

Abstract
Although a myriad of pathological responses contribute to traumatic brain injury (TBI), cerebral dysfunction has been closely linked to cell death mechanisms. A number of therapeutic strategies have been studied in an attempt to minimize or ameliorate tissue damage; however, few studies have evaluated the inherent protective capacity of the brain. Endogenous neural stem/progenitor cells (NSPCs) reside in distinct brain regions and have been shown to respond to tissue damage by migrating to regions of injury. Until now, it remained unknown whether these cells have the capacity to promote endogenous repair. We ablated NSPCs in the subventricular zone to examine their contribution to the injury microenvironment after controlled cortical impact (CCI) injury. Studies were performed in transgenic mice expressing the herpes simplex virus thymidine kinase gene under the control of the nestinδ promoter exposed to CCI injury. Two weeks after CCI injury, mice deficient in NSPCs had reduced neuronal survival in the perilesional cortex and fewer Iba-1-positive and glial fibrillary acidic protein-positive glial cells but increased glial hypertrophy at the injury site. These findings suggest that the presence of NSPCs play a supportive role in the cortex to promote neuronal survival and glial cell expansion after TBI injury, which corresponds with improvements in motor function. We conclude that enhancing this endogenous response may have acute protective roles after TBI.
Key words: : gliosis, neural stem/progenitor cell ablation, neurogenesis, neuronal survival, traumatic brain injury
Introduction
Patients with traumatic brain injury (TBI) have severe functional deficits for which there are currently few treatment options. The pathophysiological sequelae of tissue damage after TBI result from an acute and subsequent chronic phase of injury that ends in cell death and neural circuitry dysfunction. Neuronal death in the acute phase is initiated by mechanical forces, which alter cell membrane and vascular integrity, creating an injury microenvironment susceptible to necrosis and apoptosis.1–4 The secondary phase of injury occurs minutes to months after the onset of TBI whereby the release of pro-inflammatory cytokines mobilizes immune and glial cells to the injury environment causing edema and inflammation.5–10 This phase is also associated with gliosis, demyelination, and continued apoptosis. Therapeutic treatments for TBI will need to minimize progressive neuronal death and possibly replace neurons to re-create functional circuits.
Modeling stroke and TBI in animals has identified a unique response of the subventricular zone (SVZ), where endogenous neural stem/progenitor cells (NSPCs) undergo a transient expansion and begin migrating to sites of tissue damage.11–13 This enhanced neurogenic response provides a potential source of multipotent cells capable of integrating into existing neural networks to promote functional recovery.14–19 Unfortunately, this response is limited, and the degree of neuronal replacement is minimal. It is also possible that these cells may also influence the injury microenvironment. Chemical inhibition of proliferation using Ara-C in the adult ischemic brain resulted in reduced NSPC expansion that associated with increased infarct size and decreased function, supporting a role for NSPC in tissue damage after stroke.20 Ablation of adult neuroblasts in mouse stroke models also revealed greater tissue loss and behavioral deficits,21–24 although the mechanisms of how SVZ-derived cells regulate tissue responses is still unclear.
Transplantation approaches using either undifferentiated stem or stem-like cells from multiple sources could elicit both beneficial and non-beneficial central nervous system (CNS) responses on the injury milieu, because these cells produce significant amounts of growth factors, cytokines, and chemokines. Transplantation of mesenchymal stem cells was similar to ventricular neural stem cell transplants in their ability to generate anti-inflammatory response after CNS damage,25,26 which may contribute to injury progression and recovery. It is also possible that endogenous stem cells may have direct influences on the survival and proliferation of residential neural and glial cells in the injury environment, but the role of SVZ-derived NSPCs in the traumatic injured brain remains to be determined.
Therefore, we will examine the influence of adult neurogenesis on residential neurons and glial cells in the TBI cortex using nestinδ-TK+ transgenic mice to selectively ablate NSPCs.
Methods
Animals
The generation of the transgenic CD-1 mice expressing the herpes simplex virus thymidine kinase (TK) gene under control of the nestin-delta (d) promoter, nestinδ-TK- and nestinδ-TK+ mice, has been described previously.27 The nestinδ promoter has been shown to be absent from neuronal or glial populations,27 where green fluorescent protein (GFP) expression was not observed in naïve cortical tissues but was observed in perilesional regions 2 days post-controlled cortical impact (CCI) injury (DPI). Administration of ganciclovir sodium (Cytovene-IV, Roche Pharmaceuticals) to nestinδ-TK+ mice leads to phosphorylation of ganciclovir in TK expressing cells, causing cell death from inhibition of DNA synthesis. Animals were housed in a 12-h light/dark cycle with food and water ad libitum. Procedures related to animal use and care were approved by the University of Miami Animal Use and Care Committee.
Ganciclovir sodium treatment
To determine the optimal concentration of ganciclovir sodium treatment, we first performed a dosing study using 0 (vehicle, n=4), 10 (n=5), 50 (n=4), and 100 (n=4) mg/kg/d administered subcutaneously via a 2-week miniosmotic pump (Alzet model #1002; rate of 0.25 μL/h) in naïve and CCI injured mice (Supplementary Fig. 1; see online supplementary material at ftp.liebertpub.com). An optimal dose of 50 mg/kg/d ganciclovir sodium was used for the remainder of the study. To analyze the effect of ganciclovir sodium on NSPC ablation in the SVZ, mice were treated with 50 mg/kg/d ganciclovir sodium or vehicle for 2 weeks before sham surgery or CCI injury. At the time of surgery, the pump was replaced with a second pump containing 50 mg/kg/d ganciclovir sodium or vehicle, and the mice were allowed to survive for an additional 14 DPI (i.e., 4 weeks of continuous treatment; Fig. 1).
FIG. 1.
Schematic of protocol timeline used for 4 weeks of ganciclovir (Cytovene-IV) treatment, controlled cortical impact (CCI) injury, and rotarod assessment. DPI=days post-injury.
For initial pump insertion, mice were anesthetized with ketamine (75 mg/kg body weight) and xylazine (14 mg/kg body weight) by intraperitoneal injection. A small incision was made in the skin between the scapulae, and a small pocket was formed using a hemostat to separate the subcutaneous connective tissue. The pumps were inserted into the pocket with the opening of pumps pointing away from the incision. The skin incision was closed using 5-0 silk suture. Replacement pumps were inserted at the time of the sham or CCI surgery.
CCI injury
Mice were anaesthetized with ketamine (75 mg/kg body weight) and xylazine (14 mg/kg body weight) by intraperitoneal injection and positioned in a stereotaxic frame. Body temperature was monitored with a rectal probe and maintained at 37°C with a controlled heating pad set. A 5-mm craniotomy was made using a portable drill over the right parietotemporal cortex (−2.5 mm caudal and 2.0 mm lateral from bregma). The injury was generated using a 3-mm beveled stainless steel tip attached to an eCCI-6.3 device (Custom Design & Fabrication) at a velocity of 6.0 m/sec, depth of 0.5 mm, and 150 msec impact duration. Sham mice received craniotomy only. After CCI injury, the skin was sutured using 5-0 silk sutures.
Behavior
The activities of mice were observed in a standard cage containing five mice over a period of 5 min and were graded as either active or less active and their fur was identified as being groomed or not groomed. Motor function was tested on the Rotamex 4/8 accelerating rotarod before injury and at 3, 7, and 14 DPI. Groups included: (1) nestinδ-TK+ with vehicle (sham n=5, CCI injury n=5); (2) nestinδ-TK+ with ganciclovir sodium (sham n=5, CCI injury n=6); (3) nestinδ-TK- with vehicle (sham n=5, CCI injury n=7); and (4) nestinδ-TK- with ganciclovir sodium (sham n=5, CCI injury n=18). The rotating cylindrical rod was 50-mm wide and had a linear acceleration from 10 rpm to 60 rpm over a period of 10 min (i.e., rate of acceleration was 5 rpm). Mice were given four trials on each day, and the time to fall off the rotarod was averaged over the four trials. For each mouse, the post-injury motor performance was expressed as a percentage of pre-injury performance and then averaged across the mice in each group.
Histological preparation and immunohistochemistry
Bromodeoxyuridine (BrdU) (Sigma) was injected intraperitoneally (50 mg/kg of body weight) 1 h before sacrificing the mice, as described previously.28 To analyze the SVZ of mice that were sacrificed at the 2 week time point, mice were deeply anesthetized using ketamine (75 mg/kg body weight) and xylazine (14 mg/kg body weight) by intraperitoneal injection, the brains were removed, quickly frozen in OCT over isopentane on dry ice, and stored at −80°C. To analyze the injury microenvironment of mice that underwent CCI injury, 14 days later these mice were deeply anesthetized using ketamine and xylazine by intraperitoneal injection and transcardially perfused with phosphate buffered saline (PBS) and 4% paraformaldehyde (pH 7.4). The brains were stored in 4% paraformaldehyde overnight, 30% sucrose for 48 h and then frozen in OCT over isopentane on dry ice and stored at −80°C. For both fresh (2 week) and fixed (4 week) tissue, 30 μm serial frozen coronal sections were cut and stored at −80°C.
