

Abstract
Recently, parasite infections or parasite-derived products have been suggested as a therapeutic strategy with suppression of immunopathology, which involves the induction of regulatory T cells or/and T helper type 2 (Th2) responses. In a recent study, researchers reported that constructed recombinant galectin (rTl-gal) isolated from an adult worm of the gastrointestinal nematode parasite Toxascaris leonina attenuated clinical symptoms of inflammatory bowel disease in mice treated with dextran sulphate sodium. Noting the role of rTl-gal in inflammatory disease, we attempted to investigate the effect of the parasite via its rTl-gal on neuronal autoimmune disease using experimental autoimmune encephalomyelitis (EAE), a mouse inflammatory and demyelinating autoimmune disease model of human multiple sclerosis. In this model, rTl-gal-treated experimental autoimmune encephalomyelitis (EAE) mice failed to recover after the peak of the disease, leading to persistent central nervous system (CNS) damage, such as demyelination, gliosis and axonal damage. Further, rTl-gal-treated EAE mice markedly increased the number of CD45R/B220+ B cells in both infiltrated inflammation and the periphery, along with the increased production of autoantibody [anti-myelin oligodendrocyte glycoprotein (MOG)35–55] in serum at chronic stage. Upon antigen restimulation, rTl-gal treatment affected the release of overall cytokines, especially interferon (IFN)-γ and tumour necrosis factor (TNF)-α. Our results suggest that galectin isolated from a gastrointestinal parasite can deliver a harmful effect to EAE contrary to its beneficial effect on inflammatory bowel disease.
Keywords: experimental autoimmune encephalomyelitis (EAE), parasite galectin, Toxascaris leonina
Introduction
Both the incidence and prevalence of autoimmune diseases are rising rapidly worldwide, generating considerable interest in analysing the relationship between this rise in prevalence and pathogen infection. Despite decades of research, a clear causal relationship between pathogen infections and autoimmune disease remains elusive. Among various kinds of pathogens, parasites have evolved effective immune regulation strategies for immune surveillance evasion and prolonged habitation in immunocompetent hosts 1. The widely accepted hypothesis for the immune response against parasite infection involves the modified T helper type 2 (Th2) response, that biases T helper cell polarization towards Th2 or regulatory T cell subsets 2,3. Based on this theory, a substantial number of studies have investigated the immunomodulatory mechanisms of parasite-derived molecules to develop parasite-derived therapeutic agents which can suppress bystander response to self-antigens 4.
Among many candidate parasite-derived immunomodulators, parasitic galectins probably perform similar functions to vertebrate galectins, which are recognized increasingly as immunological mediators of homeostasis and disease regulation because they are conserved evolutionarily 5. In a recent study, researchers reported that constructed recombinant galectin (rTl-gal) isolated from an adult worm of the gastrointestinal nematode parasite Toxascaris leonina attenuated clinical symptoms of inflammatory bowel disease in mice treated with dextran sulphate sodium (DSS) 6. This galectin gene of T. leonina has been shown to be 35% similar to human galectin-9 and almost 90% identical with other parasite galectins. When this recombinant Tl-gal was administered to the DSS-induced colitis model, suppression of pathology was seen with the induction of interleukin (IL)-10 and transforming growth factor (TGF)-β.
Nevertheless, some researchers have suggested that galectins have evolved via independent paths in the vertebrate and invertebrate lineages 7. Although rTl-gal may have similar functions to human galectin-9 in the DSS-induced colitis model, the possibility still remains that in other circumstances rTl-gal may perform distinct functions from host galectin. In this study, in an effort to investigate the role of parasitic galectin in other neuronal autoimmune disease, we decided to examine the impact of rTl-gal during the development of a multiple sclerosis (MS) murine model of experimental autoimmune encephalomyelitis (EAE). Our results showed that, in the EAE model, rTl-gal treatment unexpectedly triggered extensive demyelination and chronic disease activity by enhancing the production of pathogenic autoantibody and proinflammatory cytokines. These data identify the role of parasitic galectin in the pathogenesis of neuronal autoimmune disease, raising the possibility that parasite-derived molecules might promote humoral autoimmunity as well as induce a Th2 response.
Materials and methods
Mice
Female C57BL/6 mice aged 6–8 weeks were purchased from Orientbio, Inc. (Sungnam, Korea). All mice were housed and bred in conventional animal facilities with an NIH-07-approved diet and maintained in a 12/12-h light/dark cycle in temperature-controlled facilities. The study reported herein conformed to the Animal Care and Use of Laboratory Animals of the Jeju National University and protocol was approved by Jeju National University Institutional Animal Care and Use Committee (accreditation no. 2012-0011). All efforts were made to minimize the number of animals used and to reduce animal suffering. Researchers and animal care staff observed and monitored all animals daily. In all cases, animal were euthanized by diethyl ether.
Induction and assessment of experimental autoimmune encephalomyelitis (EAE)
The mouse peptide autoantigen myelin oligodendrocyte glycoprotein (MOG)35–55 (M-E-V-G-W-Y-R-S-P-F-S-R-V-V-H-L-Y-R-N-G-K) was synthesized (purity > 95%) by Biosynthesis Inc. (Lewisville, TX, USA). EAE was induced in C57BL/6 mice by subcutaneous (s.c.) injection into the hind flank with 200 μg of MOG35–55 peptide in complete Freund's adjuvant (CFA) (Difco, Detroit, MI, USA) containing 500 µg of heat-inactivated Mycobacterium tuberculosis. Supplementary injections of 200 ng pertussis toxin (List Biologic, Campbell, CA, USA) were given intravenously (i.v.) on the same day and 2 days later. Clinical assessment of EAE was as follows: 0, no clinical signs; 1, flaccid tail; 2, hind limb weakness or abnormal gait; 3, complete hind limb paralysis; 4, complete hind limb paralysis with forelimb weakness or paralysis; and 5, moribund or deceased. Immunological analyses were carried out 60–62 days after immunization.
Treatment of rTl-gal
The rTl-gal used for this experiment was provided by Dr Hak Sun Yu (Department of Parasitology, Pusan National University, Republic of Korea) 6. For treatment, rTl-gal (10 μg) or phosphate-buffered saline (PBS) was injected intraperitoneally (i.p.) every day from days 3 to 9. The rTl-gal dose was chosen on the basis of our preliminary dose-finding experiment.
