid content. J Clin Endocrinol Metab 95: 1932–1938, 2010 [DOI] [PubMed] [Google Scholar]
- 173.Schrauwen-Hinderling VB, Meex RC, Hesselink MK, van de Weijer T, Leiner T, Schar M, Lamb HJ, Wildberger JE, Glatz JF, Schrauwen P, and Kooi ME. Cardiac lipid content is unresponsive to a physical activity training intervention in type 2 diabetic patients, despite improved ejection fraction. Cardiovasc Diabetol 10: 47, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, and Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res 92: e52–e59, 2003 [DOI] [PubMed] [Google Scholar]
- 175.Seiler SE, Martin OJ, Noland RC, Slentz DH, DeBalsi KL, Ilkayeva OR, An J, Newgard CB, Koves TR, and Muoio DM. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J Lipid Res 55: 635–644, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shao CH, Wehrens XH, Wyatt TA, Parbhu S, Rozanski GJ, Patel KP, and Bidasee KR. Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J Appl Physiol (1985) 106: 1280–1292, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shearer J, Ross KD, Hughey CC, Johnsen VL, Hittel DS, and Severson DL. Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab 301: E31–E39, 2011 [DOI] [PubMed] [Google Scholar]
- 178.Shen X, Zheng S, Metreveli NS, and Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55: 798–805, 2006 [DOI] [PubMed] [Google Scholar]
- 179.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM, Jr., Klein JB, and Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab 287: E896–E905, 2004 [DOI] [PubMed] [Google Scholar]
- 180.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, and Komuro I. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest 120: 1506–1514, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shimizu J, Yamashita D, Misawa H, Tohne K, Matsuoka S, Kim B, Takeuchi A, Nakajima-Takenaka C, and Takaki M. Increased O2 consumption in excitation-contraction coupling in hypertrophied rat heart slices related to increased Na+-Ca2+ exchange activity. J Physiol Sci 59: 63–74, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, and Brown DA. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol 52: 1009–1018, 2012 [DOI] [PubMed] [Google Scholar]
- 183.Stolen KQ, Kemppainen J, Ukkonen H, Kalliokoski KK, Luotolahti M, Lehikoinen P, Hamalainen H, Salo T, Airaksinen KE, Nuutila P, and Knuuti J. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol 41: 460–467, 2003 [DOI] [PubMed] [Google Scholar]
- 184.Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, and Wisloff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res 105: 527–536, 2009 [DOI] [PubMed] [Google Scholar]
- 185.Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, and Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J 272: 2684–2695, 2005 [DOI] [PubMed] [Google Scholar]
- 186.Suga H. Ventricular energetics. Physiol Rev 70: 247–277, 1990 [DOI] [PubMed] [Google Scholar]
- 187.Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, Goldblatt CS, Meyer CJ, Li X, Cai L, and Cui T. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 60: 625–633, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tang WH, Cheng WT, Kravtsov GM, Tong XY, Hou XY, Chung SK, and Chung SS. Cardiac contractile dysfunction during acute hyperglycemia due to impairment of SERCA by polyol pathway-mediated oxidative stress. Am J Physiol Cell Physiol 299: C643–C653, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Thirunavukkarasu M, Penumathsa SV, Koneru S, Juhasz B, Zhan L, Otani H, Bagchi D, Das DK, and Maulik N. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med 43: 720–729, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tjonna AE, Lee SJ, Rognmo O, Stolen TO, Bye A, Haram PM, Loennechen JP, Al-Share QY, Skogvoll E, Slordahl SA, Kemi OJ, Najjar SM, and Wisloff U. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: a pilot study. Circulation 118: 346–354, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O'Rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, and Aon MA. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 61: 3094–3105, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Turdi S, Li Q, Lopez FL, and Ren J. Catalase alleviates cardiomyocyte dysfunction in diabetes: role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci 81: 895–905, 2007 [DOI] [PubMed] [Google Scholar]
- 193.Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, and Knuuti J. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114: 2130–2137, 2006 [DOI] [PubMed] [Google Scholar]
- 194.Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, and Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta 1852: 232–242, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vazquez-Medina JP, Popovich I, Thorwald MA, Viscarra JA, Rodriguez R, Sonanez-Org , Lam L, Peti-Peterdi J, Nakano D, Nishiyama A, and Ortiz RM. Angiotensin receptor-mediated oxidative stress is associated with impaired cardiac redox signaling and mitochondrial function in insulin-resistant rats. Am J Physiol Heart Circ Physiol 305: H599–H607, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Voulgari C, Pagoni S, Vinik A, and Poirier P. Exercise improves cardiac autonomic function in obesity and diabetes. Metabolism 62: 609–621, 2013 [DOI] [PubMed] [Google Scholar]
- 197.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, and King GL. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A 94: 9320–9325, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE, and Hill JA. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156: 1179–1192, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, and Jones SP. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A 107: 17797–17802, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Weston KS, Wisloff U, and Coombes JS. High-intensity interval training in patients with lifestyle-induced cardiometabolic disease: a systematic review and meta-analysis. Br J Sports Med 48: 1227–1234, 2014 [DOI] [PubMed] [Google Scholar]
- 201.Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, Ilkun O, Theobald HA, Cooksey RC, Kandror KV, and Abel ED. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res 82: 351–360, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wright KJ, Thomas MM, Betik AC, Belke D, and Hepple RT. Exercise training initiated in late middle age attenuates cardiac fibrosis and advanced glycation end-product accumulation in senescent rats. Exp Gerontol 50: 9–18, 2014 [DOI] [PubMed] [Google Scholar]
- 203.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, and Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, and Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53: 1336–1343, 2004 [DOI] [PubMed] [Google Scholar]
- 205.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, and Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J 370: 1097–1109, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zachara NE. and Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta 1761: 599–617, 2006 [DOI] [PubMed] [Google Scholar]
- 207.Zanuso S, Jimenez A, Pugliese G, Corigliano G, and Balducci S. Exercise for the management of type 2 diabetes: a review of the evidence. Acta Diabetol 47: 15–22, 2010 [DOI] [PubMed] [Google Scholar]
- 208.Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, and Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res 89: 148–156, 2011 [DOI] [PubMed] [Google Scholar]
- 209.Zou MH, Shi C, and Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 109: 817–826, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]