

Abstract
The tumour necrosis factor (TNF)-α-induced proteins (TNFAIP)9 and TNFAIP3 play an important pathogenic role in murine arthritis. To clarify their pathophysiological roles in patients with rheumatoid arthritis (RA), we examined their expression and localization in peripheral blood mononuclear cells (PBMC). TNFAIP9 and TNFAIP3 mRNA expression was determined in PBMC of RA patients and healthy subjects (control). Flow cytometry was used to analyse the main TNFAIP9- and TNFAIP3-expressing cell populations. TNFAIP9 and TNFAIP3 mRNA expression levels were examined in vitro on CD14+ cells stimulated with TNF-α and lipopolysaccharide (LPS). The expression levels of TNFAIP9 and TNFAIP3 mRNA were also measured before and 12 weeks after treatment with tocilizumab and abatacept. TNFAIP9 expression was significantly higher, while TNFAIP3 expression was lower in PBMC of RA (n = 36) than the control (n = 24) (each P < 0·05). TNFAIP9 was expressed on CD14+ cells, especially in human leucocyte antigen D-related (HLA-DR)+CD14brightCD16−cells, while TNFAIP3 was expressed mainly on CD3+ T cells. TNF-α and LPS induced TNFAIP9 and TNFAIP3 in human CD14+monocytes in vitro. Treatment with tocilizumab (n = 13), but not abatacept (n = 11), significantly reduced TNFAIP9 mRNA expression in PBMC, which was associated with reduction in the number of circulating CD14bright monocytes. The expression of TNFAIP9 in CD14+ cells was specifically elevated in patients with RA, regulated by TNF-α and LPS, and suppressed by tocilizumab, while TNFAIP3 in PBMC showed different localization and induction patterns.
Keywords: monocytes, pathogenesis, rheumatoid arthritis, TNFAIP9, TNF-α-induced proteins
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease that targets mainly multiple joints. Cytokines [e.g. tumour-necrosis factor (TNF)-α and interleukin (IL)-6] play a crucial role in the pathogenesis of RA, and many studies have reported the efficacy of treatments that target such molecules 1–3. TNF-α induces numerous proteins during the inflammatory process. In recent years some of these proteins, so-called TNF-α-induced proteins (TNFAIPs), have gained a great deal of interest. One of these proteins is TNFAIP9, or TNF-α-induced adipose-related protein (TIARP), the human homologue of six-transmembrane epithelial antigen of prostate 4 (STEAP4). We have reported previously the up-regulation of TNFAIP9 in CD11b+ splenocytes and joints of glucose-6-phosphate isomerase (GPI)-induced arthritis, and that TNFAIP9-deficient (TNFAIP9–/–) mice developed spontaneous enthesitis and synovitis with overproduction of IL-6 4,5. Moreover, it was suggested that TNFAIP9 regulate arthritis negatively by suppressing IL-6 production and signalling and TNF-α-induced nuclear factor (NF)-κB signalling. However, the expression and function of TNFAIP9 in human RA remain unclear.
Another TNFAIP, TNFAIP3, also known as A20, is a cytoplasmic zinc finger protein expressed in all types of cells. TNFAIP3 also regulates NF-κB negatively by de-ubiquitinating specific signalling molecules 6. TNFAIP3-deficient mice are hypersensitive to TNF and die prematurely due to severe multi-organ inflammation and cachexia 7. Matmati et al. 8 reported that myeloid-specific TNFAIP3-deficient mice spontaneously developed severe polyarthritis resembling human RA. These findings suggest that TNFAIP3 expressed on monocytes and macrophages also acts as a negative regulator of arthritis.
Thus, these two TNFAIPs seem to play a pivotal role in arthritis in mice. In this regard, we have recently summarized and discussed the similarities and differences between these two proteins 9. The expression levels of TNFAIP9 in the spleen and joints are the reverse of those of TNFAIP3 in GPI-induced arthritis, and some association between the two has been observed in macrophages 9. To our knowledge, however, the molecular expression, localization, fluctuation and comparison of TNFAIP3 and TNFAIP9 in humans remain elusive. The present study was designed to delineate the expression of TNFAIP3 and TNFAIP9 in human peripheral blood mononuclear cells (PBMC) and to define the similarities and differences between the two proteins.