Cell culture and proliferation assay
Cells from the spleens were isolated and prepared in single-cell suspensions, as described previously 8. Briefly, splenocytes were isolated mechanically through a 40 μm strainer. After red blood cell lysis with ammonium chloride potassium (ACK) lysis buffer for 10 min, cells were washed with Dulbecco's PBS (DPBS; Gibco-BRL, Life Technologies, Grand Island, NY, USA). Spleen mononuclear cells were then suspended in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum (Gibco-BRL) and 1% antibiotics (100 U/ml penicillin–streptomycin; Gibco-BRL).
To perform the proliferation assay, 4 × 105 spleen mononuclear cells per well were incubated in 200 µl culture medium with MOG35–55 peptide (10 µg/ml), proteolipid protein (PLP)139–151 peptide (10 µg/ml) or concanavalin A (ConA) (5 µg/ml; Sigma, St Louis, MO, USA) at 37 °C in 5% CO2 for 3 days, and then pulsed with 1 μCi of [3H]-methylthymidine (specific activity 42 Ci/mmol; Amersham, Arlington Heights, IL, USA) for the final 18 h of culture. Stimulation with ConA was used as a positive control. Cells were harvested onto glass fibre filters, and the amount of thymidine incorporated into the DNA was measured by liquid scintillation spectrometer (Wallac Micro Beta® TriLux; Perkin Elmer, Waltham, MA, USA).
Preparation of tissue and histological analysis
Spinal cords were collected and fixed in 10% formalin and embedded in paraffin to obtain 3-μm-thick sections. Sections were stained with haematoxylin and eosin (H&E) to assess routine histology and inflammation and with Luxol Fast Blue to analyse demyelination. The extent of inflammation was estimated with three to four areas per tissue section of each mouse (at ×100 magnification), and expressed as mean ± standard error of the mean (s.e.m.). Histological findings were graded into four categories (1, leptomeningeal infiltration; 2, mild perivascular cuffing; 3, extensive perivascular cuffing; 4, extensive perivascular cuffing and severe parenchymal cell infiltration) 9. Demyelination in the spinal cords was scored as follows 10: 1, traces of subpial demyelination; 2, marked subpial and perivascular demyelination; 3, confluent perivascular or subpial demyelination; 4, massive perivascular and subpial demyelination involving one half of the spinal cord with the presence of cellular infiltrates into central nervous system (CNS) parenchyma; and 5, extensive perivascular and subpial demyelination involving the whole cord section with presence of cellular infiltrates into CNS parenchyma.
Immunofluorescent staining and immunohistochemistry
Paraffin-embedded spinal cord sections were deparaffinized and rehydrated using standard methods. For detection of lymphocyte and macrophage immunoreactivity, rabbit anti-mouse CD3e (1 : 50, BD Biosciences, San Jose, CA, USA), biotin-conjugated anti-mouse CD45R/B220 (1 : 100, BD Biosciences) and rat anti-mouse F4/80 (1 : 100, Biolegend, San Diego, USA) were used. Biotinylated goat anti-rabbit immunoglobulin (Ig)G or rabbit anti-rat IgG followed by avidin–biotin–peroxidase complexes (Vector Laboratories, Burlingame, CA, USA) were used as secondary antibodies. Controls with secondary antibody alone were negative throughout. Horseradish peroxidase (HRP) binding sites were detected with 3,3'-diaminobenzidine tetrachloride (DAB; Vector Laboratories) and counterstained with haematoxylin. Quantifying positive cells stained by immunohistochemistry was performed using ImageJ 1.38× software (National Institutes of Health, Bethesda, MD, USA) under a high-power field (HPF) light microscope (×400).
Gliosis was examined using mouse anti-mouse glial fibrillary acidic protein (GFAP) (1 : 500; Novus Biologicals, San Diego, CA, USA). For secondary antibodies, antibodies conjugated directly with fluorescein isothiocyanate (FITC) at a dilution of 1 : 100; anti-mouse IgG (Santa Cruz Biotechnologies, Santa Cruz, CA, USA) were used. Controls with secondary antibody alone were negative throughout. Sections were counterstained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI, 1 : 2000; Sigma) and observed under a fluorescent microscope (LeicaDM LB2; Leica, Wetzler, Germany) with appropriate filters. Images were obtained using an Olympus DP-72 microscope camera (Olympus, Tokyo, Japan) and assembled using Adobe Photoshop™ software. Quantitative analysis was performed using ImageJ software. Two to three sections per animal were examined under a lower-magnification microscope (×100 magnification) and the three lesions showing the most representative expression from sections were analysed under higher magnification (×400 magnification).
To detect axonal damage, sections were allowed to react with mouse anti-mouse non-phosphorylated neurofilament-H (NF-H) antibody (SMI-32, 1 : 1000; Convance Inc., Princeton, NJ, USA). Biotinylated horse anti-mouse IgG followed by avidin–biotin–peroxidase complexes (Vector Laboratories) were used as secondary antibodies. HRP binding sites were detected with DAB (Vector Laboratories) and counterstained with haematoxylin. Slides were examined using a light microscope (LeicaDM LB2; Leica), and positive areas were measured by ImageJ software.
Flow cytometric analysis
Single-cell suspensions of spleen (1 × 106 cells) were stained with monoclonal antibodies (mAbs) directly labelled with one of the following fluorescent tags: FITC, phosphatidyl ethanolamine (PE), CD3 (145-2c11), CD4 (H129.19), CD8 (53–6.7), CD45 (Ly-5), CD11b (M1/70) (BD Biosciences, San Jose, CA, USA), CD25 (PC61.5), CD45R/B220 (RA3-6B2) and CD11c (HL3) (eBioscience, San Diego, CA, USA). Appropriate isotype controls (rat IgG1, rat IgG2a, rat IgG2b or hamster IgG1; Biolegend) were always included. Twenty thousand viable cells per mouse (as determined by light-scatter profiles) were analysed using a BD FACSCalibur™ flow cytometer (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Spleen mononuclear cell suspensions containing 4 × 105 cells in 200 µl culture medium were placed in 96-well round-bottomed microtitre plates with or without 10 μg/ml MOG35–5511. Supernatants were collected after 48 h, and the production of interferon (IFN)-γ, tumour necrosis factor (TNF)-α, IL-10 (BioSource International, Camarillo, CA, USA), IL-1β, IL-17 (eBioscience) and IL-4 (R&D Systems, Minneapolis, MN, USA) was measured using mouse cytokine-specific ELISA kits according to the manufacturer's instructions. Mice serum was analysed for the detection of MOG35–55-specific IgG by ELISA kit (AnaSpec, Fremont, CA, USA), according to the manufacturer's instructions.