To analyze the cellular composition of the SVZ and perilesional region of the cortex, other sections were permeabilized and immunohistochemically stained with the primary antibodies anti-GFP (1:500; Molecular Probes), anti-nestin (1:500; Neuromics), anti-Ki67 (1:25; Dako), anti-glial fibrillary acidic protein (GFAP) (1:1000; Dako), anti-Iba1 (1;1000; Wako), anti-NeuN (1:500; Millipore), anti-doublecortin (anti-DCX) (1:200; Santa-Cruz) and anti-polysialylated-neural cell adhesion molecule (PSA-NCAM) (1;1000; Millipore) overnight at 4°C.
For Ki67 staining on fixed tissue, sections underwent antigen retrieval with 10 mM sodium citrate buffer, pH 6.0, where slides were placed in 200 mL buffer and heated twice for 4 min in a 1500 W microwave oven. For BrdU staining, sections were first incubated in 2 N HCl for 1 h at 37°C, then washed with 0.1 M borate buffer, pH 8.5. All sections were washed three times in PBS, incubated in fluorescent secondary antibodies (Molecular Probes) for 30 min at room temperature, washed an additional three times in PBS, and cover-slipped in mounting medium containing DAPI (Invitrogen).
Collection of conditioned NSPC tissue culture medium
NSPCs were isolated from the SVZ of adult wild type CD-1 mice (2 to 4 months old) and grown as described previously.29 Once NSPC cultures were confluent, the conditioned NSPC media (NSCM) was collected and stored at −80°C. On the day of use, this media was defrosted and used as 1:1 or diluted 1/2, 1/20, or 1/200 in NSPC media not incubated with cells. To remove NSPC debris, some undiluted NSCM media was centrifuged at 1500 rpm for 5 min at 4°C.
Astrocyte culture, scratch wound assay, cell death assay and immunocytochemistry
For the astrocyte scratch wound assay, brains from newborn wildtype (P1-P3) CD-1 mice were simultaneously dissected under sterile conditions using Hanks' balanced salt solution media containing 10 mM HEPES. The cortices were isolated and combined, tissue samples were dissociated mechanically and triturated in DMEM containing 10% fetal bovine serum (Invitrogen), before being filtered through a 40-μm cell strainer and then a 10-μm cell strainer. The freshly dissected mouse astrocytes were plated onto uncoated 10-cm plates at 100,000 cells/mL and grown to confluence for 14 days at 37°C in a humidified atmosphere containing 5% CO2. To reduce microglial growth, the culture medium was changed every other day.
On confluency, cells were directly transferred into uncoated 24-well plates for scratch wound assays at a density of 1×105 cells per well. After 4 to 6 days in 24-well plates, astrocyte monolayers were scratched with a sterile 200 μL pipette tip. Wells were washed twice with sterile PBS, then either DMEM containing 10% FCS, unconditioned NSCM, or conditioned NSCM (1:1 or diluted 1:2, 1:20, or 1:200 with unconditioned NSCM) with or without centrifugation (1500 rpm, 5 min, 4°C) were added to the plates and incubated for an additional 48 h, after which the cells were fixed with 4% paraformaldehyde for 10 min. The cells were then permeabilized and incubated in the primary antibodies anti-GFAP (1;1000; Dako), anti-Ki-67 (1:200; Dako), anti-nestin (1:500; Neuromics), anti-vimentin (1:100; Santa-Cruz), anti-O1 and anti-O4 (1:2; kind gifts from Dr. Pat Wood), anti-NG2 (1:500; Millipore), anti-Iba-1 (1:1000; Wako) for 30 min at room temperature on an orbital shaker.
The cells were subsequently washed three times in PBS, incubated with appropriate secondary fluorescent antibodies (Molecular Probes) and DAPI (Santa-Cruz) for 30 min at room temperature on an orbital shaker, and washed a final three times with PBS before imaging. The scratch wound assay consisted of at least three independent experiments in triplicate per treatment group.
For cell death assays, astrocytes were cultured from nestinδ-TK- and nestinδ-TK+ mice as indicated above. On confluency, cells were directly transferred onto uncoated 96-well plates at a density of 5×104 cells per well. After 2 days in culture, ganciclovir sodium (0–720 μM) or staurosporine (100 nM or 10 μM; Biolmol) was added and incubated an additional 48 h. This concentration and time were chosen because 2–10 μM ganciclovir reduces 30–70% cell viability in vitro.30,31 Cells were further treated with 0.05 μM Sytox Red (Life Technologies) and 10 μg/mL Hoechst 33258. Ten minutes later, the live/dead cells were automatically counted using Cellomics ArrayScan VTI Live HCS reader (Thermo Scientific) at 37°C and 5% CO2. For the cell death assay, the experiment consisted of a minimum of six wells for each treatment group.
Histological analysis
For in vivo analysis, photomicrographs of sections containing the entire rostrocaudal extent of the injury site using cresyl violet sections were taken on an Olympus Bx50 microscope using an Olympus SC30 camera with Olympus “AnalySIS getIT!” software. The distance between the most caudal and rostral sections was calculated to determine the rostral-caudal injury length. Cortical tissue sparing was assessed by contouring the volume of remaining ipsilateral and contralateral cortical tissue using MicroBrightField StereoInvestigator 10.30.1 software package and using MicroBrightField NeuroLucida Explorer 11.03 calculating the volume of remaining ipsilateral cortex as a percentage of the contralateral cortical area.
Stereology
For in vivo analysis, sections at the rostral extent of the SVZ were collected from nestinδ-TK+ (n=5) and nestinδ-TK- (n=4) treated with 50 mg/kg/d ganciclovir sodium for 2 weeks before injury, and in nestinδ-TK+ (n=6) and nestinδ-TK- (n=6) mice at 14 DPI. SVZ proliferation and NPSC ablation was assessed by stereological assessment of the total number of BrdU-, nestin-, and PSA-NCAM-positive cells, respectively, using a motorized Zeiss Axio-phot microscope, Optronix Microfire camera, and MicroBrightField StereoInvestigator 10.30.1 software package.
To assess neuroblast, neuronal, astrocyte, and microgial/macrophage cell numbers in the perilesional region and corpus callosum (CC), nonbiased cell number estimations were performed on the five most central rostrocaudal sections around the injury epicenter (as determined using cresyl violet stained sections), which were 30 μm thick and 180 μm apart. For NeuN, DCX and PSA-NCAM cortical counts, a contour of approximately 250 μm deep was placed over the cortex on either side of the injury site at 5X magnification, and a grid of 125×125 μm was placed over this area, with a counting frame of 50×50 μm. For DCX and PSA-NCAM counts in the CC, the area of the CC was outlined (approximately 150–200 μm wide) at 5X magnification, and a grid of 125×125 μm was placed over this area, with a counting frame of 50×50 μm.
For astrocytes and microglia/macrophage counts, contours of approximately 700 μm or 250 μm deep, respectively, were made around the entire injury site, and a grid of 400×400 μm or 200×200 μm, respectively, was placed over this area, each with a counting frame of 50×50 μm. To assess proliferation, the SVZ was outlined to create a contour and a grid of 40×40 μm was placed over this area, with a counting frame of 20×20 μm. The number of NeuN, DCX, PSA-NCAM, GFAP, Iba-1, BrdU, and nestin-positive cells with an identifiable nucleus were randomly counted using the optical fractionator at 63X magnification.
For in vitro analysis, photomicrographs of the scratch wound (1 mm2 area centered on the scratch midline) were taken at 10X (which captured both sides of the scratch area) on a Zeiss Axiovert200 microscope with an Axiocam MRm camera using Axiovision 4.8 software and converted to 8 bit grayscale images. For counting the number of DAPI- and Ki-67-positive nuclei, the images were inverted in ImageJ software and the ITCN automated counter plug-in was used to count cells. The ITCN plug-in was first optimized to ensure every nucleus was counted once, which was validated approximately every 20–30 photomicrographs. For each photograph, the number of Ki67-positive nuclei was calculated as a percentage of the number of DAPI-positive nuclei.
