

Abstract
Chronic relapsing experimental autoimmune encephalomyelitis (crEAE) in mice recapitulates many of the clinical and histopathological features of human multiple sclerosis (MS), making it a preferred model for the disease. In both, adaptive immunity and anti-myelin T cells responses are thought to be important, while in MS a role for innate immunity and complement has emerged. Here we sought to test whether complement is activated in crEAE and important for disease. Disease was induced in Biozzi ABH mice that were terminated at different stages of the disease to assess complement activation and local complement expression in the central nervous system. Complement activation products were abundant in all spinal cord areas examined in acute disease during relapse and in the progressive phase, but were absent in early disease remission, despite significant residual clinical disease. Local expression of C1q and C3 was increased at all stages of disease, while C9 expression was increased only in acute disease; expression of the complement regulators CD55, complement receptor 1-related gene/protein y (Crry) and CD59a was reduced at all stages of the disease compared to naive controls. These data show that complement is activated in the central nervous system in the model and suggest that it is a suitable candidate for exploring whether anti-complement agents might be of benefit in MS.
Keywords: chronic relapsing, complement, experimental autoimmune encephalomyelitis (EAE), multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by demyelination, neurodegeneration and axonal loss, causing severe and progressive disability 1. Although MS can manifest in a number of clinical forms, more than 80% of patients develop relapsing–remitting disease usually followed by a chronic progressive phase 2. Current anti-inflammatory or immunomodulatory treatments, while partially protective in the relapsing–remitting stage of the disease, have a limited or no effect on the development of neurodegeneration and disability 3–7. Therefore, it is apparent that a successful treatment must target the inflammatory and neurodegenerative component of the disease. The complement system is a key component of innate immunity 8 and has been implicated in the pathogenesis of inflammatory 9–11 and neurodegenerative diseases 12–16, indicating that complement may be a good target to slow, halt or reverse both processes. Notably, a role for complement in MS has been suggested based upon evidence that complement is activated and deposited in the MS brain 17–23, and it is a critical effector in the acute experimental autoimmune encephalomyelitis model of the disease 24–27. However, the extent and nature of complement activation and its contribution to the progressive phase of the disease remain controversial and difficult to investigate in acute models that do not replicate the natural course of the human disease.
The chronic relapsing experimental autoimmune encephalomyelitis (crEAE) model in Biozzi ABH mice, induced by immunization with myelin proteins or syngeneic spinal cord homogenate, reproduces many of the clinical and pathological hallmarks of MS, including relapsing–remitting episodes with inflammatory-mediated demyelination and progressive disease with axonal and neuronal loss 28,29. Roles of complement in this model have not been explored previously, limiting its usefulness in testing potential therapeutic approaches targeting complement. To address this gap we have explored roles of complement in the model. We first assessed whether Biozzi ABH mice have an intact and functional complement system, an important first step given the frequency of complement deficiency in inbred mouse strains 30,31. We then performed a regional and temporal analysis of complement synthesis, deposition and activation in crEAE. Finally, we tested the effect of inhibition of classical pathway-driven complement activation on complement deposition and clinical disease in the acute phase. We found that complement activation and deposition of C3 fragments and membrane attack complex (MAC) occurred in the model and reflected the clinical course of the disease, absent in remission. Complement protein expression was generally up-regulated, while complement regulator expression was decreased in disease compared to naive controls. The data demonstrate that complement is activated in the acute, relapse and progressive phases and suggest that crEAE is a suitable model to test potential anti-complement approaches of relevance to MS.
Materials and methods
Mice
Biozzi ABH and C57BL/6 mice were bred in-house and kept in groups of four to six in cages at a temperature of 20ºC on a 12 : 12-h light : dark cycle. They were allowed free access to food and water for the entire duration of the study and provided with wetted mash as necessary. Animals were monitored for microbiological status according to the Federation of European Laboratory Animal Science Associations (FELASA) recommendations. Experimental protocols relating to animal studies that conform with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines have been reported previously 32, and complied with national and local guidelines for the care of experimental animals.
Haemolytic assay
Blood samples were collected from tail veins of five male and five female C57BL/6 and five male and five female Biozzi ABH mice and transferred immediately on ice. Serum was separated and stored at −80ºC. Complement (C) haemolytic activity was assayed by standard haemolytic assays utilizing antibody-sensitized rabbit erythrocytes, as described previously 33. The absorbance of supernatants was measured at 415 nm and percentage haemolysis calculated by standard methods 34.