Statistical analysis
The results were presented as the mean ± s.e.m. for each group. One-way analysis of variance (anova) was used to determine statistical difference between groups. Clinical scores and histological score were analysed using the non-parametric Mann–Whitney U-test. A P-value less than 0·05 was considered statistically significant.
Results
rTl-gal aggravates the clinical severity of EAE
To determine whether or not injection of rTl-gal can affect the occurrence of EAE, C57BL/6 mice were immunized with MOG33–35 in complete Freund's adjuvant (CFA) and monitored for clinical disease development. Mice treated with the rTl-gal developed more severe disease than did the controls (Fig. 1). The disease incidence was 100% in both groups, and the day of disease onset was similar between the two groups (Table1). No significant difference in clinical disease severity was observed between rTl-gal-treated and control mice, starting from 13 days post-immunization (dpi), and through the first disease peak and up to approximately 25 dpi. Control mice reached the disease peak at 18 dpi and exhibited a partial remission until 30 dpi. In contrast, rTl-gal-treated mice failed to recover after the initial disease peak, leading to sustained neurological impairment that started at approximately 26 dpi and was maintained up to 60 dpi. As EAE is thought to be mediated by infiltrating peripheral myelin-reactive T cells into the CNS, we analysed whether rTl-gal treatment affected autoreactive T cell responses in the spleen of MOG35–55-induced EAE mice. Initially, splenic mononuclear cells from mice at the peak (14–18 dpi) and chronic (60–62 dpi) stages of EAE were restimulated with MOG35–55 peptide and proliferation was measured using [3H]-thymidine incorporation. Noticeably, at chronic stage, the proliferation of splenic mononuclear cells from rTl-gal-treated EAE mice displayed more than a 90% increase in response to MOG35–55 peptide (Fig. 1b). However, splenic mononuclear cells from rTl-gal-treated and vehicle-treated mice at peak stage proliferated similarly, consistent with their clinical score (Supporting information, Fig. S1c).
Fig 1.
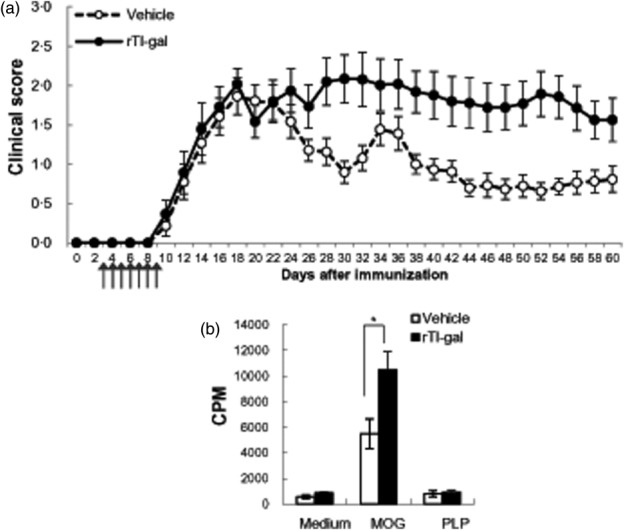
rTl-gal treatment increased the clinical symptoms of experimental autoimmune encephalomyelitis (EAE). (a) Clinical scores of myelin oligodendrocyte glycoprotein (MOG)-induced EAE in recombinant Toxascaris leonine galectin (rTl-gal)- and vehicle-treated mice. rTl-gal showed higher mean clinical scores than vehicle-treated control mice. A representative of six independent experiments is shown, in which each data point represents the mean ± standard error of the mean (s.e.m.) of 27 vehicle and 20 rTl-gal-treated mice. Arrows indicate days of rTl-gal injection. (b) rTl-gal treatment stimulated the proliferation of splenic mononuclear cells to MOG35–55 restimulation at chronic stage. Proliferative response to the antigens [MOG35–55, proteolipid protein (PLP)138–151 or concanavalin A (ConA)] was assessed in triplicate wells for each experiment. Background proliferation was 729 ± 245 counts per minute (cpm) and concanavalin A (ConA)-induced proliferation was 42 986 ± 5737 cpm. Data are the mean ± s.e.m. of cpm and are representative of three independent experiments. Statistical evaluation was performed to compare the experimental groups and corresponding control groups, respectively. *P < 0·05.
Table 1.
Summary of active clinical parameters
| Clinical status | |||
|---|---|---|---|
| Treatment | Incidence of EAE (%) | Days of onset | Mean of maximal score |
| Vehicle | 100 (27/27) | 13.2 ± 0.6 | 2.9 ± 0.2 |
| rTl-gal | 100 (20/20) | 13.5 ± 0.8 | 3.3 ± 0.1* |
Experimental autoimmune encephalomyelitis (EAE) clinical parameters were observed in myelin oligodendrocyte glycoprotein (MOG)-induced EAE in recombinant Toxascaris leonine galectin (rTl-gal)-treated and vehicle-treated mice. Each number represents the mean ± standard error of the mean of six independent experiments. Statistical difference of P < 0·05 (*) for rTl-gal-treated mice versus vehicle controls is indicated.
rTl-gal exacerbates neuropathology of MOG-immunized mice
Infiltration of autoreactive immune cells into the CNS results in inflammation and demylination, alterations in neuronal function and axonal damage. At the first peak of disease (14–18 dpi), marked immune cell infiltration and demyelination (Supporting information, Fig. S1a) were present in both rTl-gal-treated and control mice, respectively. The inflammatory scores which indicated the diffuse infiltration by mixed macrophages, T and B lymphocytes into CNS in the lumbar spinal cord were similar between the two groups (Supporting information, Fig. S1b). These findings were consistent with clinical severity at the peak stage of the EAE. However, demyelination in the lumbar spinal cords of EAE mice was slightly more severe in the group treated with rTl-gal, even though the difference was not statistically significant (Supporting information, Fig. S1B). Demyelination induced by rTl-gal treatment was marked at the chronic stage (60–62 dpi) (Fig. 2). The demyelination score in dorsal, ventral and lateral columns of chronic stage of rTl-gal-treated EAE mice was profoundly increased, with a slight difference of inflammation between the two groups (Fig. 2i). These findings demonstrate that the attenuation of EAE remission by rTl-gal treatment is caused mainly by severe demyelination.
Fig 2.