Astrocyte reactivity at the scratch-wound edge was determined by GFAP density, whereby images were thresholded and the area fraction of pixels positive for GFAP-labeling was measured in ImageJ. For each photograph, GFAP expression levels (arb. units) where measured in fractional areas to give an average GFAP intensity. DAPI-positive cells were also counted for each photograph to provide GFAP intensity/DAPI cell value. Values for each photograph were then averaged per well to provide an average GFAP intensity/DAPI cell value.
Statistical analysis
All data were assessed for homogeneity of variance, after which statistical analysis was performed. Histological differences were assessed using the Student t test, and behavioral differences (intra- and intergroup analysis) were assessed using two-way Repeated measures analysis of variance with Student-Newman-Keuls method post hoc test in SigmaPlot 13.0 where significance was <0.05. Data in figures are expressed as mean±standard error of the mean.
Results
Nestinδ-TK transgenic mice show transgene expression in NSPCs and neuroblasts
In the current study, we took advantage of transgenic mice that selectively express GFP under the nestinδ promoter in NSPCs that reside in neurogenic regions of the adult brain.27 To examine the distribution of NSPCs in sham and CCI injured mice, we evaluated the immunohistochemical distribution of GFP-labeled NSPCs using an anti-GFP antibody. We observed significant and selective expression of GFP in the SVZ, rostral migratory stream (RMS), olfactory bulb (OB), and dentate gyrus (DG) of the hippocampus in the sham mice (Fig. 2A, inset). Co-labeling studies show that anti-DCX labeled neuroblasts (red) are almost exclusively found in the neurogenic regions similar to GFP-labeled NSPCs, whereas anti-GFAP labeled astrocyte-like stem cells and mature astrocytes (blue) are observed in the SVZ and tissues surrounding these neurogenic regions, respectively (Fig. 2A). High-magnification images show cellular localization of NSPCs (green), neuroblasts (red), and astrocytes (blue) in the RMS (Fig. 2A1), SVZ (Fig. 2A2), dorsal ventricular wall (Fig. 2A3), and dorsal cortical tissues (Fig. 2A4).
FIG. 2.

Green fluorescent protein (GFP) transgene expression in nestinδ-TK+ mice shows increased expression outside neurogenic regions at 2 DPI. (A) Photomicrographs show anti-GFP staining (inset) and anti-GFP (green), anti-doublecortin (DCX) (red) and anti-glial fibrillary acidic protein (GFAP) (blue) staining in the subventricular zone (SVZ), rostral migratory stream (RMS), dorsal ventricle wall, and regions outside neurogenic zones. High-magnification images show GFP labeling alone and merged with DCX and GFAP labeling in RMS (A1), SVZ (A2), dorsal ventricle wall (A3), but not subcortical regions (A4) in sham mice. (B) Photomicrographs at 2 DPI show anti-GFP staining (inset) and anti-DCX/anti-GFAP costaining in both neurogenic regions as well as outside regions including corpus callosum, subcortical tissues. and striatum. Arrowhead depicts GFP-labeled cells negative for DCX and GFAP in the cortex. High-magnification images show GFP labeling alone and merged with DCX and GFAP labeling in RMS (B5), SVZ (B6), dorsal ventricle wall (B7), and subcortical regions (B8). Scale bar=400 μm in low-magnification images and 12 μm in high-magnification images. Color image is available online at www.liebertpub.com/neu
In the sham brain, NSPCs and neuroblasts were mainly observed in neurogenic tissues, and few if any were seen in the surrounding cortex or striatum. Sham injuries resulted in mild vascular disruption and minor tissue damage, where small numbers of GFP-labeled cells were observed in non-neurogenic regions compared with none in naïve mice.27 At 2 days after CCI injury (DPI), we observed greater overall numbers of GFP-labeled NSPCs compared with sham control mice in regions inside and outside the neurogenic zones that include CC and cortical tissues (Fig. 2B). High-magnification images show that within the SVZ and RMS we observed some cells that co-label with GFP and DCX, or GFP and GFAP (Fig. 2B5–B7); however, outside these regions, DCX/GFP co-labeling was not observed (Fig. 2, arrowheads) and GFAP was not observed with either DCX or GFP expressing cells (Fig. 2B8).
Our observed reductions in GFP-labeled neuroblasts likely result from the down-regulation of nestin after NSPC differentiation. Interestingly, undifferentiated GFP-labeled cells exiting the RMS have processes that extend from this region, supporting a perpendicular migratory trajectory to cell polarities in the normal RMS (Fig. 2B5; arrow). These findings provide support for the specificity of the nestinδ promoter, where NSPC and not mature astrocytes express GFP as previously shown.27 Further, this suggests that migratory NSPCs can remain undifferentiated, and both NSPCs and neuroblasts have the potential to migrate into damaged tissues.
Ganciclovir sodium treatment ablates NSPCs in the SVZ of nestinδ-GFP-TK transgenic mice
The antiviral drug ganciclovir sodium (Cytovene-IV) exists in an un-ionized form, compared with ganciclovir, with increased solubility at physiological pH and body temperature. Ganciclovir sodium is rapidly phosphorylated by viral TK and is incorporated into the DNA where it inhibits DNA synthesis, ultimately leading to cell toxicity and death.32 We administered ganciclovir sodium (50 mg/kg, subcutaneously, mini-osmotic pumps) to adult mice with or without the TK transgene (i.e., nestinδ-TK+ and nestinδ-TK-, respectively) for 2 weeks to ablate proliferating NSPCs within the SVZ. This dose was based on our observations that higher concentrations of ganciclovir sodium (i.e., 100 mg/kg) resulted in reduced grooming behavior before injury and reduced rotarod locomotor function after a CCI injury model of TBI, in nestinδ-TK- mice, compared with vehicle treated nestinδ-TK- mice (Supplementary Fig. 1A, B; see online supplementary material at ftp.liebertpub.com). The 2-week time course was based on initial preliminary studies showing treatment with 50 mg/kg reduces the number of proliferating cells in the SVZ.
To confirm whether ganciclovir sodium administration leads to fewer proliferating NSPCs, we quantified the number of proliferating cells in the SVZ and the number of neuroblasts in a region where neuroblasts accumulate before migrating to the OB via the RMS (Fig. 3A). We observed 51% fewer BrdU-labeled cells in nestinδ-TK+ mice (14,252±3194 cells), compared with nestinδ-TK- mice (28,842±987 cells) after ganciclovir sodium treatment (Fig. 3B–D), which corresponded to significant reductions (55%) in the number of nestin-labeled cells (Fig. 3E–K). Similarly, reduced Ki-67 labeling was observed in the SVZ-RMS transition region in nestinδ-TK+ mice (Fig. 3T), but not nestinδ-TK- mice (Fig. 3P). Two weeks of suppression of NSPCs showed a trend toward fewer numbers of polysialylated-neural cell adhesion molecule (PSA-NCAM)-labeled cells in the SVZ-RMS transition region (Fig. 3L–O, Q–S). Together, these results support a model where 50 mg/kg ganciclovir sodium treatment for 2 weeks results in approximately 50% reduction of proliferating NSPCs in the SVZ and fewer neuroblasts entering the RMS.
FIG. 3.