Induction and scoring of crEAE
Disease was induced in 8–12-week-old female Biozzi ABH mice by subcutaneous injection into the flank of syngeneic spinal cord homogenate emulsified in complete Freund's adjuvant (Sigma-Aldrich, St Loius, MO, USA) on days 0 and 7, as described previously 28,32. Each animal received 1 mg of spinal cord homogenate and 60 mg mycobacteria [Mycobacterium tuberculosis H37Ra, M. butyricum (4 : 1); Difco, BD Biosciences, San Jose, CA, USA] per injection. Body weight and clinical signs were assessed daily, as described previously 33,34, using the following scoring system: 0, normal; 1, loss of tail tone; 2, impaired righting reflex; 3, partial hind limb paralysis, with 1 limb affected; 4, complete hind limb paralysis, with both limbs affected; and 5, moribund. Remission from the active phase of disease was defined as the resolution of clinical paralysis, weight gain and stabilization of the neurological deficit 35–37. Relapse was defined as an increase in clinical score of at least 1 point, together with development of paresis (typically score 3 or above) associated with weight loss 33. Results are shown as the mean daily clinical scores ± standard error of the mean (s.e.m.) and mean maximum clinical scores; in some instances average scores were calculated for a given disease period, resulting in a measure of the length of time an animal had spent with active clinical disease.
Experimental groups and tissue processing
Spinal cords from naive control Biozzi ABH mice (n = 7) and mice immunized with spinal cord homogenate were harvested at various times post-immunization (p.i.); 19 days p.i. (n = 9) corresponding to the initial acute paralytic attack; 28 days p.i. (n = 7) during the first remission; 35 days p.i. (n = 6) corresponding to the first relapse; and 75 days p.i. (n = 7) at the start of the progressive phase 32. Mice were deeply anaesthetized then killed by transcardiac perfusion with phosphate-buffered saline (PBS); the spinal columns were excised and dissected into cervical, thoracic and lumbar regions, separately embedded in octreotide (OCT) on dry ice and stored at −80 °C for further immunohistological analysis. Four naive and four mice at acute disease stage were deeply anaesthetized then killed by transcardiac perfusion with PBS followed by perfusion with 4% paraformaldehyde; the spinal columns were excised and dissected into cervical, thoracic and lumbar regions, processed for embedding into paraffin for further in-situ hybridization analysis.
In-situ hybridization
Seven micron paraffin sections of naive (n = 4) and acute crEAE (n = 4) mouse spinal cords were mounted on Superfrost Plus glass slides (Knittel Glass, Braunschweig, Germany). In-situ hybridization for C3 was performed using 5′fluorescein-labelled 19-mer anti-sense oligonucleotide containing locked nucleic acid (LNA) and 2′OME RNA moieties (C3: 5'-TucTccAccAccGuuTccC-3'); capitals indicate LNA, lower case indicates 2′OME RNA. The oligonucleotide was synthesized by Ribotask ApS (Odense, Denmark). Oligonucleotide [1 μM in hybridization mix; 4 M urea, 600 mM NaCl, 10 mM HEPES buffer, pH 7.5, 1 mM ethylenediamine tetracetic acid (EDTA), ×5 Denhardt's reagent] was incubated on 7 µm sections of paraffin-embedded materials at 55 °C for 60 min. After hybridization, tissue sections were washed consecutively for 5 min each with ×2 saline sodium citrate (SSC), ×0·5 SSC and ×0·2 SSC at 55 °C. The probes were detected using anti-fluorescein-Fab fragments coupled to alkaline phosphatase (1 : 1000; Roche, Basel, Switzerland) for 1 h. The signal was visualized using the Vector blue AP substrate kit (Vector Laboratories, Burlingame, CA, USA). Mismatch anti-sense probes were used as negative controls. Tissue was photographed using a light microscope (Olympus BX41TF, Zoeterwoude, the Netherlands) and images processed with Cell D software (Olympus, Zoeterwoude, the Netherlands).
Immunohistochemistry
Transverse sections of frozen OCT-embedded spinal cords of 7 µm thickness were cut and fixed in cold acetone for 10 min. Endogenous peroxidases were blocked in 0·03% H2O2 in PBS, and non-specific binding sites were blocked with 10% normal goat serum (NGS) in PBS. Slides were then incubated with either 2 µg/ml monoclonal rat anti-mouse C3/C3b/iC3b/C3d (clone 11H9; Hycult Biotechnology, Uden, the Netherlands) or 2 µg/ml affinity purified polyclonal rabbit anti-rat C9/MAC (made in-house using standard immunization procedures) diluted in PBS containing 1% bovine serum albumin (BSA). Slides were washed, then incubated with the appropriate secondary (goat anti-rat or goat anti-rabbit biotinylated antibody; VectorLabs, Peterborough, UK) diluted 1 : 200 in PBS/1% BSA, washed again and incubated with peroxidase-labelled polystreptavidin (Sigma-Aldrich, St Louis, MO, USA; 1 : 400 in PBS/1% BSA). Sections incubated with secondary conjugate or isotype alone were included as negative controls. To visualize peroxidase activity, the slides were incubated in 3,3-diaminobenzidine tetrahydrochloride (DAB) (DAB Peroxidase Substrate Kit; VectorLabs) followed by counterstaining with haematoxylin. Slides were dehydrated in a series of ascending concentrations of ethanol and mounted in Pertex (Histolab, Gothenburg, Sweden). Images were captured with a light microscope (Olympus BX41TF) and percentage of the area stained positive measured using the Cell D software (Olympus).