Neuropathological changes within representative lumbar spinal cord sections obtained from recombinant Toxascaris leonine galectin (rTl-gal)-treated and control (vehicle-treated) mice. Inflammatory infiltration and a large plaque of demyelination were seen in rTl-gal-treated experimental autoimmune encephalomyelitis (EAE) mice at chronic stage (60–62 dpi) (e–h). Images in (b–d) and (f–h) are higher magnifications of the boxed portions in images (a) and (e), respectively. Bars = 60 µm in (b–d) and (f–h). (i) Mice were subjected to histopathological assay. Each group consisted of three animals. Sections were analysed for haematoxylin and eosin (H&E) and luxol fast blue staining to detect inflammatory infiltration and demyelination, respectively. Values represent the mean ± standard error of the mean (s.e.m.). Statistical difference of P < 0·05 (*) for rTl-gal-treated mice versus controls is indicated.
rTl-gal increases the infiltration of CD45R/B220+ B cells into CNS, not CD3+ T cells and F4/80+ macrophage, at the chronic stage of EAE
To verify the specific cell population whose infiltration was altered by rTl-gal treatment, we assessed the immunoreactivity of CD3+ T cells, CD45R/B220+ B cells and F4/80+ macrophages in the spinal cords of each group. In rTl-gal-treated mice, the infiltration of CD45R/B220+ B cells was increased markedly compared to control mice (Fig. 3b,e,k), a result which mirrored the enhancement of demyelination by rTl-gal treatment. However, there was no significant difference in CD3+ T cell (Fig. 3a,d,j) and F4/80+ macrophage (Fig. 3c,f,l) infiltration between the two groups. Demyelination was typically observed more than inflammation at the chronic stage of disease (60–62 dpi), which raised the possibility that increasing B cell infiltration by rTl-gal treatment was associated with demyelination and more severe clinical disease scores seen in this phase.
Fig 3.

Recombinant Toxascaris leonine galectin (rTl-gal) increased the infiltration of CD45R/B220+ cells into the central nervous system (CNS) at chronic stage. The immunohistochemical images depicted CD3+ (a,d), CD45R/B220+ (b,e) and F4/80+ (c,f) cells in lumbar spinal cords of rTl-gal-treated and vehicle-treated experimental autoimmune encephalomyelitis (EAE) mice, respectively. Haematoxylin staining was performed to illustrate nuclei. Controls with secondary antibody alone were negative throughout (g–i). Quantification of CD3+ (j), CD45R/B220+ (k) and F4/80+ (l) infiltrated cells per high-power field (HPF, magnification × 400) was performed using three lesions showing the most representative expression from each mouse. Results are representative of more than three independent experiments. Bars = 60 µm.
rTl-gal induces the astrogliosis and axonal damage of spinal cord of EAE mice at the chronic stage
Demyelination is commonly accompanied by both gliosis and axonal damage. To investigate if rTl-gal affected glial cells in the CNS as well as demyelination, we examined the immunoreactivity of GFAP and ionizing calcium binding adaptor molecule-1 (Iba-1). We observed significantly increased GFAP+ astrocytes in spinal cord of rTl-gal-treated EAE mice compared with vehicle-treated mice (Fig. 4a), but there was no significant difference in Iba-1 immunoreactivity on microglia between the two groups (Fig. 4b). These findings indicate that the demyelination induced by rTl-gal treatment may be associated with astrogliosis in the CNS during MOG-induced EAE.
Fig 4.
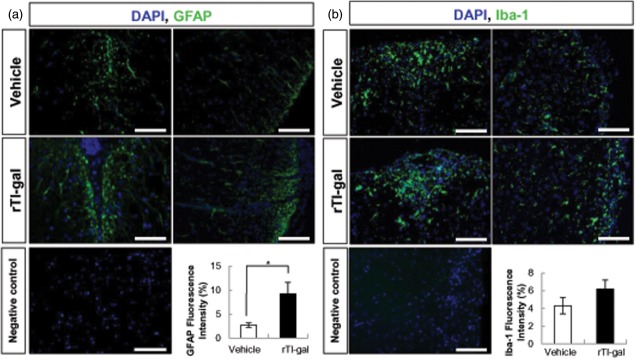
Recombinant Toxascaris leonine galectin (rTl-gal) increased astrogliosis in spinal cords at chronic stage. Spinal cord sections from vehicle- or rTl-gal-treated EAE mice were stained for glial fibrillary acidic protein (GFAP) (a, astrocytes) and Iba-1 (b, microglia). Quantification of fluorescence intensity demonstrates rTl-gal-treated mice had more GFAP reactivity than vehicle-treated mice. 4',6-diamidino-2-phenylindole dihydrochloride (DAPI) staining was used for indicating nuclei. Results are representative of more than three independent experiments. Values represent the mean ± standard error of the mean (s.e.m.). Statistical difference of P < 0·05 (*) for rTl-gal-treated mice versus controls is indicated. Bars = 60 µm.
Axonal damage is one of the major criteria which correlate with clinical scores in chronic EAE. Based on the results indicating that rTl-gal increased both demyelination and astrogliosis, we assumed that rTl-gal treatment might induce axonal damage in the CNS of EAE mice. In rTl-gal-treated EAE mice, a significant number of axons reacted to NF-H antibody, a marker of axonal damage, in both white (Fig. 5c,f) and grey matter (Fig. 5b,e) of spinal cord. Quantitative analysis confirmed that rTl-gal treatment caused a significant 1.86-fold increase in the intensity of the NF-H (Fig. 5g). These results indicate that irreversible axonal damage induced by rTl-gal treatment results in inhibition of disease remission.
Fig 5.
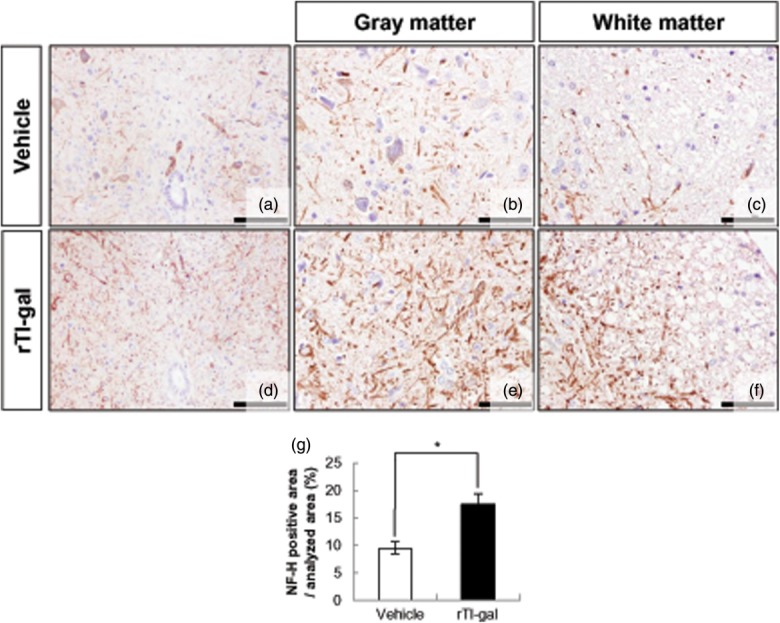
Recombinant Toxascaris leonine galectin (rTl-gal) treatment induced axonal dystrophy in spinal cords at chronic stage. Immunohistochemistry was performed with SMI-32 antibody against non-phosphorylated neurofilament-H (NF-H) in the spinal cords of mice treated with either vehicle or rTl-gal at the chronic (60–62 dpi) stage, respectively. Axonal damage of spinal cord was severe in both grey and white matter of rTl-gal-treated mice (e,f) compared to control mice (b,c). (a,d) Lower magnification view of spinal cord. Results are representative of more than three independent experiments. (g) Quantification of the NF-H-positive area shows that the rTl-gal-treated group had more NF-H intensity than the vehicle-treated group. Values represent the mean ± standard error of the mean (s.e.m.). Statistical difference of P < 0·05 (*) for rTl-gal-treated mice versus controls is indicated. Bars = 60 µm in (b–c) and (e–f) and 120 µm in (a–d).