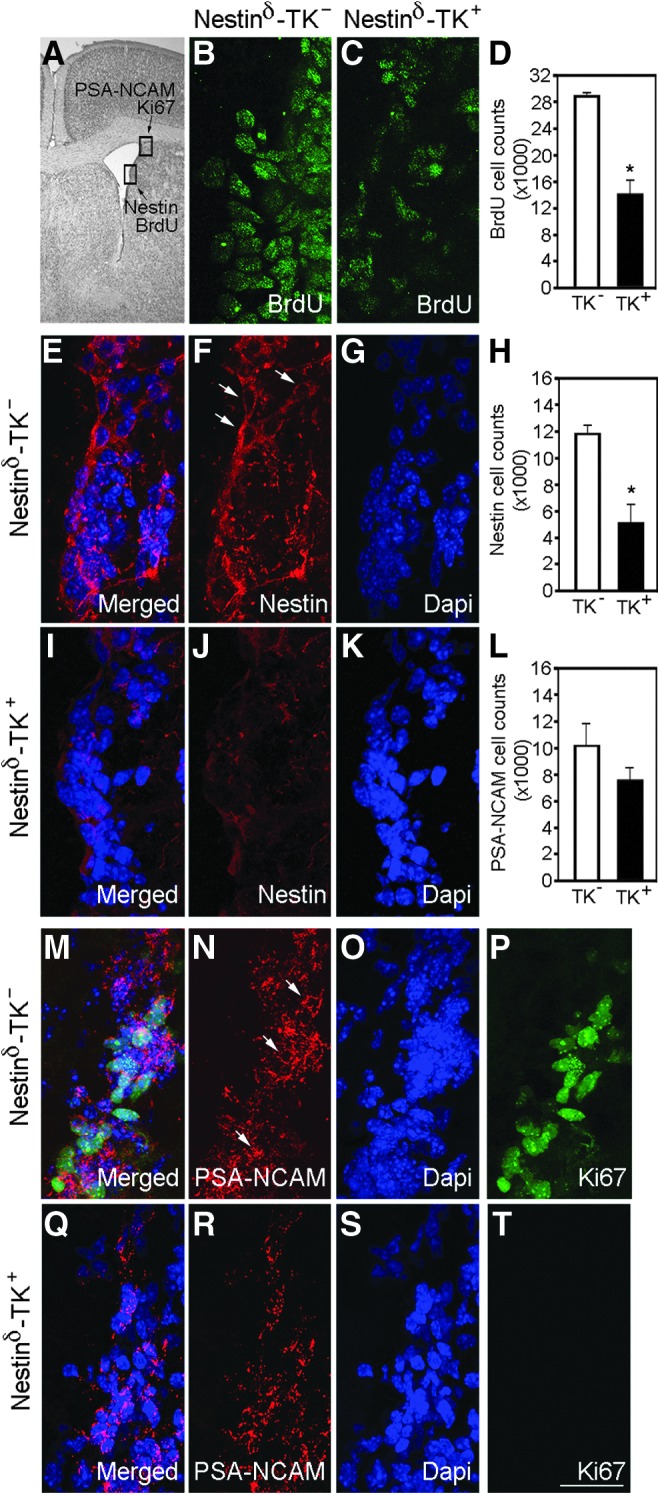
Ganciclovir sodium treatment for 2 weeks reduces neural stem/progenitor cell (NSPC) numbers in the subventricular zone (SVZ) of nestinδ-TK+ mice (n=5) compared with nestinδ-TK- mice (n=4). (A) Black and white photomicrograph of a cresyl violet stained brain section showing the lateral ventricle. Boxes indicate location where photomicrographs examine nestin and bromodeoxyuridine (BrdU) staining in SVZ (B–K) and polysialylated-neural cell adhesion molecule (PSA-NCAM) and Ki67 staining SVZ- rostral migratory stream (RMS) transition zone (L–T). Photomicrographs show BrdU-positive cells in the SVZ of ganciclovir sodium treated nestinδ-TK- mice (B), but fewer cells in nestinδ-TK+ mice (C), with graph showing differences in the number of BrdU-positive cells (D) (n=6). Photomicrographs show the number of nestin-positive cells in the SVZ of ganciclovir sodium treated nestinδ-TK- mice (E–G) compared with nestinδ-TK+ mice (I–K). Graph shows a reduction in the number of nestin-positive NSPCs in the SVZ of nestinδ-TK+ mice (H) (n=6). Photomicrographs show the number of PSA-NCAM- and Ki67-positive cells in the SVZ of ganciclovir sodium treated nestinδ-TK- (M–P) and nestinδ-TK+ (Q–T) mice, with the graph showing reduced number of PSA-NCAM-positive cells in nestinδ-TK+mice (L). Scale bar in T=20 μm. *p<0.05. Color image is available online at www.liebertpub.com/neu
NSPC ablation in the SVZ reduces neuroblast migration into perilesional regions after TBI
To determine whether NSPCs migrate outside the neurogenic regions after injury, we quantified the number of neuroblasts using two markers, PSA-NCAM and DCX, in the CC and perilesional regions of the cortex at 14 DPI (Fig. 4). After 2 weeks of initial ganciclovir sodium administration, we performed a CCI injury with sustained infusion of 50 mg/kg ganciclovir sodium for an additional 2 weeks (Fig. 1). After 4 weeks of ganciclovir sodium treatment, we observed similar reductions in proliferation of NSPCs in the SVZ of nestinδ-TK+ mice compared with 2 weeks administration (not shown). In addition, no abnormalities were observed in grooming behavior or motor function at the 4-week time point in nestinδ-TK- mice (Supplementary Fig. 1B; see online supplementary material at ftp.liebertpub.com).
FIG. 4.

Neural stem/progenitor cell (NSPC) ablation reduces the number of migrating neuroblasts at 14 days post-injury (DPI). Polysialylated-neural cell adhesion molecule (PSA-NCAM)- (A) and doublecortin (DCX)-labeled (B) cells in the cortex and corpus callosum (CC) after controlled cortical impact (CCI) injury. Graphs show reductions in the numbers of PSA-NCAM- and DCX-positive cells in the ipsilateral perilesional CC and cortex at 14 DPI in ganciclovir sodium treated nestinδ-TK+ mice compared with nestinδ-TK- mice (n=6 each). *p<0.05, ***p<0.001. (C) Cresyl violet staining of CCI-injured mouse with box showing approximate location of images of panels D–O. (D, E) DCX and PSA-NCAM labeled cortical tissue from naïve mice (approximately similar stereological coordinates to the perilesional region of CCI-injured mice) showing an absence of DCX-labeled cells and minimal PSA-NCAM labeling. (F, G) High-magnification images of ganciclovir sodium treated nestinδ-TK+ mice show no reactivity with DCX and little reactivity with PSA-NCAM in CCI injured tissues. High magnification images of DCX (H, I, L, M) and PSA-NCAM (J, K, N, O) co-labeled with the neuronal marker NeuN and glial marker glial fibrillary acidic protein, respectively, in the CCI-injured cortex. Arrows depict representative DCX- or PSA-NCAM-positive cells in perilesional cortex, and scale bar for high magnification images=25 μm. Color image is available online at www.liebertpub.com/neu
In the CC and perilesional region of CCI-injured ganciclovir sodium treated nestinδ-TK+ mice, we observed approximately 80% and 69% fewer PSA-NCAM-positive cells, respectively, compared with the number observed in the same regions of ganciclovir sodium treated nestinδ-TK- mice (Fig. 4A, G). While PSA-NCAM is a marker of migrating NSPCs, in the injured adult cortex, it labels a subset of astrocytes and mature neurons (Fig. 4J, K, N, O)33,34; therefore, we also examined neuroblast numbers by labeling with DCX. We observed little to no cross-reactivity with mature neurons or glial (Fig. 4H, I, L, M). In the CC and perilesional region of CCI-injured ganciclovir sodium treated nestinδ-TK+ mice, we also observed 80% and 90% fewer DCX-labeled cells compared with ganciclovir sodium treated nestinδ-TK- mice (Fig. 4B, F). We observed no DCX+ and few PSA-NCAM+ cells (1–2 cells/tissue section) in sham or naïve (Fig. 4D, E) tissues. In summary, the reduction in neuroblast numbers in the CC and cortex after NSPC ablation may represent a lower production of neuroblasts from early undifferentiated, dividing NSPCs leaving fewer available to migrate.
NSPC ablation reduces residential cell numbers in the injured cortex
To provide evidence that nestin-expressing cells in the adult CNS play a role in injury progression, we examined whether NSPC depletion led to alterations in CCI-mediated pathology. We examined cortical tissues by quantifying the number of neurons and glia in perilesional regions. Quantitative analysis performed 2 weeks after CCI injury revealed no significant differences in the percent volume of spared cortical tissue (compared with contralateral noninjured cortex) or injury length between nestinδ-TK+ and nestinδ-TK- mice (Fig. 5A–D). Conversely, stereological analysis of cells residing in the perilesional regions of the cortex revealed significant differences in neuronal and glial numbers, which was not observed in the contralateral cortex (not shown).
FIG. 5.

Neural stem/progenitor cell (NSPC) ablation reduces neuronal, astrocytic, and macrophage/microglia cell numbers in the perilesional cortex at 14 days post-injury. Photomicrographs illustrate cresyl violet-stained sections of nestinδ-TK- (A) and nestinδ-TK+ mice (B), where quantification shows no difference in the percent volume of spared cortical tissue compared with contralateral cortex (C) or injury length (D) between nestinδ-TK- and nestinδ-TK+ mice (n=6–8). Photomicrographs show a reduced number of NeuN-positive neurons in the perilesional cortex of nestinδ-TK+ mice (F), compared with nestinδ-TK- mice (E) with graph showing significant reductions (G) (n=6). Photomicrographs show a reduced number of glial fibrillary acidic protein (GFAP)-positive astrocytes in the perilesional cortex of nestinδ-TK+ mice (I) compare with nestinδ-TK- mice (H) with graph showing significant reductions (J) (n=6). (K, L) High magnification photomicrographs of boxes in panels H and I, respectively. Photomicrographs show a reduced number of Iba-1-positive microglia/macrophages in the perilesional cortex of nestinδ-TK+ mice (O) compared with nestinδ-TK- mice (N) with graph showing significant reductions (M) (n=6). E, F, H, I, N, and O: Scale bar=50 μm; K–L: Scale bar=20 μm. *p<0.05. Color image is available online at www.liebertpub.com/neu
Specifically, the cortex of ganciclovir sodium treated nestinδ-TK+ mice had approximately 20% fewer NeuN-positive neurons (143±6 cells per [100 μm]3), compared with the cortex of nestinδ-TK- mice (176±9 cells per [100 μm]3) (Fig. 5E–G) and approximately 30% fewer GFAP-positive astrocytes (96±8 cells per [100 μm]3 and 133±7 cells per [100 μm]3, respectively; Fig. 5H–J). Interestingly, although fewer astrocytes were observed after NSPC ablation, the remaining astrocytes in nestinδ-TK+ mice appeared more hypertrophic, compared with the astrocytes in nestinδ-TK- mice (Fig. 5K, L), suggesting that NSPCs may influence astrocyte survival, proliferation, differentiation, and/or reactivity after injury.