RNA isolation and quantitative polymerase chain reaction (PCR)
Total RNA was extracted from spinal cord using the GeneElute Mammalian Total RNA Miniprep kit (Sigma-Aldrich). cDNAs were synthesized using TaqMan reverse transcription reagents (Applied Biosystems, Warrington, UK). To quantify copies of the gene of interest, purified RNAs were reverse-transcribed and labelled with iQ SYBR Green Supermix (Bio-Rad, Hemel Hempstead, UK), then analysed on the Mini Opticon Taqman (Bio-Rad) using optimized primer pairs. To analyse the data, the comparative Ct method (ΔΔCt) was used 38. All PCR data were normalized to the expression of the housekeeping gene beta (β)-actin. At least two independent experiments were carried out in triplicate for each cDNA analysed.
Statistical analysis
Two-way analysis of variance (anova) with Bonferroni correction was performed for the haemolytic assay. One-way anova with Bonferroni correction was performed for quantification of the immunohistochemistry and for the quantitative PCR (qPCR) analysis of complement components and regulators. EAE scores were assessed using Mann–Whitney U-test statistics. Data were considered statistically significant when P < 0·05 at 95% confidence level.
Results
ABH mice have a functional C system
There were no data in the literature assessing complement activity in Biozzi ABH mice; given the known frequency of complement deficits in inbred mouse strains 30,31, it was considered critical to test the mice first. Male and female mice were assessed separately because complement levels in females are low compared to males in several mouse strains. In male mice, serum haemolytic activity in Biozzi ABH was identical to that in C57BL/6 mice, known to have robust complement activity (Fig. 1a); in female mice, haemolytic activity in Biozzi ABH serum showed lytic activity but at levels around 50% of C57BL/6 serum (Fig. 1b). Despite this reduced level of haemolytic complement, female mice were chosen for testing in the model because of better disease reproducibility in published studies.
Fig 1.
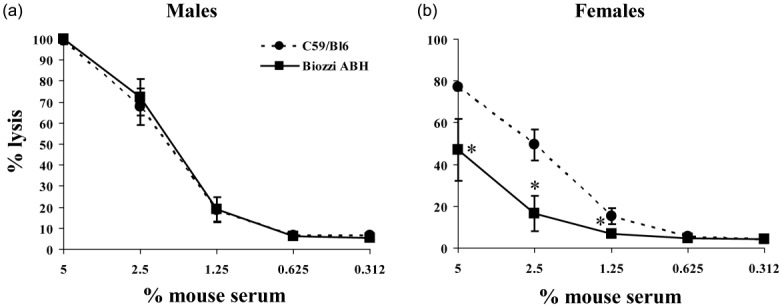
Complement haemolytic activity. Serum complement haemolytic activity in male (a) and female (b) Biozzi ABH mice. Antibody-sensitized rabbit erythrocytes were incubated with dilutions of serum from Biozzi ABH (squares) or C57/Bl6 (round) mice (n = 5 per group) and haemolytic activity was measured. Classical pathway activity was reduced to about 40–60% in females Biozzi ABH serum compared to female C57/Bl6 serum, whereas the serum complement haemolytic activity of Biozzi ABH male mice did not differ from that of C57/Bl6 males. The asterisks (*) indicate statistically significant differences determined by two-way analysis of variance (anova).
ABH mice develop relapsing–remitting progressive disease
Female Biozzi ABH mice immunized with spinal cord homogenate showed a clinical course similar to that reported previously 39 (Fig. 2). The first clinical signs of disease were seen on day 12 post-immunization (p.i.). By day 13, disease had reached an average clinical score of 3 (partial hind limb paralysis), plateaued with scores above 3 until day 19, then recovered rapidly with clinical score reaching close to 1 by day 24 and remaining in remission up to day 32. Relapse was rapid, with the average score exceeding 3·5 on day 33 and peaking close to 4 on day 35. Disease then settled into a progressive picture with an average score of around 3 for the remainder of the experiment. Weight loss mirrored clinical disease, dropping to around 83% of initial weight at day 15 in the acute phase, then slowly recovering, only to fall again at the onset of relapse (Fig. 2).
Fig 2.