rTl-gal treatment increases the population of CD45R/B220+ B cells in the periphery
Because rTl-gal treatment increased the infiltration of B cells in CNS of EAE-affected mice, we investigated whether rTl-gal also affected the mononuclear cell population in the periphery. Flow cytometric analysis revealed that there was no significant difference in other immune cell subpopulations (CD3+CD4+ cells, CD3+CD8+ cells, CD45+CD11b+ cells and CD45+CD11c+ cells) between the two groups. Only the frequency of splenic CD45R/B220+ B cells was up-regulated significantly in rTl-gal-treated EAE mice compared to that in vehicle-treated mice (Fig. 6a,b), suggesting that rTl-gal treatment induces the expansion of B cells in the periphery as well as the CNS.
Fig 6.

Recombinant Toxascaris leonine galectin (rTl-gal) treatment up-regulated the population of CD45R/B220-positive B cells in the periphery. (a) Splenic mononuclear cells at chronic stage (60–62 dpi) from mice treated with either vehicle or rTl-gal were analysed by flow cytometry for CD3+CD4+ T cells, CD3+CD8+ T cells, CD45+CD45R/B220+ B cells, CD45+CD11b+ macrophages and CD45+CD11c+ dendritic cells, respectively. Dot-plots shown represent one of three separate experiments with similar observations. (b) The mean numbers of each cell population of splenic mononuclear cells were plotted. *Statistically significant difference (P < 0·05 by Student's t-test) from vehicle-treated EAE mice.
As many immune regulatory molecules from parasites are reported to stimulate the expansion of CD4+CD25+ regulatory T cells 1, we assessed the possibility that rTl-gal might affect the population of regulatory T cells (Tregs) by quantifying the number of Tregs in spleen of both groups. As a result, comparison of rTl-gal-treated and vehicle-treated mice showed no difference in the frequency of CD4+CD25+ Tregs (Supporting information, Fig. S2b). These results show that rTl-gal increases the population of CD45R/B220+ B cells in the periphery by acting parasitic galectin without altering Treg activity.
rTl-gal treatment modifies cytokine production in EAE mice at the chronic stage
To delineate the immune mechanism underlying increased EAE severity in rTl-gal-treated mice, cytokine production was evaluated at the chronic stage. Splenic mononuclear cells were collected at chronic disease, restimulated with MOG35–55 and the cytokine profiles in the culture supernatants were measured. The release of IFN-γ and TNF-α increased significantly in rTl-gal-treated mice compared with controls (Fig. 7a). Furthermore, the production of other proinflammatory (IL-17 and IL-1β) and anti-inflammatory (IL-4 and IL-10) cytokines was increased in the rTl-gal treated group, although the changes were not statistically significant. These alterations by rTl-gal treatment were not observed at the peak (14–18 dpi) stage of disease (Supporting information, Fig. S1d).
Fig 7.
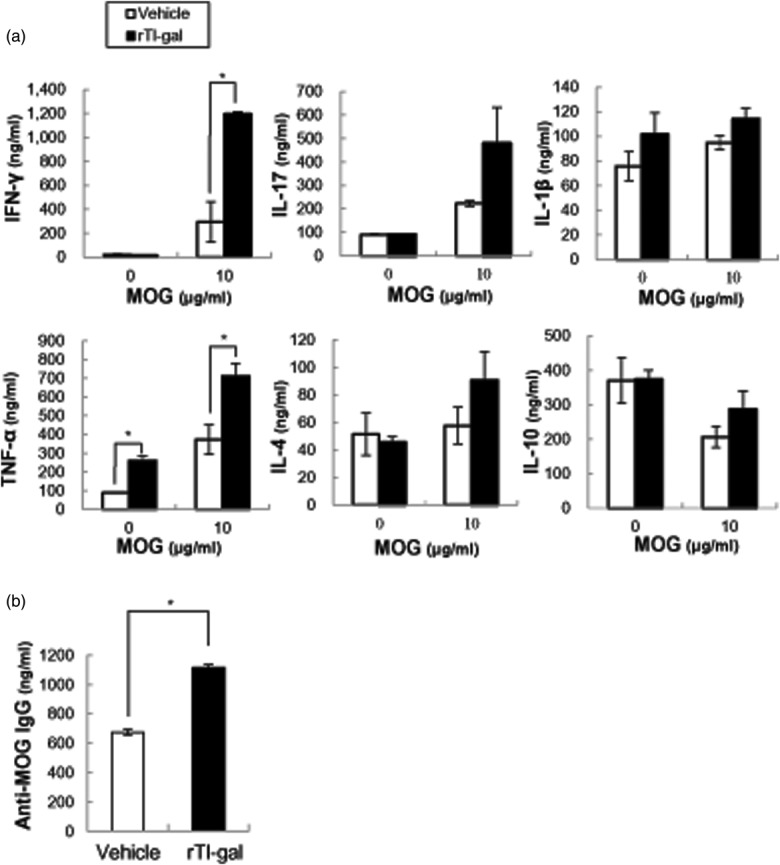
Proinflammatory cytokine and anti-myelin oligodendrocyte glycoprotein (MOG)35–55 immunoglobulin (IgG) were increased in recombinant Toxascaris leonine galectin (rTl-gal)-treated mice compared to controls. (a) Single-cell suspension of spleen was prepared from mice treated with either vehicle or rTl-gal at chronic stage (60–62 dpi). The cell suspension was cultured with or without MOG35–55 (10 μg/ml) for 48 h and the production of cytokines [interferon (IFN)-γ, interleukin (IL)-17, IL-1β, tumour necrosis factor (TNF)-α, IL-4 and IL-10) was measured by enzyme-linked immunosorbent assay (ELISA). (b) ELISA detection of MOG-specific IgG in serum at chronic experimental autoimmune encephalomyelitis (EAE). Representative results from at least three experiments are shown. *Statistically significant difference (P < 0·05) from vehicle-treated EAE mice.