We also quantitated the number of macrophage/microglia-positive cells in the perilesional region using Iba-1 labeling and stereological measurements. Similar to astrocytes, we observed a 30% reduction in the numbers of Iba-1-labeled macrophage/microglia in ganciclovir sodium treated nestinδ-TK+ mice, compared with nestinδ-TK- mice (81±3 cells per [100 μm]3 and 111±8 cells per [100 μm]3, respectively; Fig. 5M–O). Similar to the astrocytic population, the remaining Iba-1-positive cells in the ganciclovir sodium treated nestinδ-TK+ mice appeared more hypertrophied and amoeboid-shaped resembling reactive microglia (not shown); however, it is difficult to distinguish between reactive microglia and infiltrating macrophages because most antibodies, including Iba-1, label both cell types.
Taken together, these findings suggest that ablation of nestin-expressing cells in the adult brain attenuates neuronal survival and reduces glial numbers in the perilesional region, as well as playing a role in glial hypertrophy post-CCI injury.
NSPC-conditioned media increases astrocyte proliferation and reduces GFAP expression in culture
To examine whether NSPCs release soluble factors that directly influence astrocyte proliferation and reactivity, a scratch wound assay was performed on cultured cortical astrocytes exposed to either NSCM or unconditioned media (Fig. 6). Initially, we evaluated astrocyte purity using immunohistochemical markers to a variety of non-neuronal cell types. Astrocytes showed expression of the early glial progenitor cell marker nestin (Fig. 6B, F), and more mature astrocyte makers, GFAP (Fig. 6C, G) and vimentin (Supplementary Fig. 2A, B; see online supplementary material at ftp.liebertpub.com), while few to no cells expressed the oligodendrocyte markers 01 and 04, the microglial/macrophage marker Iba-1, or the glial precursor marker NG2 (Supplementary Fig. 2C-J; see online supplementary material at ftp.liebertpub.com). This expression pattern supports previous studies on cultured astrocyte purity,35,36 and demonstrates that our astrocyte cultures are >95% pure.
FIG. 6.

Conditioned neural stem/progenitor cell (NSPC) media (NSCM) enhances cortical astrocyte proliferation and reduces glial fibrillary acidic protein (GFAP) expression after scratch wound injury in culture. Photomicrographs show typical astrocytes observed at the edge of the scratch wound stained with Ki-67, nestin, and GFAP after being cultured for 48 h in unconditioned media (A–D) or undiluted NSCM (E–H). Proliferating Ki-67–positive astrocytes predominantly co-label with nestin and not GFAP. (J) Graph represents increased Ki-67 staining of astrocytes at the edge of the scratch wound after being cultured with NSCM or centrifuged NSCM for 48 h. (K) Graph shows reduced GFAP expression of astrocytes at the edge of the scratch wound after being cultured with NSCM or centrifuged NSCM for 48 h. n=3 independent experiments in triplicate per group. Scale bar=200 μm. *p<0.05, **p<0.01, **p<0.001. Color image is available online at www.liebertpub.com/neu
To determine whether NSPC secretions regulate cortical astrocyte proliferation, we labeled the astrocytes with Ki-67. The majority of Ki-67-positive nuclei (Fig. 6A, D, E, H) were observed in cells expressing high levels of nestin (Fig. 6D, H), although a small number of Ki-67-positive cells also co-labeled with GFAP (Fig. 6D, H). We did not observe Ki-67-labeled nuclei in cells that were GFAP/vimentin-positive and nestin-negative. At the scratch wound edge, defined as a 1 mm2 area centered on the scratch midline, Ki-67 labeling increased after exposure to NSCM, at concentrations of 1/20 and higher, compared with unconditioned media (Fig. 6J). In addition, undiluted (1/1) NSCM decreased astrocyte reactivity at the scratch wound edge as assessed by reduced intensity of GFAP expression (Fig. 6K) as well as reducing astrocyte hypertrophy. These findings support the possibility that NSPC secretions may promote proliferation of cortical glial cells in perilesional regions after CCI injury.
To further demonstrate that ganciclovir sodium administration does not ablate potential off target nestin-positive reactive astrocytes or astrocyte progenitor cells in the perilesional region after CCI injury, we examined the influence of ganciclovir sodium treatment on cultured nestinδ-TK+ and nestinδ-TK- cortical astrocytes in a scratch wound assay. We observed less than 5% cell death of nestinδ-TK+ or nestinδ-TK- cortical astrocytes treated with 0 to 720 mM of ganciclovir sodium compared with vehicle controls (Supplementary Fig. 3; see online supplementary material at ftp.liebertpub.com). Staurosporine (0.1 and 10 mM) was applied as a positive control for cell death, where increased cell death was observed in both nestinδ-TK+ and nestinδ-TK- astrocytes. These studies further support the specificity of the nestinδ promoter in NSPCs and suggest that the reduced number of GFAP cells after CCI injury did not result from increased astrocyte or astrocyte precursor cell death.
NSPC ablation attenuates motor function after CCI injury
To evaluate whether NSPCs are involved in motor responses, we assessed the animals' motor performance using an accelerating rotarod before CCI injury and at 3, 7, and 14 DPI. To exclude potential gene, drug, or surgical effects on mouse behavior, we examined the performance of sham-injured mice with vehicle or ganciclovir sodium treatment. Although we observed no significant differences between groups, we did observe significant differences in group and time interaction for both sham and CCI injury (Fig. 7A, B). The Student-Newman-Keuls method also revealed significant intragroup differences in motor responses compared with pre-injury performance (designated with an asterisk), and intragroup differences between 3 DPI and 7 or 14 DPI (designated with a pound sign) (Fig. 7A–C). This is most apparent when pre-injury latencies were normalized, where intragroup improvements were observed in all groups except ganciclovir sodium treated nestinδ-TK+ mice, resulting in significantly worse motor performance at 7 DPI compared with control groups (designed with a dollar sign) (Fig. 7C). This suggests that ablation of NPSCs reduces motor recovery after CCI injury.
FIG. 7.