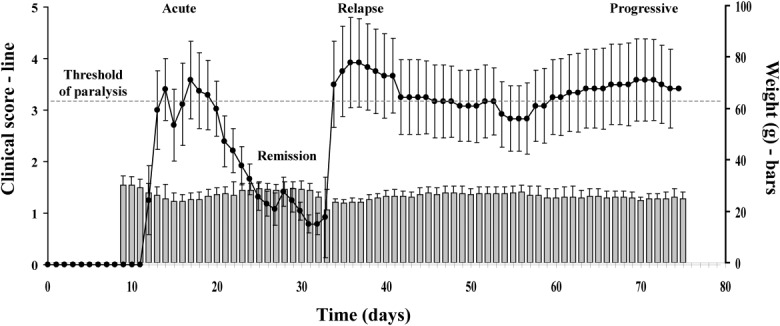
Clinical course of relapsing–progressive experimental autoimmune encephalomyelitis (EAE). The relapsing–progressive EAE course in Biozzi ABH mice is illustrated by the average clinical disease scores (black line, left axis). There is near-complete recovery (remission) from clinical signs after the acute stage. Following an induced relapse, animals enter a chronic plateau disease phase (progressive) from which recovery from paralysis (clinical score of 3 or higher) does not occur. Weight loss is observed at the beginning of the acute phase and at the stage of induced relapse in this model (grey bars, right axis).
C is activated during acute, relapsing and progressive phases of crEAE
C3 fragment deposition was absent or trace in naive Biozzi ABH mice and during disease remission but was strong in all cord areas sampled at the acute, relapsing and progressive disease stages (Fig. 3). C3 fragment staining was present in similar amounts within white and grey matter at all stages. Staining for MAC deposition as a measure of terminal pathway complement activation was completely absent in naive mice and in disease remission, but was detected in all spinal cord regions in acute disease (white matter predominant), progressive disease (grey matter predominant) and relapse (equally distributed in white and grey matter) (Fig. 4).
Fig 3.
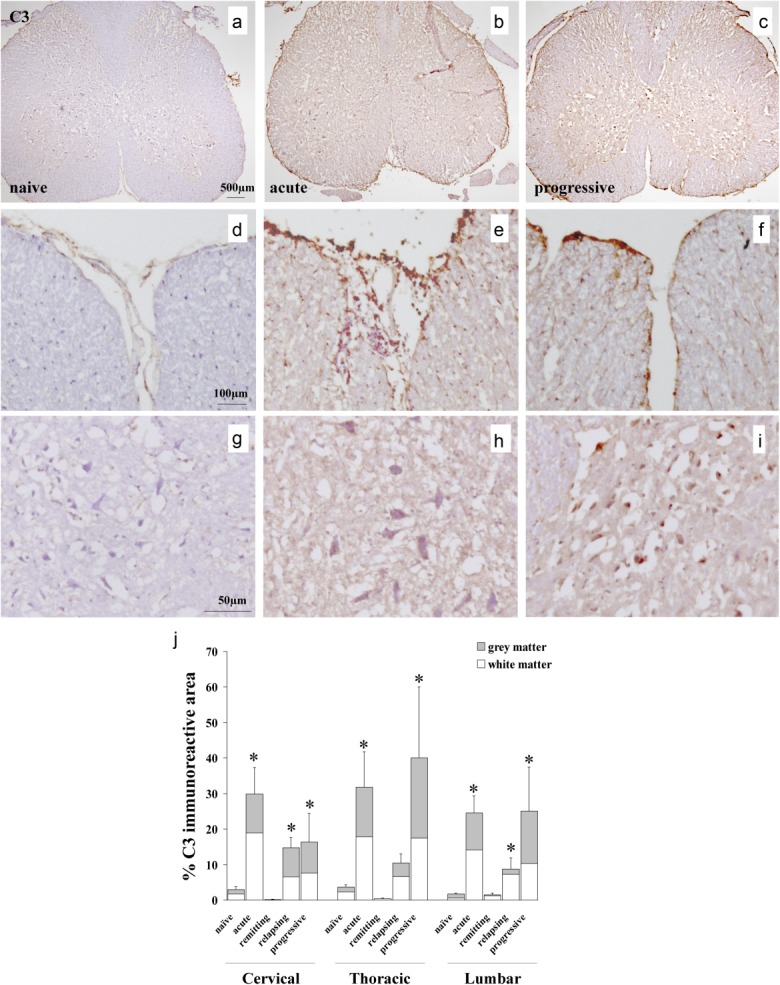
C3 deposition in relapsing–progressive experimental autoimmune encephalomyelitis (EAE). (a–i) Representative immunohistochemical stainings in spinal cord sections from naive and chronic relapsing experimental autoimmune encephalomyelitis (crEAE) mice, showing C3 staining in meninges, white matter (e,f) and grey matter (h,i) during acute and disease progressive disease. Spinal cords from naive mice were negative for C3 (a,d,g). (j) Quantification of C3 (clone 11H9) immunostaining in cross-sections of cervical, thoracic and lumbar spinal cord regions from naive mice and during acute, remitting, relapsing and progressive disease stages of crEAE. Values are expressed as mean ± standard deviation. The asterisks (*) indicate statistically significant differences determined by one-way analysis of variance (anova) (P ≤ 0·05).