rTl-gal-treated EAE mice have increased levels of autoantibody in the serum
Because rTl-gal-treated EAE mice showed up-regulation of the B cell response and demyelination, the level of anti-MOG autoantibodies in the serum was measured by examining the presence of MOG-specific IgG in sera harvested at the chronic stage. It emerged that anti-MOG IgG was increased significantly in the serum of rTl-gal-treated EAE mice compared to vehicle-treated mice (Fig. 7b).The up-regulation of anti-MOG IgG production by rTl-gal treatment was not observed at the peak (14–18 dpi) stage of disease (Supporting information, Fig. S1d). These results suggest that the autoantibody production triggered by EAE induction in rTl-gal-treated mice is maintained, even at later time-points post-immunization, and high levels of anti-MOG autoantibody are probably responsible for perpetuating the B cell response and demyelination.
Discussion
The association between parasite infection and autoimmune disease has interested researchers for many years. This is mainly because parasite infections typically have immunosuppressive activity by triggering Th2/Treg inducing immune responses. Many parasites and parasite-derived products have been reported to improve the symptoms of autoimmune disease, whereas others seem to contribute to generation or exacerbation 12,13.
Galectins are a family of animal lectins with affinity to β-galactose-containing oligosaccharides. These are currently recognized as important immune mediators through regulating the homeostasis and functioning of the immune cells 14. Because galectins are conserved evolutionarily, they have been found in a number of parasites as well as in humans. As no function has yet been attributed to parasite galectins, it has been speculated that they may be involved in host–parasite interaction 15. For example, a galectin from Angiostronglylus cantonensis plays a role in the activation of microglia, but the mechanism is unknown 16. Moreover, current evidence suggests that parasite galectins participate actively in the infection process as pattern-recognition receptors (PRRs) 17,18.
In one study, the chemokinetic activity of the infective third stage larvae of Haemonchus contortus was mediated by the nematode galectins in a carbohydrate-dependent manner and was speculated to mimic the action of host galectin-9 19. Similarly, constructed recombinant galectin (rTl-gal) isolated from an adult worm of the gastrointestinal nematode parasite T. leonina attenuated the clinical symptoms of the DSS colitis mouse model 6. Also, this gene has been shown to have 35% similarity with human galectin-9. The galectin-9 has been known as a ligand for the Th1- and Th17-specific cell-surface molecule Tim-3, and has been conjectured to function in regulating Th1 and Th17 responses. Furthermore, recombinant galectin-9 reduced disease severity and mortality in mice with EAE 20. For this reason, we first expected that rTl-gal would also attenuate EAE severity but, unexpectedly, rTl-gal treatment did not alter the frequency of Tim-3-positive cells (data not shown). The population change of regulatory T cells was also not detected, either in rTl-gal-treated EAE mice (Supporting information, Fig. S2) or the rTl-gal treated DSS-colitis model 6. These results raise the possibility that rTl-gal may perform distinct functions from host galectin.
EAE is the most commonly used animal model for the human inflammatory demyelinating disease, MS. Like MS, EAE is characterized by CNS inflammation and demyelination and follows the disease course of acute, relapsing–remitting (RR) or chronic stages. The clinical heterogeneity of these diseases reflects pathological heterogeneity in terms of relative proportions of the main pathological features such as inflammation, demyelination, axonal loss and gliosis. For example, inflammatory infiltrates with various immune cells and active demyelination are observed mainly at the acute monophasic disease lesion. When this lesion becomes chronic, significant myelin loss and gliosis, known as ‘plaque', are shown distinctively. Finally, irreversible axonal loss leads to long-term disability and progression 21. This heterogeneity in EAE is caused not only by genetic and/or antigenic difference, but by the degree of autoreactivity at the early stages of disease. It has been demonstrated that the development of chronic EAE phenotype is driven by processes that occur during the early stage by the direct comparison of RR-EAE and chronic-EAE generated in the same strain with the same antigen 22. It has also been shown that inflammatory infiltration, demyelination and axonal loss are greater in chronic-EAE than in RR-EAE during the course of disease and sustained in the later stage of chronic-EAE 22. It is therefore likely that the degree of autoimmunity, demyelination and axonal loss are decisive factors to determine whether or not the disease progresses to the chronic sustained stage.
Additionally, B cells are thought to contribute to the pathogenesis of EAE through producing autoantibodies that amplify demyelination via opsonization of myelin, although macrophage and T cells are the main cells in the inflammatory infiltrate 23. For this reason, autoantibodies in blood are considered to be associated with the development of demyelination and consequently raise the possibility the disease becomes chronic. Therefore, there is a strong possibility that rTl-gal treatment-induced B cell pathogenesis in EAE is caused by the high concentration of anti-myelin antibodies, although there is no direct evidence of whether rTl-gal affects B cell directly.
The current study found that rTl-gal-treated EAE mice developed a chronic sustained disease with demyelination and irreversible axonal loss. Most significantly, we found that rTl-gal treatment resulted in large populations of B cells in both the periphery and the CNS, as well as high serum titres of MOG-specific antibodies. These results are in agreement with a recent report that autoantibody production plays an important role in the progression of demyelination, which leads to axonal damage 24,25. It is known that axonal loss is the pathological substrate of permanent disability in chronic EAE 26. Hence, it could conceivably be hypothesized that rTl-gal treatment may promote the secretion of autoantibody from B cells and consequently trigger the chronic disease activity which induces demyelination and irreversible axonal loss.