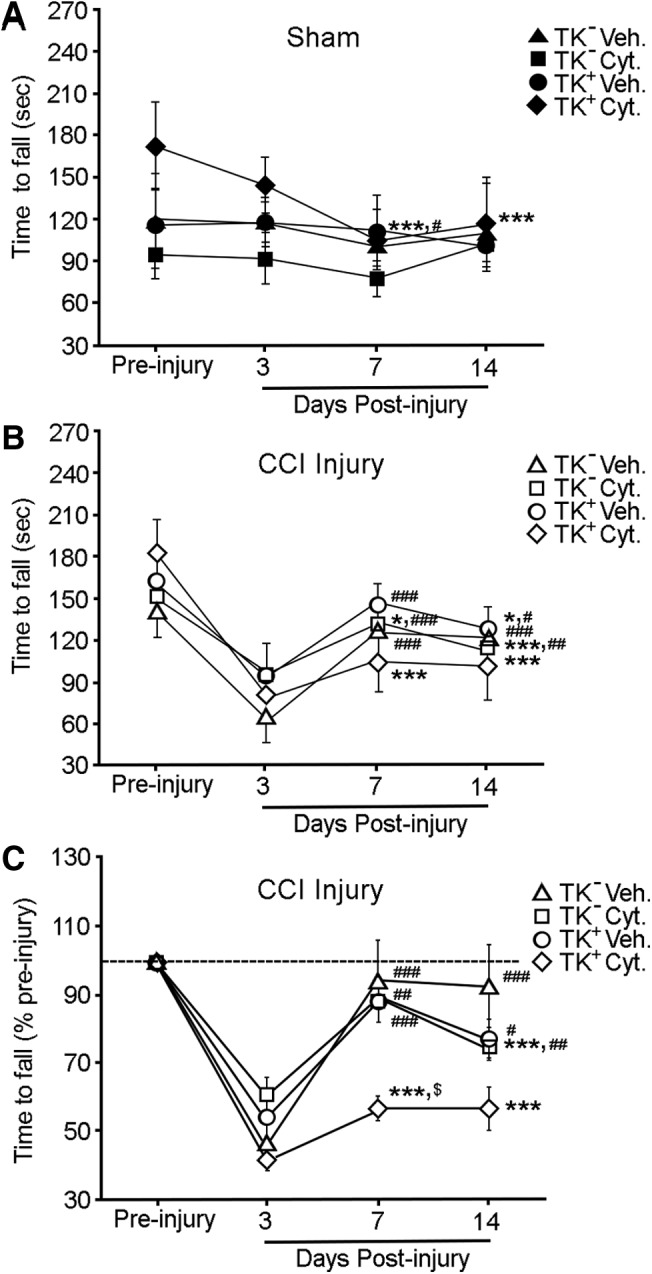
Neural stem/progenitor cell (NSPC) depletion reduces motor recovery in ganciclovir sodium treated nestinδ-TK+ mice. (A) Graph shows time to fall (sec) from the accelerating rotarod of vehicle or ganciclovir sodium treated sham-injured nestinδ-TK- and nestinδ-TK+ mice (n=5 per group; two-way repeated measures analysis of variance [ANOVA]; day p<0.001, group and day interaction p=0.014). (B) Graph shows time to fall (sec) from rotarod of vehicle and ganciclovir sodium treated controlled cortical impact (CCI)-injured nestinδ-TK- and nestinδ-TK+ mice (two-way repeated measures ANOVA; day p<0.001, group and day interaction p=0.012). (C) Graph compares time to fall from rotarod of vehicle and Cytovene-IV treated nestinδ-TK- and nestinδ-TK+ CCI injured mice when pre-injury times are standardized to 100%. Ganciclovir sodium treated nestinδ-TK+ injured mice were unable to improve their ability to walk on the rotarod at 7 and 14 days post-injury (DPI) compared with 3 DPI, resulting in the ganciclovir sodium treated nestinδ-TK+ mice having significantly worse motor performance than other groups ($) (two-way repeated measures ANOVA day p<0.001, group and day interaction p=0.017). Student-Newman-Keuls method showed variance between time points 3, 7, and 14 DPI compared with pre-injury (*), and variances of 7 or 14 DPI compared with 3 DPI (#). *,#,$ p<0.05; **,## p<0.01; ***,### p<0.001.
Discussion
After the discovery of endogenous NSPCs in the adult brain in the 1960s, scientists have proposed that the CNS might retain the capacity to self-renew after injury. In fact, studies have provided evidence that NSPCs migrate to damaged tissues and differentiate into neurons if given enough time.14 Strategies to enhance neurogenesis may improve the number of cells that integrate into the damaged circuitry; however, cell replacement would likely still take place over months. Our studies support an acute neuroprotective role for migrating SVZ-derived NSPCs and/or neuroblasts after CCI injury.
We used transgenic mice expressing the viral TK gene under control of the nestinδ promoter to specifically ablate NSPCs and elucidate their role and/or their progenies role on progressive cortical tissue damage and function in the acute phase of TBI. We demonstrated that NSPC ablation causes a reduction in the number of neuroblasts migrating toward the injury, resulting in fewer residential neurons and glial cells in perilesional regions of the CCI injured cortex. This reduction in neurons and glia is associated with increased astrogliosis and reduced locomotor recovery. These findings support the beneficial influences of NSPCs after TBI suggesting that endogenous NSPCs represent therapeutic targets to minimize injury progression.
One of the most intriguing findings of this study is the possibility that NSPCs support residential neuronal survival in the injured cortex. Although it may be possible that the increased number of neurons and glial cells in normal mice represent, in part, a migrating population of SVZ-derived NSPCs that differentiate into neurons and/or glial cells, the short time course and large number of cells renders this possibility unlikely. Arvidsson and colleagues suggest that migrating NSPCs take 1 to 3 months to differentiate into neurons,14 and even then these new neurons replace only 0.2% of the dying neuronal population.
Our studies show that at 2 DPI, NSPCs and neuroblasts are likely migrating outside the SVZ and RMS to striatal and cortical tissues, but these distances are not great. GFP-expressing NSPCs seem to have greater migratory potentials, and by 14 DPI many more neuroblasts were observed in the cortical penumbra, which may possibly result from their in situ differentiation from migrating NSPCs. Regardless, the numbers of DCX-labeled neuroblasts are far below the 20% deficits observed in mature neurons and glial cells in the absence of NPSC after CCI injury. Thus, it is more likely that migration of endogenous NSPCs are stabilizing the injury milieu enabling neuroprotection and proliferation of cortical glial cells.
There are a number of factors to consider in evaluating the role of NSPCs and neuroblasts in tissue stabilization. First, our observation that approximately 2500 DCX-labeled cells per mm3 present in perilesional tissues suggests that even relatively few cells have important influences on tissue stability. Li and associates20 examined the effects of NSPCs on neuroprotection following cortical stroke, and demonstrated that cultured NSPCs secrete brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) protecting cortical neurons from cell death. Neurotrophins, such as BDNF, have well-established actions on cell survival, growth, and differentiation in both the developing and injured adult nervous systems.37–39 Likewise, VEGF is a growth factor known for its neurogenic, angiogenic, and neuroprotective roles, which clearly influence the TBI microenvironment.40–42 Care must be taken, however, when translating in vitro results into the in vivo injury environment, because a sole study performed in the hippocampus shows NSPCs cease expressing VEGF upon differentiation into transit-amplifying cells.43 Therefore, further studies are needed to confirm the trophic responses of migrating NSPCs in injured tissues.
Alternatively, other mechanisms could be responsible for enabling cortical neuroprotection, such as infiltrating NSPC may stabilize injured cortical tissues by regulating glial cell proliferation and reactivity. Astrocytes express VEGF,43–45 and increased numbers of astrocytes may lead to synergistic enhancement in trophic support. Conversely, reactive cortical astrocytes after injury have been shown to reduce VEGF expression,46,47 suggesting that the increased numbers and reduced reactivity of astrocytes in the presence of NSPCs would provide a more protective environment. Our in vitro scratch wound model and in vivo TBI model support these conclusions.
Alternatively, SVZ-derived astrocytes have been shown to migrate into damaged cortical tissues by 14 DPI to stabilize microvascular hemorrhage.48,49 Our observations that astrocyte numbers were attenuated by 14 DPI in NSPC ablated mice supports this possibility; however, the number of migrating SVZ-derived cells is insufficient to account for the overall reductions in penumbra astrocytes supporting a more diverse NSPC role. We also observed reduced microglia/macrophage numbers in the perilesional region in the absence of NSPCs. Despite microglia and macrophages being involved in the induction of pro-inflammatory cascades, microglia also secrete numerous neuroprotective cytokines and trophic factors after injury.7,50,51 It is therefore reasonable to suggest that microglia may be responsive to the presence of NSPCs, and overall increases in NPSCs lead to increased proliferation of local glial cells that may be beneficial in minimizing acute injury progression.
In recent years, animal models have begun to elucidate the neurological relevance of increased neurogenesis in pathological conditions. Specific transgenic ablation of endogenous NSPCs provides an important tool for examining the influence of NSPCs and their progeny on functional recovery after TBI. In models of stroke, ablation of either NSPCs or neuroblasts resulted in prolonged deficits in motor recovery over 2 months.20–24 In a CCI model of TBI, the same nestinδ-TK+ mice were used to examine behavioral responses after NSPC ablation, and significant learning and memory deficit were found without motor dysfunction.52 Our observed deficits in motor function are likely from a more severe injury (6 m/sec vs. 4.4 m/sec by Blaiss and colleagues52) and longer assessment periods.
Rotarod motor assessments after CCI have a typical pattern where at 3 DPI there is a dramatic reduction in time spent on the rotating rod, which is not observed in naïve or sham animals and may result from acute injury-related physiology. This deficit is stabilized by 7 DPI, where motor performance is usually between 10 and 20% below pre-injury levels. Interestingly, ganciclovir sodium treated nestinδ-TK+ mice were the only group to show no significant improvement from 3 to 14 DPI, suggesting that ablating NSPCs reduces neuronal survival that ultimately influences motor functions.