Materials and methods
Subjects and blood samples
Peripheral blood samples were obtained from 36 patients with RA and 24 healthy subjects (HS) for quantitative real-time reverse transcription–polymerase chain reaction (qRT–PCR). All RA patients were being treated with disease-modifying anti-rheumatic drug (DMARDs) and/or prednisolone, but none had received biological agents. RA disease activity was assessed by the disease activity score in 28 joints (DAS28)–C-reactive protein (CRP) 10. The characteristics of the patients and HS are shown in Table1.
Table 1.
Characteristics of the study subjects
| Healthy subjects (n = 24) | RA patients (n = 36) | |
|---|---|---|
| Females, n (%) | 20 (83%) | 31 (86%) |
| Males, n (%) | 4 (17%) | 5 (14%) |
| Age (years) | 38 ± 11 | 55 ± 10 |
| Disease duration (years) | 9 ± 8 | |
| Steinbrocker stage, n (1/2/3/4) | 4/16/5/11 | |
| Steinbrocker functional class, n (1/2/3/4) | 7/17/11/1 | |
| DAS28–CRP | 3·9 ± 1·0 | |
| Erythrocyte sedimentation rate (mm/h) | 28·9 ± 16·8 | |
| C-reactive protein (mg/dl) | 1·4 ± 1·2 | |
| Rheumatoid factor (IU/ml) | 138·7 ± 409·2 | |
| Matrix metalloproteinase 3 (ng/ml) | 243·7 ± 193·1 | |
| Anti-CCP antibodies positive, n | 24 (unknown: 12) | |
| Prednisolone use, n (%, mean dose) | 31 (86%, 6·2 ± 2·8 mg/day) | |
| Methotrexate use, n (%, mean dose) | 36 (100%, 8 ± 1 mg/week) | |
| Use of biological agents, n (%) | 0 (0%) |
Values are mean ± standard deviation or numbers/percentages. CRP = C-reactive protein. RA = rheumatoid arthritis; DAS 28 = disease activity score in 28 joints; CCP = cyclic citrullinated peptide.
Peripheral blood samples were also obtained from 23 RA patients (a part of 36 patients, above) who had not been treated with biological agents and 13 HS for analysis of TNFAIP9 mRNA expression in CD14+ cells and analysis of the CD14+ cell subpopulation. To analyse the effects of biological agents, we collected new blood samples from 11 RA patients who were treated with tocilizumab and 13 RA patients who were treated with abatacept. DAS 28–CRP was calculated at baseline and after 12 weeks of treatment, and the response to treatment was determined according to the response criteria of the European League Against Rheumatism (EULAR) 10. The baseline characteristics of the 24 patients are shown in Table2.
Table 2.
Characteristics of study subjects treated with biological agents
| Tocilizumab (n = 11) | Abatacept (n = 13) | |
|---|---|---|
| Females, n | 10 | 12 |
| Age (years) | 57 ± 13 | 49 ± 16 |
| Disease duration (years) | 13 ± 15 | 6 ± 12 |
| Steinbrocker stage, n (1/2/3/4) | 2/3/3/3 | 3/5/3/2 |
| DAS28–CRP | 4·0 ± 0·7 | 3·4 ± 1·6 |
| Prednisolone use, n (mean dose) | 9 (6·4 ± 3·1 mg/day) | 8 (9·8 ± 3·5 mg/day) |
| Methotrexate use, n (mean dose) | 6 (9·7 ± 3·0 mg/week) | 4 (9·0 ± 4·2 mg/week) |
Data are mean ± standard deviation. Abbreviations as in Table1.