As activated Th1 cells (IFN-γ-producing cells) and Th17 cells (IL-17-producing cells) are thought to be the main culprits in EAE/MS, immune deviation strategy is often used for MS treatment 27. In a similar fashion, there is a strong possibility that parasites or parasite-derived products are used as treatments, because parasites were generally thought to suppress Th1 cells while inducing Th2 cells and Tregs. However, it has been found that some parasitic products can induce other immune responses than Th2 responses 28, and might also act like allergens 29. In our experiments, rTl-gal treatment strongly affected the effector arm of the immune response, IFN-γ and TNF-α. It also slightly affected other cytokines, such as proinflammatory cytokines (IL-17 andIL-1β), as well as anti-inflammatory cytokines (IL-4 and IL-10), suggesting that rTl-gal may contribute to overall immune activation. Supporting this view are reports showing that, in non-human primate EAE, the induction of Th2 response to myelin antigens may exacerbate autoimmunity by enhancing the production of pathogenic autoantibodies 30. Therefore, rTl-gal seems to be linked to neuronal autoimmune disease development, although the mechanism by which cells and receptors are related to rTl-gal-induced disease severity is as yet unknown.
In conclusion, we demonstrate that parasite galectin isolated from T. leonina enhances EAE severity and antibody production resulting in the attenuation of EAE remission. Although further studies are required to delineate definitively the mechanism of how the parasite galectin exacerbates disease, this research suggests that not all parasite-based therapy is safe and not all parasites have the ability to suppress inflammatory disease.
Acknowledgments
This research was supported by a Mid-career Researcher Program (no. 2012R1A2A2A01011645) and Global PhD Fellowship Program (no. 2012H1A2A1010108) through a National Research Foundation (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) of Korea. The authors wish to thank Tae-Hoon Chung for editorial assistance.
Author contributions
S. J. B. designed the study, performed experiments, analysed the data and wrote the manuscript. D. H., G. A., J. C., A. K. and S. K. P. performed experiments and analysed the data. H. S. Y. participated in its co-ordination and revised the manuscript. Y. J. designed the study and revised the manuscript. All authors have read and approved the final manuscript.
Disclosures
None to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Fig. S1. The neuropathogical change and immune response at peak stage were not different between the two groups. (a) Luxol fast blue: haematoxylin and eosin (H&E) staining of recombinant Toxascaris leonine galectin (rTl-gal)-treated and vehicle-treated spinal cords at peak (14–18 dpi) stage of experimental autoimmune encephalomyelitis (EAE). Bars = 60 µm. (b) The inflammation and demyelination scores in dorsal, ventral and lateral columns at peak (14–18 dpi) stage of rTl-gal-treated and vehicle-treated EAE mice. Values represent the mean ± standard error of the mean (s.e.m.). (c) Proliferation of T lymphocytes isolated from spleens at peak (14–18 dpi) stage of EAE, cells were incubated for 72 h with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (10 µg/ml), proteolipid protein (PLP)139–151 peptide (10 µg/ml) or concanavalin A (ConA). Proliferation was evaluated by thymidine incorporation measured during the last 18 h of culture. Data are mean ± s.e.m. of counts per minute (cpm) and are representative of three independent experiments. (d) Enzyme-linked immunosorbent assay (ELISA) determination of interferon (IFN)-γ, interleukin (IL)-17, IL-1β, tumour necrosis factor (TNF)-α, IL-4 and IL-10 in supernatant of splenic mononuclear cells isolated at peak (14–18 dpi) stage of EAE and restimulated with MOG35–55 peptide (10 µg/ml). Values represent the mean ± s.e.m. and are representative of three independent experiments. (e) Serum was harvested at peak (14–18 dpi) stage of EAE and the levels of anti-MOG35–55 immunoglobulin (Ig)G were quantified through ELISA. Values represent the mean ± s.e.m. and are representative of two independent experiments.
Fig. S2. Recombinant Toxascaris leonine galectin (rTl-gal) treatment did not affect the regulatory T cells. (a) Spinal cord sections from vehicle- or rTl-gal-treated experimental autoimmune encephalomyelitis (EAE) mice were stained for CD4 (green colour) and CD25 (red colour). Bars = 60 µm). (b) rTl-gal treatment did not affect the population of CD4+CD25+ regulatory T cells. Splenic mononuclear cells were analysed by flow cytometry for CD4+CD25+ on days 60–62 dpi of mice treated with either vehicle or rTl-gal. Dot-plots represent one of three independent experiments with similar observations and the mean population of each cell was plotted in the graph. (c) Real-time polymerase chain reaction (PCR) on cDNA isolated from spinal cords and spleens of vehicle- or rTl-gal-treated EAE mice was performed with forkhead box protein 3 primers. Expression data were normalized relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and fold change was compared to vehicle-treated mice.
References
- Sorci G, Cornet S, Faivre B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens. 2013;2:71–91. doi: 10.3390/pathogens2010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. 2013;13:607–14. doi: 10.1038/nri3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JA, Friberg IM, Little S, Bradley JE. Review series on helminths, immune modulation and the hygiene hypothesis: immunity against helminths and immunological phenomena in modern human populations: coevolutionary legacies? Immunology. 2009;126:18–27. doi: 10.1111/j.1365-2567.2008.03010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSorley HJ, Hewitson JP, Maizels RM. Immunomodulation by helminth parasites: defining mechanisms and mediators. Int J Parasitol. 2013;43:301–10. doi: 10.1016/j.ijpara.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Young AR, Meeusen EN. Galectins in parasite infection and allergic inflammation. Glycoconj J. 2002;19:601–6. doi: 10.1023/B:GLYC.0000014091.00844.0a. [DOI] [PubMed] [Google Scholar]
- Kim J-Y, Cho MK, Choi SH, et al. Inhibition of dextran sulfate sodium (DSS)-induced intestinal inflammation via enhanced IL-10 and TGF-β production by galectin-9 homologues isolated from intestinal parasites. Mol Biochem Parasitol. 2010;174:53–61. doi: 10.1016/j.molbiopara.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Dodd RB, Drickamer K. Lectin-like proteins in model organisms: implications for evolution of carbohydrate-binding activity. Glycobiology. 2001;11:71R–79R. doi: 10.1093/glycob/11.5.71r. [DOI] [PubMed] [Google Scholar]