Fig 4.
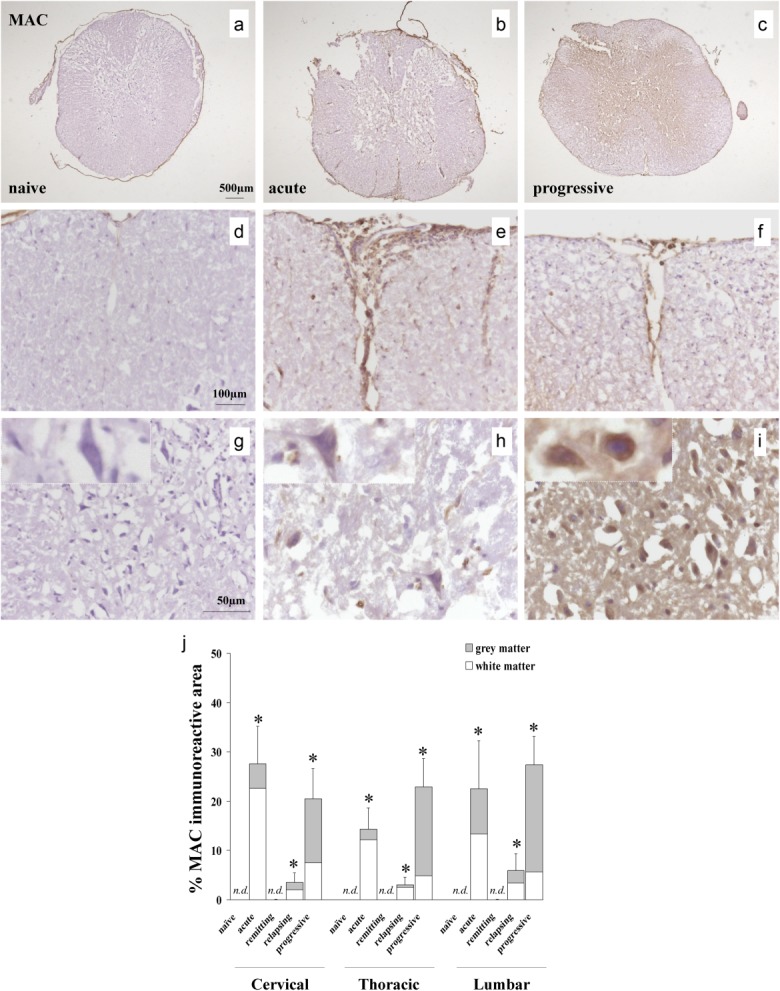
Membrane attack complex (MAC) deposition in relapsing–progressive experimental autoimmune encephalomyelitis (EAE). (a–i) Representative immunohistochemical stainings in spinal cord sections from naive and chronic relapsing experimental autoimmune encephalomyelitis (crEAE) mice, showing MAC staining in meninges and white matter during acute disease (e), whereas abundant MAC staining localizes in grey matter (i) during progressive disease. Spinal cords from naive mice were negative for MAC (a,d,g). (j) Quantification of MAC immunostaining in cross-sections of cervical, thoracic and lumbar spinal cord regions from naive mice and during acute, remitting, relapsing and progressive disease stages of crEAE. Values are expressed as mean ± standard deviation. The asterisks (*) indicate statistically significant differences determined by one-way analysis of variance (anova) (P ≤ 0·05).
Altered expression of C components and regulators during crEAE
Expression of C components C1q, C3 and C9, and C regulators decay-accelerating factor (DAF), complement receptor 1-related gene/protein y (Crry) and CD59 were assessed by quantitative PCR in RNA from spinal cords sampled at all disease stages (Fig. 5). C1q and C3 expression was elevated significantly at all disease stages compared to naive controls, while C9 expression was elevated in acute disease but reduced significantly at all other disease stages compared to controls (Fig. 5 a–c). Expression of DAF was reduced significantly compared to controls at all disease stages (Fig. 5d); Crry expression was reduced significantly at all stages of disease, except in acute disease (Fig. 5e); CD59 expression was reduced significantly at all disease stages in relapsing and progressive stages (Fig. 5f).
Fig 5.