All RA patients fulfilled the American College of Rheumatology (ACR) 1987 criteria for the classification of RA 11. All subjects provided informed consent and the relevant ethics review committee approved the study. PBMC were separated from blood using Ficoll density centrifugation.
qRT–PCR
Total RNA was extracted with Isogen (Nippon Gene, Toyama, Japan) using the protocol provided by the manufacturer, and cDNA was synthesized using a reverse transcription kit. RT–PCR was performed using the SYBR Green I method with the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Data of the controls and patients with RA were calculated by the ΔΔCt method, while the other data were analysed using prepared standard curves. All data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers used in real-time PCR were the following: TNFAIP9 forward, 5′-AAGCAATTCATGAAGCCTGAAGCTA-3′, TNFAIP9 reverse; 5′GGCCAAAGGTCAGGTCTGGA-3′, TNFAIP3 forward, 5′-TGCTGCCCTAGAAGTACAATAGGAA-3′, TNFAIP3 reverse, 5′-GCAGCTGGTTGAGTTTATGCAAG-3′; GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′, GAPDH reverse, 5′-TGGTGAAGACGCCAGTGGA-5′.
Flow cytometry
PBMC (5 × 105 cells) obtained from the control subjects and RA were washed with phosphate-buffered saline (PBS) containing 2·5% fetal bovine serum (FBS) and 0·02% sodium azide, and stained with monoclonal antibodies against CD14 [antigen-presenting cells (APC); Biolegend, San Diego, CA, USA], CD16 [phycoerythrin (PE); Biolegend] and human leucocyte antigen D-related (HLA-DR) [peridinin chlorophyll/cyanin 5·5 (PerCp/Cy5·5); Biolegend], CD3 (PerCP; Biolegend) and CD20 (APC; Biolegend). Intracellular staining was performed using the BD Cytofix/Cytoperm method according to the manufacturer's instructions, and then cells were stained with 0·5 μg anti-human TNFAIP9 antibody (clone: 418714; R&D Systems, Minneapolis, MN, USA) or 0·5 μg anti-TNFAIP3 antibody (clone: 59A426; Abcam, Cambridge, MA, USA) conjugated using Zenon Alexa Fluor 488 anti-mouse immunoglobulin (Ig)G labelling kit (Invitrogen, Eugene, OR, USA). As isotype control, purified mouse IgG2b (BD Pharmingen, San Jose, CA, USA) and mouse IgG1 (Biolegend) were used. Data were analysed on a fluorescence activated cell sorter (FACS)Calibur and Verse using CellQuest software (BD Bioscience, San Jose, CA, USA) and FlowJo software (TreeStar, Inc., Ashland, OR, USA). The mean fluorescence intensity (MFI) ratio was calculated by the MFI of the sample divided by the MFI of isotype control. Monocyte populations were identified as HLA-DR-expressing CD14+ cells, and divided into three subsets according to the expression levels of CD14 and CD16; namely, CD14brightCD16−, CD14brightCD16+ and CD14dimCD16+ cells.
In-vitro cell culture studies
CD14+ cells were purified using anti-CD14 magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) and a magnetic cell separator (MACS) from PBMC obtained from the control subjects and RA patients. CD14+ cells (5 × 104 cells/200 μl) were cultured in α-minimum essential medium (α-MEM) plus 10% fetal bovine serum (FBS) in the presence or absence of TNF-α (R&D Systems) and lipopolysaccharide (LPS) (Sigma Aldrich, St Louis, MO, USA), and collected 1, 3, 6, 12 and 24 h after stimulation to determine the mRNA expression levels of TNF-α, IL-6, TNFAIP9 and TNFAIP3.
Statistical analysis
Data are expressed as mean ± standard deviation (s.d.) or mean ± standard error of the mean (s.e.m.). The Mann–Whitney U-test was used to test for differences in various parameters between the control and RA groups. Differences between the baseline and post-treatment with biological agents were analysed using Wilcoxon's signed-rank test. A P-value less than 0·05 denoted the presence of a statistically significant difference.
Results
TNFAIP9 and TNFAIP3 mRNA expression levels
We examined the mRNA expression levels of TNFAIP9 and TNFAIP3 in PBMC of 36 RA patients and 24 HS (controls) using qPCR. TNFAIP9 expression was significantly higher in RA (median: 1·4) compared with normal subjects (median: 0·9, P < 0·05). In contrast, TNFAIP3 expression was lower in RA (median: 1·0) compared with HS (median: 1·8, P < 0·05) (Fig. 1).
Fig 1.