Although SVZ-derived NSPCs are the likely candidate cells for influencing cortical motor responses, we cannot rule out the influence of other motor centers. The SVZ lies alongside the striatum, which has been known to play a role in motor initiation.53–56 In 2006, Dang and coworkers53 reported that minor changes in striatal neuronal morphology can severely impair motor function, and more recently it was shown that ablating SVZ-derived NSPCs causes perturbations in striatal spontaneous excitatory post-synaptic currents enhancing glutamatergic-mediated excitotoxicity.57 This effect was linked to NSPCs secreting endogenous cannabinoids into the striatum. In the current study, it is plausible that SVZ-derived NSPCs may play critical roles in modulating striatal synaptic connections in both injured and noninjured conditions. Differences in pre-training motor performance were observed between ganciclovir sodium treated nestinδ-TK+ mice and all other groups, where 1–2 weeks of NSPC ablation trends toward improved noninjured motor functions. At present, it is unclear how ablating NSPC might alter pre-training motor performance, but we cannot exclude pre-injury roles for NSPCs in the striatum or more distant regions, such as the amygdala and cerebellum, which may also have nestin-expressing cells and regulate motor coordination.58–60
Conclusion
NSPCs play important roles in the cortex after TBI, and the recruitment of SVZ-derived NSPCs and their neuroblast progeny to the perilesional region may influence the injury microenvironment by promoting neuronal survival, local glial proliferation, reduced gliosis, and motor recovery.
Supplementary Material
Acknowledgments
We would like to thank Jerome Ricard for critical reading of this manuscript.
This work was supported by NIH/NINDS NS049545 & NS30291 (DJL), DOD W81XWH-05-1-0061 (DJL), NS007459 & NS064699 (MHT) and the Miami Project to Cure Paralysis.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Raghupathi R. (2004). Cell death mechanisms following traumatic brain injury. Brain Pathol 14, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo C., Jiang J., Lu Y., and Zhu C. (2002). Spatial and temporal profile of apoptosis following lateral fluid percussion brain injury. Chin. J. Traumatol. 5, 24–27 [PubMed] [Google Scholar]
- 3.Lotocki G., de Rivero Vaccari J.P., Perez E.R., Sanchez-Molano J., Furones-Alonso O., Bramlett H.M., and Dietrich W.D. (2009). Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J. Neurotrauma 26, 1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Connor C.A., Cernak I., and Vink R. (2006). The temporal profile of edema formation differs between male and female rats following diffuse traumatic brain injury. Acta Neurochir. Suppl. 96, 121–124 [DOI] [PubMed] [Google Scholar]
- 5.Ekmark-Lewen S., Lewen A., Israelsson C., Li G.L., Farooque M., Olsson Y., Ebendal T., and Hillered L. (2010). Vimentin and GFAP responses in astrocytes after contusion trauma to the murine brain. Restor. Neurol. Neurosci. 28, 311–321 [DOI] [PubMed] [Google Scholar]
- 6.Homsi S., Piaggio T., Croci N., Noble F., Plotkine M., Marchand-Leroux C., and Jafarian-Tehrani M. (2010). Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J. Neurotrauma 27, 911–921 [DOI] [PubMed] [Google Scholar]
- 7.Morganti-Kossman M.C., Lenzlinger P.M., Hans V., Stahel P., Csuka E., Ammann E., Stocker R., Trentz O., and Kossmann T. (1997). Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry 2, 133–136 [DOI] [PubMed] [Google Scholar]
- 8.Ghirnikar R.S., Lee Y.L., and Eng L.F. (1998). Inflammation in traumatic brain injury: role of cytokines and chemokines. Neurochem. Res. 23, 329–340 [DOI] [PubMed] [Google Scholar]
- 9.Lossinsky A.S., and Shivers R.R. (2004). Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol. Histopathol. 19, 535–564 [DOI] [PubMed] [Google Scholar]
- 10.Taupin P. (2008). Adult neurogenesis, neuroinflammation and therapeutic potential of adult neural stem cells. Int. J. Med. Sci. 5, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thored P., Arvidsson A., Cacci E., Ahlenius H., Kallur T., Darsalia V., Ekdahl C.T., Kokaia Z., and Lindvall O. (2006). Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 24, 739–747 [DOI] [PubMed] [Google Scholar]
- 12.Thored P., Wood J., Arvidsson A., Cammenga J., Kokaia Z., and Lindvall O. (2007). Long-term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke 38, 3032–3039 [DOI] [PubMed] [Google Scholar]
- 13.Chen X.H., Iwata A., Nonaka M., Browne K.D., and Smith D.H. (2003). Neurogenesis and glial proliferation persist for at least one year in the subventricular zone following brain trauma in rats. J. Neurotrauma 20, 623–631 [DOI] [PubMed] [Google Scholar]
- 14.Arvidsson A., Collin T., Kirik D., Kokaia Z., and Lindvall O. (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970 [DOI] [PubMed] [Google Scholar]
- 15.Teramoto T., Qiu J., Plumier J.C., and Moskowitz M.A. (2003). EGF amplifies the replacement of parvalbumin-expressing striatal interneurons after ischemia. J. Clin Invest. 111, 1125–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y.P., Lang B.T., Baskaya M.K., Dempsey R.J. and Vemuganti R. (2009). The potential of neural stem cells to repair stroke-induced brain damage. Acta Neuropathol. 117, 469–480 [DOI] [PubMed] [Google Scholar]
- 17.Lai B., Mao X.O., Xie L., Jin K., and Greenberg D.A. (2008). Electrophysiological neurodifferentiation of subventricular zone-derived precursor cells following stroke. Neurosci. Lett. 442, 305–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arias-Carrion O., Hernandez-Lopez S., Ibanez-Sandoval O., Bargas J., Hernandez-Cruz A., and Drucker-Colin R. (2006). Neuronal precursors within the adult rat subventricular zone differentiate into dopaminergic neurons after substantia nigra lesion and chromaffin cell transplant. J. Neurosci. Res. 84, 1425–1437 [DOI] [PubMed] [Google Scholar]
- 19.Guerra-Crespo M., Gleason D., Sistos A., Toosky T., Solaroglu I., Zhang J.H., Bryant P.J., and Fallon J.H. (2009). Transforming growth factor-alpha induces neurogenesis and behavioral improvement in a chronic stroke model. Neuroscience 160, 470–483 [DOI] [PubMed] [Google Scholar]
- 20.Li B., Piao C.S., Liu X.Y., Guo W.P., Xue Y.Q., Duan W.M., Gonzalez-Toledo M.E., and Zhao L.R. (2010). Brain self-protection: the role of endogenous neural progenitor cells in adult brain after cerebral cortical ischemia. Brain Res. 1327, 91–102 [DOI] [PubMed] [Google Scholar]
- 21.Sun F., Wang X., Mao X., Xie L., and Jin K. (2012). Ablation of neurogenesis attenuates recovery of motor function after focal cerebral ischemia in middle-aged mice. PloS One 7, e46326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Mao X., Xie L., Sun F., Greenberg D.A., and Jin K. (2012). Conditional depletion of neurogenesis inhibits long-term recovery after experimental stroke in mice. PloS One 7, e38932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin K., Wang X., Xie L., Mao X.O., and Greenberg D.A. (2010). Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc. Natl. Acad, Sci. U S A 107, 7993–7998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun C., Sun H., Wu S., Lee C.C., Akamatsu Y., Wang R.K., Kernie S.G., and Liu J. (2013). Conditional ablation of neuroprogenitor cells in adult mice impedes recovery of poststroke cognitive function and reduces synaptic connectivity in the perforant pathway. J. Neurosci. 33, 17314–17325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S.T., Chu K., Jung K.H., Kim S.J., Kim D.H., Kang K.M., Hong N.H., Kim J.H., Ban J.J., Park H.K., Kim S.U., Park C.G., Lee S.K., Kim M., and Roh J.K. (2008). Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain 131, 616–629 [DOI] [PubMed] [Google Scholar]
- 26.Galindo L.T., Filippo T.R., Semedo P., Ariza C.B., Moreira C.M., Camara N.O., and Porcionatto M.A. (2011). Mesenchymal stem cell therapy modulates the inflammatory response in experimental traumatic brain injury. Neurol. Res. Int. 2011, 564089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu T.S., Zhang G., Liebl D.J., and Kernie S.G. (2008). Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J Neurosci. 28, 12901–12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricard J., Salinas J., Garcia L., and Liebl D.J. (2006). EphrinB3 regulates cell proliferation and survival in adult neurogenesis. Mol. Cell Neurosci. 31, 713–722 [DOI] [PubMed] [Google Scholar]