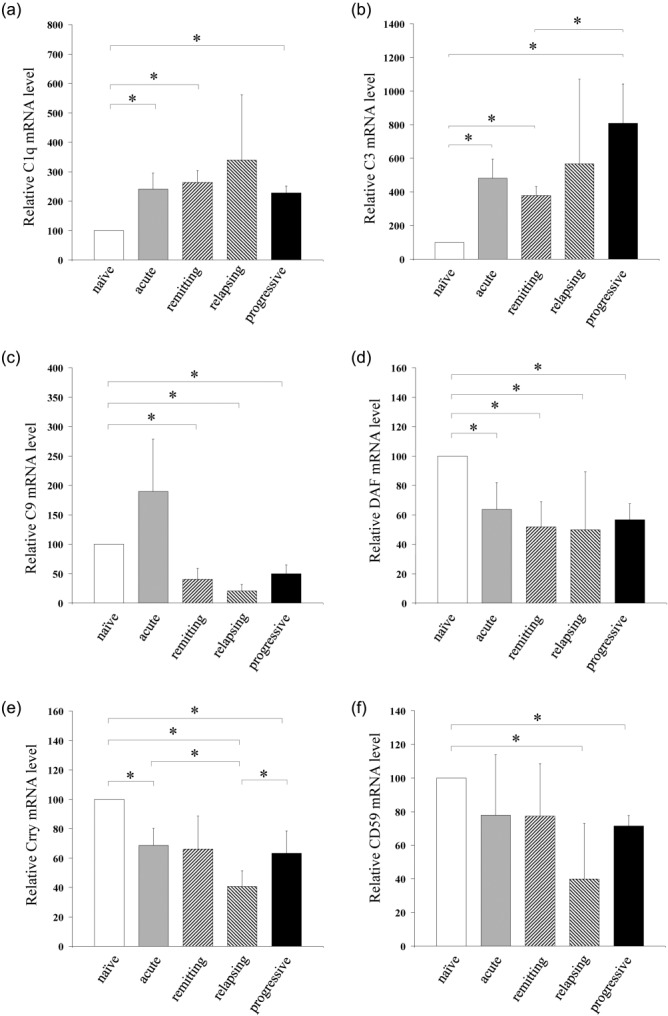
Complement expression in relapsing–progressive experimental autoimmune encephalomyelitis (EAE). Relative mRNA expression of C components C1q (a), C3 (b) and C9 (c) and regulators decay-accelerating factor (DAF) (d), complement receptor 1-related gene/protein y (Crry) (e) and CD59 (f) in spinal cord of naive Biozzi ABH mice and during the acute, remitting, relapsing and progressive phases of chronic relapsing experimental autoimmune encephalomyelitis (crEAE). Values are normalized to the expression of β-actin and given as percentage (means ± standard deviation) of naive control levels. The asterisks (*) indicate statistically significant differences determined by one-way analysis of variance (anova).
To test which cells produce the C components which were found to be up-regulated in the mouse spinal cord by quantitative PCR, we performed in-situ hybridization for C3 mRNA, the key central component of the C system (Fig. 6). C3 message was up-regulated in neurones in the spinal cords of mice in the acute stage of crEAE compared to naive controls, proving evidence for local synthesis of C3 by neurones during disease.
Fig 6.
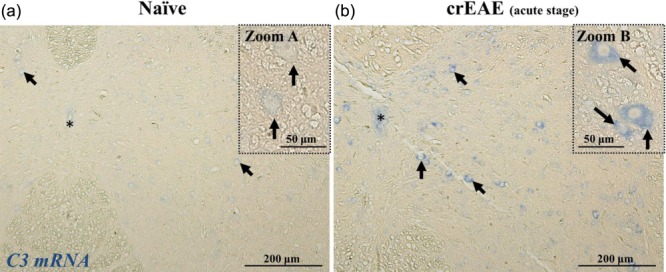
C3 is produced locally by neurones in chronic relapsing experimental autoimmune encephalomyelitis (crEAE). In-situ hybridization for C3 mRNA on spinal cords from naive mice (a, representative of four mice) and crEAE mice at acute stage of disease (b, representative of four mice), showing the C3 mRNA signal (in blue) in neurones (arrows and zoom), as inferred by the morphology and large nuclei. Note the light blue staining in neurones of naive mouse spinal cord (zoom A), whereas a strong blue staining is detected in the cytoplasm of neurones in the spinal cord of crEAE mice at acute stage of disease (zoom B). Images are taken at equivalent spinal cord locations of naive and crEAE mice. The central canal is indicated by the asterisk (*). Scale bar A and B, 200 μm; zoom A and B, 50 μm.