- Bing SJ, Ha D, Kim MJ, et al. Geraniin down regulates gamma radiation-induced apoptosis by suppressing DNA damage. Food Chem Toxicol. 2013;57:147–53. doi: 10.1016/j.fct.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Hwang I, Ahn G, Park E, Ha D, Song J-Y, Jee Y. An acidic polysaccharide of Panax ginseng ameliorates experimental autoimmune encephalomyelitis and induces regulatory T cells. Immunol Lett. 2011;138:169–78. doi: 10.1016/j.imlet.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Zappia E, Casazza S, Pedemonte E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–61. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- Jee Y, Piao WH, Liu R, et al. CD4+CD25+ regulatory T cells contribute to the therapeutic effects of glatiramer acetate in experimental autoimmune encephalomyelitis. Clin Immunol. 2007;125:34–42. doi: 10.1016/j.clim.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Erb KJ. Can helminths or helminth-derived products be used in humans to prevent or treat allergic diseases? Trends Immunol. 2009;30:75–82. doi: 10.1016/j.it.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Pinelli E, Brandes S, Dormans J, Gremmer E, Van Loveren H. Infection with the roundworm Toxocara canis leads to exacerbation of experimental allergic airway inflammation. Clin Exp Allergy. 2008;38:649–58. doi: 10.1111/j.1365-2222.2007.02908.x. [DOI] [PubMed] [Google Scholar]
- Liu F-T. Regulatory roles of galectins in the immune response. Int Arch Allergy Immunol. 2005;136:385–400. doi: 10.1159/000084545. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Gruppi A. Galectins as immunoregulators during infectious processes: from microbial invasion to the resolution of the disease. Parasite Immunol. 2005;27:103–14. doi: 10.1111/j.1365-3024.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- Liu L, He H-J, Lv Z, et al. The mRNA level of the galectin-10 of Angiostrongylus cantonensis induced by reactive oxygen stress. Parasitol Res. 2013;112:933–43. doi: 10.1007/s00436-012-3210-5. [DOI] [PubMed] [Google Scholar]
- Van den Berg TK, Honing H, Franke N, et al. LacdiNAc-glycans constitute a parasite pattern for galectin-3-mediated immune recognition. J Immunol Baltim 1950. 2004;173:1902–7. doi: 10.4049/jimmunol.173.3.1902. [DOI] [PubMed] [Google Scholar]
- Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. 2009;7:424–38. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DG, Wildblood LA, Inglis NF, Jones DG. Characterization of a galectin-like activity from the parasitic nematode, Haemonchus contortus, which modulates ovine eosinophil migration in vitro. Vet Immunol Immunopathol. 2008;122:138–45. doi: 10.1016/j.vetimm.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) Br J Pharmacol. 2011;164:1079–106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berard JL, Wolak K, Fournier S, David S. Characterization of relapsing–remitting and chronic forms of experimental autoimmune encephalomyelitis in C57BL/6 mice. Glia. 2010;58:434–45. doi: 10.1002/glia.20935. [DOI] [PubMed] [Google Scholar]
- Batoulis H, Recks MS, Addicks K, Kuerten S. Experimental autoimmune encephalomyelitis – achievements and prospective advances. APMIS. 2011;119:819–30. doi: 10.1111/j.1600-0463.2011.02794.x. [DOI] [PubMed] [Google Scholar]
- Berer K, Wekerle H, Krishnamoorthy G. B cells in spontaneous autoimmune diseases of the central nervous system. Mol Immunol. 2011;48:1332–7. doi: 10.1016/j.molimm.2010.10.025. [DOI] [PubMed] [Google Scholar]
- Ohtani S, Kohyama K, Matsumoto Y. Autoantibodies recognizing native MOG are closely associated with active demyelination but not with neuroinflammation in chronic EAE. Neuropathology. 2011;31:101–11. doi: 10.1111/j.1440-1789.2010.01131.x. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197:373–85. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Batoulis H, Recks MS, Addicks K, Kuerten S. Experimental autoimmune encephalomyelitis – achievements and prospective advances. APMIS. 2011;119:819–30. doi: 10.1111/j.1600-0463.2011.02794.x. [DOI] [PubMed] [Google Scholar]
- Fallon PG, Mangan NE. Suppression of TH2-type allergic reactions by helminth infection. Nat Rev Immunol. 2007;7:220–30. doi: 10.1038/nri2039. [DOI] [PubMed] [Google Scholar]
- Donnelly S, Dalton JP, Loukas A. Proteases in helminth- and allergen- induced inflammatory responses. In: Capron M, Trottein F, editors. Chemical immunology and allergy. Karger: Basel; 2005. pp. 45–64. [DOI] [PubMed] [Google Scholar]
- Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274:2054–7. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The neuropathogical change and immune response at peak stage were not different between the two groups. (a) Luxol fast blue: haematoxylin and eosin (H&E) staining of recombinant Toxascaris leonine galectin (rTl-gal)-treated and vehicle-treated spinal cords at peak (14–18 dpi) stage of experimental autoimmune encephalomyelitis (EAE). Bars = 60 µm. (b) The inflammation and demyelination scores in dorsal, ventral and lateral columns at peak (14–18 dpi) stage of rTl-gal-treated and vehicle-treated EAE mice. Values represent the mean ± standard error of the mean (s.e.m.). (c) Proliferation of T lymphocytes isolated from spleens at peak (14–18 dpi) stage of EAE, cells were incubated for 72 h with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (10 µg/ml), proteolipid protein (PLP)139–151 peptide (10 µg/ml) or concanavalin A (ConA). Proliferation was evaluated by thymidine incorporation measured during the last 18 h of culture. Data are mean ± s.e.m. of counts per minute (cpm) and are representative of three independent experiments. (d) Enzyme-linked immunosorbent assay (ELISA) determination of interferon (IFN)-γ, interleukin (IL)-17, IL-1β, tumour necrosis factor (TNF)-α, IL-4 and IL-10 in supernatant of splenic mononuclear cells isolated at peak (14–18 dpi) stage of EAE and restimulated with MOG35–55 peptide (10 µg/ml). Values represent the mean ± s.e.m. and are representative of three independent experiments. (e) Serum was harvested at peak (14–18 dpi) stage of EAE and the levels of anti-MOG35–55 immunoglobulin (Ig)G were quantified through ELISA. Values represent the mean ± s.e.m. and are representative of two independent experiments.
Fig. S2. Recombinant Toxascaris leonine galectin (rTl-gal) treatment did not affect the regulatory T cells. (a) Spinal cord sections from vehicle- or rTl-gal-treated experimental autoimmune encephalomyelitis (EAE) mice were stained for CD4 (green colour) and CD25 (red colour). Bars = 60 µm). (b) rTl-gal treatment did not affect the population of CD4+CD25+ regulatory T cells. Splenic mononuclear cells were analysed by flow cytometry for CD4+CD25+ on days 60–62 dpi of mice treated with either vehicle or rTl-gal. Dot-plots represent one of three independent experiments with similar observations and the mean population of each cell was plotted in the graph. (c) Real-time polymerase chain reaction (PCR) on cDNA isolated from spinal cords and spleens of vehicle- or rTl-gal-treated EAE mice was performed with forkhead box protein 3 primers. Expression data were normalized relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and fold change was compared to vehicle-treated mice.