- 29.Theus M.H., Ricard J., and Liebl D.J. (2012). Reproducible expansion and characterization of mouse neural stem/progenitor cells in adherent cultures derived from the adult subventricular zone. Curr. Protoc. Stem Cell Biol. Chapter 2, Unit 2D 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neves S.S., Sarmento-Ribeiro A.B., Simoes S.P., and Pedroso de Lima M.C. (2006). Transfection of oral cancer cells mediated by transferrin-associated lipoplexes: mechanisms of cell death induced by herpes simplex virus thymidine kinase/ganciclovir therapy. Biochim. Biophys. Acta 1758, 1703–1712 [DOI] [PubMed] [Google Scholar]
- 31.Chiu C.C., Li C.H., Fuh T.S., Chen W.L., Huang C.S., Chen L.J., Ung W.H., and Fang K. (2005). The suppressed proliferation and premature senescence by ganciclovir in p53-mutated human non-small-lung cancer cells acquiring herpes simplex virus-thymidine kinase cDNA. Cancer Detect. Prev 29, 286–293 [DOI] [PubMed] [Google Scholar]
- 32.Wei S.J., Chao Y., Hung Y.M., Lin W.C., Yang D.M., Shih Y.L., Ch'ang L.Y., Whang-Peng J., and Yang W.K. (1998). S- and G2-phase cell cycle arrests and apoptosis induced by ganciclovir in murine melanoma cells transduced with herpes simplex virus thymidine kinase. Exp. Cell Res. 241, 66–75 [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Climent M.A., Guirado R., Castillo-Gomez E., Varea E., Gutierrez-Mecinas M., Gilabert-Juan J., Garcia-Mompo C., Vidueira S., Sanchez-Mataredona D., Hernandez S., Blasco-Ibanez J.M., Crespo C., Rutishauser U., Schachner M., and Nacher J. (2011). The polysialylated form of the neural cell adhesion molecule (PSA-NCAM) is expressed in a subpopulation of mature cortical interneurons characterized by reduced structural features and connectivity. Cereb. Cort. 21, 1028–1041 [DOI] [PubMed] [Google Scholar]
- 34.Budinich C.S., Chen H., Lowe D., Rosenberger J.G., Bernstock J.D., and McCabe J.T. (2012). Mouse brain PSA-NCAM levels are altered by graded-controlled cortical impact injury. Neural Plast. 2012, 378307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eliasson C., Sahlgren C., Berthold C.H., Stakeberg J., Celis J.E., Betsholtz C., Eriksson J.E., and Pekny M. (1999). Intermediate filament protein partnership in astrocytes. J. Biol. Chem. 274, 23996–24006 [DOI] [PubMed] [Google Scholar]
- 36.Ahlemeyer B., Kehr K., Richter E., Hirz M., Baumgart-Vogt E., and Herden C. (2013). Phenotype, differentiation, and function differ in rat and mouse neocortical astrocytes cultured under the same conditions. J. Neurosci. Methods 212, 156–164 [DOI] [PubMed] [Google Scholar]
- 37.Snapyan M., Lemasson M., Brill M.S., Blais M., Massouh M., Ninkovic J., Gravel C., Berthod F., Gotz M., Barker P.A., Parent A., and Saghatelyan A. (2009). Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 29, 4172–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petridis A.K,. and El Maarouf A. (2011). Brain-derived neurotrophic factor levels influence the balance of migration and differentiation of subventricular zone cells, but not guidance to the olfactory bulb. J. Clin. Neurosci. 18, 265–270 [DOI] [PubMed] [Google Scholar]
- 39.Heymach J.V., Jr., and Barres B.A. (1995). Neurobiology. Neuronal self-reliance. Nature 374, 405–406 [DOI] [PubMed] [Google Scholar]
- 40.Nishijima K., Ng Y.S., Zhong L., Bradley J., Schubert W., Jo N., Akita J., Samuelsson S.J., Robinson G.S., Adamis A.P., and Shima D.T. (2007). Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 171, 53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O. , Shohami E., Alexandrovich A.G., and Leker R.R. (2012). Subacute treatment with vascular endothelial growth factor after traumatic brain injury increases angiogenesis and gliogenesis. Neuroscience 202, 334–341 [DOI] [PubMed] [Google Scholar]
- 42.Ma Y., Liu W., Wang Y., Chao X., Qu Y., Wang K., and Fei Z. (2011). VEGF protects rat cortical neurons from mechanical trauma injury induced apoptosis via the MEK/ERK pathway. Brain Res. Bull. 86, 441–446 [DOI] [PubMed] [Google Scholar]
- 43.Bernal G.M., and Peterson D.A. (2011). Phenotypic and gene expression modification with normal brain aging in GFAP-positive astrocytes and neural stem cells. Aging Cell 10, 466–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proia P., Schiera G., Mineo M., Ingrassia A.M., Santoro G., Savettieri G., and Di Liegro I. (2008). Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 21, 63–67 [PubMed] [Google Scholar]
- 45.Suzuki R., Okuda M., Asai J., Nagashima G., Itokawa H., Matsunaga A., Fujimoto T., and Suzuki T. (2006). Astrocytes co-express aquaporin-1, −4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. Suppl. 96, 398–401 [DOI] [PubMed] [Google Scholar]
- 46.Salhia B., Angelov L., Roncari L., Wu X., Shannon P., and Guha A. (2000). Expression of vascular endothelial growth factor by reactive astrocytes and associated neoangiogenesis. Brain Res. 883, 87–97 [DOI] [PubMed] [Google Scholar]
- 47.Krum J.M., and Rosenstein J.M. (1998). VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp. Neurol. 154, 57–65 [DOI] [PubMed] [Google Scholar]
- 48.Faiz M., Acarin L., Villapol S., Schulz S., Castellano B., and Gonzalez B. (2008). Substantial migration of SVZ cells to the cortex results in the generation of new neurons in the excitotoxically damaged immature rat brain. Mol. Cell. Neurosci. 38, 170–182 [DOI] [PubMed] [Google Scholar]
- 49.Benner E.J., Luciano D., Jo R., Abdi K., Paez-Gonzalez P., Sheng H., Warner D.S., Liu C., Eroglu C., and Kuo C.T. (2013). Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497, 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giunti D., Parodi B., Usai C., Vergani L., Casazza S., Bruzzone S., Mancardi G., and Uccelli A. (2012). Mesenchymal stem cells shape microglia effector functions through the release of CX3CL1. Stem Cells 30, 2044–2053 [DOI] [PubMed] [Google Scholar]
- 51.Takahashi K., Rochford C.D., and Neumann H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaiss C.A., Yu T.S., Zhang G., Chen J., Dimchev G., Parada L.F., Powell C.M., and Kernie S.G. (2011). Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J. Neurosci. 31, 4906–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang M.T., Yokoi F., Yin H.H., Lovinger D.M., Wang Y., and Li Y. (2006). Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc. Nat. Acad. Sci. U S A 103, 15254–15259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cromwell H.C., and Berridge K.C. (1996). Implementation of action sequences by a neostriatal site: a lesion mapping study of grooming syntax. J. Neurosci. 16, 3444–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doyon J., Owen A.M., Petrides M., Sziklas V., and Evans A.C. (1996). Functional anatomy of visuomotor skill learning in human subjects examined with positron emission tomography. Eur. J. Neurosci. 8, 637–648 [DOI] [PubMed] [Google Scholar]
- 56.Nakano K. (2000). Neural circuits and topographic organization of the basal ganglia and related regions. Brain Dev. 22, Suppl 1, S5–S16 [DOI] [PubMed] [Google Scholar]
- 57.Butti E., Bacigaluppi M., Rossi S., Cambiaghi M., Bari M., Cebrian Silla A., Brambilla E., Musella A., De Ceglia R., Teneud L., De Chiara V., D'Adamo P., Garcia-Verdugo J.M., Comi G., Muzio L., Quattrini A., Leocani L., Maccarrone M., Centonze D., and Martino G. (2012). Subventricular zone neural progenitors protect striatal neurons from glutamatergic excitotoxicity. Brain 135, 3320–3335 [DOI] [PubMed] [Google Scholar]
- 58.Ehninger D., Wang L.P., Klempin F., Romer B., Kettenmann H., and Kempermann G. (2011). Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res. 345, 69–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rani S.B., Mahadevan A., Anilkumar S.R., Raju T.R., and Shankar S.K. (2006). Expression of nestin—a stem cell associated intermediate filament in human CNS tumours. Indian J. Med. Res. 124, 269–280 [PubMed] [Google Scholar]
- 60.Mizuno Y., Ohama E., Hirato J., Nakazato Y., Takahashi H., Takatama M., Takeuchi T., and Okamoto K. (2006). Nestin immunoreactivity of Purkinje cells in Creutzfeldt-Jakob disease. J. Neurol. Sci. 246, 131–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.