Discussion
Chronic relapsing EAE is considered to be a T cell-driven disease that mirrors many of the clinical and pathological features of MS 40. However, it is now clear that multiple immune effectors, including complement and other innate systems, play important roles in the propagation of disease in MS and represent a potential target for therapy 41. To understand the relevance of crEAE to MS it is therefore necessary to explore the contribution of complement activation to disease in the model. Disease was induced using established protocols and reproduced the temporal evolution of early clinical relapses into a progressive disease stage, mimicking key clinical features of MS, as reported previously 29,39,42. Complement activation and expression assessed in spinal cord at different stages in its course showed that complement activation, demonstrated by staining for C3 activation products and the MAC, occurs in all cord regions during acute, relapse and progressive disease but is virtually absent in the brief remission phase. The lack of evidence of ongoing complement activation in remission despite residual clinical disease was remarkable; indeed, in other models and MS tissues it has been suggested that complement activation products, particularly C3 fragments, remain detectable long after the clinical disease has resolved 19,20. These data suggest, first, that complement activation is diminished markedly in remission, and secondly, that complement activation products are cleared rapidly from the CNS. Reduced complement activation could be caused by removal of the initiating trigger, decreased availability of complement proteins or increased availability of complement regulatory components; to test the latter two possibilities, expression of complement components C1q, C3 and C9 as well as regulators of the C3 convertase (DAF, Crry) and the MAC (CD59a) in spinal cord at different disease stages was measured. Expression of C1q and C3 was increased at all stages of disease, including during remission, while C9 expression was elevated in acute disease but reduced significantly at all other disease stages; in-situ hybridization showed that for the most abundant component, C3, essential for all complement pathways, increased C3 mRNA expression at the acute stage of crEAE was localized in neurones, proving evidence of an intrinsic immune response within the CNS compartment. In addition, expression of DAF, Crry and CD59a was decreased in remission and generally at all stages of disease; these data make it unlikely that decreased availability of components or increased availability of complement regulators was responsible for the observed decrease in complement deposition in remission. The precise drivers of complement activation in crEAE are unclear; anti-myelin autoantibodies have been described in the model, although their pathological relevance was questioned 43. We have detected anti-myelin protein antibodies as early as day 10 post-disease initiation (data not shown) and in preliminary experiments have tested whether the regulator of the antibody-triggered classical pathway impacts upon disease in the model; initial results suggest little impact in acute disease. Regardless of the mode of activation, complement C3 fragment and MAC deposition was seen. Clearance of C3 fragments and MAC involves a combination of complement regulators, receptors and phagocytic cells. Microglia are present and activated in acute and chronic stages of crEAE and are known to express C3 fragment receptors that can drive opsonic clearance; it is possible that efficient removal of complement deposits contributes to the development of remission.
Our data demonstrate that complement is activated abundantly and through to completion in the crEAE model at key stages of the disease. There is an urgent need to further explore pathways to complement activation and test the effects of inhibition using different agents. The initial phase of EAE is typically inflammatory, driven by T cell activity and induces limited destruction of myelin and nerves 28,36. Once antibody responses are formed they can contribute to damage via complement-mediated mechanisms 44. Therefore it will also be important to assess the role of complement at different disease stages in order to fully ascertain the value of this model as a test bed for anti-complement approaches to therapy of MS.
Acknowledgments
This work was supported by a Wellcome Trust VIP award no. 084542 to V. R.
Disclosures
The authors have no competing interests to declare.
References
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Vukusic S. The clinical course of multiple sclerosis. Handb Clin Neurol. 2014;122:343–69. doi: 10.1016/B978-0-444-52001-2.00014-5. [DOI] [PubMed] [Google Scholar]
- Coles AJ, Wing MG, Molyneux P, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol. 1999;46:296–304. doi: 10.1002/1531-8249(199909)46:3<296::aid-ana4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Filippi M, Rovaris M, Rice GP, et al. The effect of cladribine on T(1) ‘black hole' changes in progressive MS. J Neurol Sci. 2000;176:42–4. doi: 10.1016/s0022-510x(00)00303-8. [DOI] [PubMed] [Google Scholar]
- Filippi M, Rocca MA, Pagani E, et al. European study on intravenous immunoglobulin in multiple sclerosis: results of magnetization transfer magnetic resonance imaging analysis. Arch Neurol. 2004;61:1409–12. doi: 10.1001/archneur.61.9.1409. [DOI] [PubMed] [Google Scholar]
- Molyneux PD, Kappos L, Polman C, et al. The effect of interferon beta-1b treatment on MRI measures of cerebral atrophy in secondary progressive multiple sclerosis. European Study Group on Interferon beta-1b in secondary progressive multiple sclerosis. Brain. 2000;123:2256–63. doi: 10.1093/brain/123.11.2256. [DOI] [PubMed] [Google Scholar]
- Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5:343–54. doi: 10.1016/S1474-4422(06)70410-0. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lambris JD. Complement and dysbiosis in periodontal disease. Immunobiology. 2012;217:1111–6. doi: 10.1016/j.imbio.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rozelle AL, Lepus CM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–9. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kohl J. A complex role for complement in allergic asthma. Expert Rev Clin Immunol. 2010;6:269–77. doi: 10.1586/eci.09.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Sta M, Sylva-Steenland RM, Casula M, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42:211–20. doi: 10.1016/j.nbd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011;121:1429–44. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43:465–71. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50:646–57. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Parratt JD, Cho ES, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann Neurol. 2009;65:32–46. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Brink BP, Veerhuis R, Breij EC. van der V, Dijkstra CD, Bo L. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J Neuropathol Exp Neurol. 2005;64:147–55. doi: 10.1093/jnen/64.2.147. [DOI] [PubMed] [Google Scholar]
- Ingram G, Loveless S, Howell OW, et al. Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun. 2014;2:53. doi: 10.1186/2051-5960-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, McGeer PL. Complement activated C4d immunoreactive oligodendrocytes delineate small cortical plaques in multiple sclerosis. Exp Neurol. 2002;174:81–8. doi: 10.1006/exnr.2001.7851. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. 2002;168:458–65. doi: 10.4049/jimmunol.168.1.458. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Neal JW, Griffiths MR, et al. Deficiency of the complement regulator CD59a enhances disease severity, demyelination and axonal injury in murine acute experimental allergic encephalomyelitis. Lab Invest. 2004;84:21–8. doi: 10.1038/labinvest.3700015. [DOI] [PubMed] [Google Scholar]
- Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152:5477–84. [PubMed] [Google Scholar]
- Szalai AJ, Hu X, Adams JE, Barnum SR. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol. 2007;44:3132–6. doi: 10.1016/j.molimm.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, O'Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol. 1990;28:261–70. doi: 10.1016/0165-5728(90)90019-j. [DOI] [PubMed] [Google Scholar]
- Hampton DW, Anderson J, Pryce G, et al. An experimental model of secondary progressive multiple sclerosis that shows regional variation in gliosis, remyelination, axonal and neuronal loss. J Neuroimmunol. 2008;201–202:200–11. doi: 10.1016/j.jneuroim.2008.05.034. [CrossRef] [DOI] [PubMed] [Google Scholar]
- Bhole D, Stahl GL. Molecular basis for complement component 6 (C6) deficiency in rats and mice. Immunobiology. 2004;209:559–68. doi: 10.1016/j.imbio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Wetsel RA, Fleischer DT, Haviland DL. Deficiency of the murine fifth complement component (C5). A 2-base pair gene deletion in a 5'-exon. J Biol Chem. 1990;265:2435–40. [PubMed] [Google Scholar]
- Ruseva MM, Hughes TR, Donev RM, et al. Crry deficiency in complement sufficient mice: C3 consumption occurs without associated renal injury. Mol Immunol. 2009;46:803–11. doi: 10.1016/j.molimm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Al-Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S, Baker D. Practical guide to the induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. Mult Scler Rel Dis. 2012;1:29–38. doi: 10.1016/j.msard.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Morgan BP. Measurement of complement hemolytic activity, generation of complement-depleted sera, and production of hemolytic intermediates. Methods Mol Biol. 2000;150:61–71. doi: 10.1385/1-59259-056-X:61. [DOI] [PubMed] [Google Scholar]
- Baker D, Butler D, Scallon BJ, O'Neill JK, Turk JL, Feldmann M. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF receptor-immunoglobulin fusion proteins. Eur J Immunol. 1994;24:2040–8. doi: 10.1002/eji.1830240916. [DOI] [PubMed] [Google Scholar]
- O'Neill JK, Baker D, Turk JL. Inhibition of chronic relapsing experimental allergic encephalomyelitis in the Biozzi AB/H mouse. J Neuroimmunol. 1992;41:177–87. doi: 10.1016/0165-5728(92)90068-v. [DOI] [PubMed] [Google Scholar]
- Pryce G, O'Neill JK, Croxford JL, et al. Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J Neuroimmunol. 2005;165:41–52. doi: 10.1016/j.jneuroim.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Donev RM, Morgan BP. A quantitative method for comparison of expression of alternatively spliced genes using different primer pairs. J Biochem Biophys Methods. 2006;66:23–31. doi: 10.1016/j.jbbm.2005.11.001. [CrossRef] [DOI] [PubMed] [Google Scholar]
- Hampton DW, Serio A, Pryce G, et al. Neurodegeneration progresses despite complete elimination of clinical relapses in a mouse model of multiple sclerosis. Acta Neuropathol Commun. 2013;1:84. doi: 10.1186/2051-5960-1-84. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin Exp Immunol. 2009;155:128–39. doi: 10.1111/j.1365-2249.2008.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SJ, Lee J, Nikodemova M, Fabry Z, Duncan ID. Quantification of myelin and axon pathology during relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. J Neuropathol Exp Neurol. 2009;68:616–25. doi: 10.1097/NEN.0b013e3181a41d23. [CrossRef] [DOI] [PubMed] [Google Scholar]
- Whitham RH, Nilaver G, Bourdette DN, Seil FJ. Serum anti-myelin antibodies in chronic relapsing experimental allergic encephalomyelitis. J Neuroimmunol. 1988;18:155–70. doi: 10.1016/0165-5728(88)90063-x. [DOI] [PubMed] [Google Scholar]
- Morris MM, Piddlesden S, Groome N, Amor S. Anti-myelin antibodies modulate experimental allergic encephalomyelitis in Biozzi ABH mice. 1997;25:168S. doi: 10.1042/bst025168s. Biochem Soc Trans. [DOI] [PubMed] [Google Scholar]