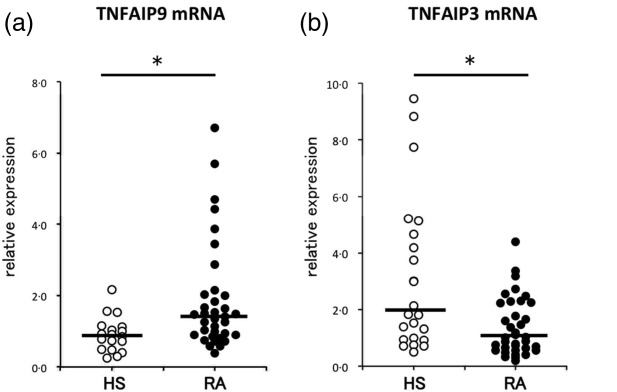
Comparison of (a) tumour necrosis factor (TNF)-α-induced proteins (TNFAIP)9 and (b) TNFAIP3 mRNA expression levels in peripheral blood mononuclear cells of healthy subjects (HS, n = 24) and patients with rheumatoid arthritis (RA, n = 36) by real-time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The thick horizontal lines represent the median values. *P < 0·05 by Mann–Whitney U-test.
CD14+ monocytes are dominant cell population that express TNFAIP9
We analysed the dominant cell population that expressed TNFAIP9 and TNFAIP3 in peripheral blood using flow cytometry. Blood samples were obtained from 13 patients with RA and 10 HS. In both groups, TNFAIP9 was expressed mainly in CD14+ cells, but not in lymphocyte subsets (Fig. 2a, left, upper panel with representative histogram). Conversely, TNFAIP3 was detected in lymphocyte subsets, especially in the CD3+ population of both RA and controls (Fig. 2a, left, lower panel), in agreement with the findings of a previous study 12. These expression patterns were almost comparable between RA and HS (individual plots were shown in Fig. 2a, right panel). We also compared TNFAIP9 and TNFAIP3 mRNA expression in CD14+ cells of RA (n = 23) and HS (n = 13). As anticipated, TNFAIP9 expression in CD14+ cells was significantly higher in RA than controls (Fig. 2b). In contrast, TNFAIP3 expression in CD14+ cells was not significantly different between the two groups. These findings demonstrated the different localization of TNFAIP9 and TNFAIP3 in peripheral blood and the abundance of TNFAIP9 in CD14+ cells.
Fig 2.
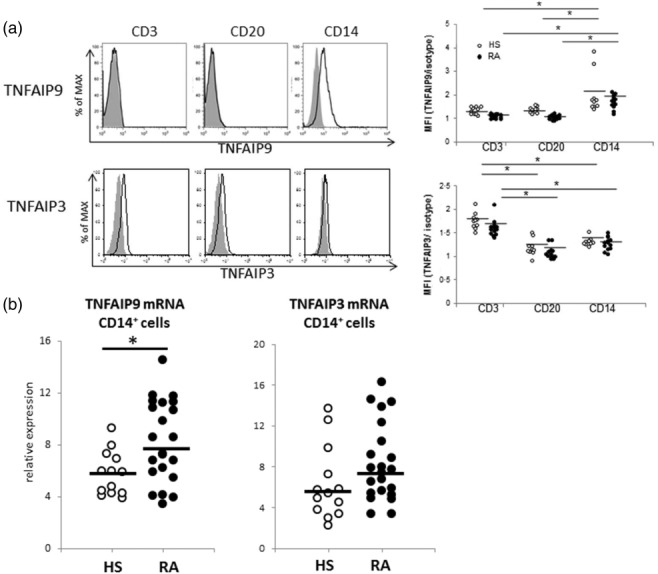
Localization of tumour necrosis factor (TNF)-α-induced proteins (TNFAIP)9 and TNFAIP3 in peripheral blood mononuclear cells (PBMC). TNFAIP9 and TNFAIP3 expression in peripheral blood mononuclear cells were examined by flow cytometry. (a) Expression of TNFAIP9 and TNFAIP3 on CD3, CD20 and CD14 cells indicated by histograms. Left panel indicates fluorescence activated cell sorter (FACS) data of representative TNFAIP9-positive rheumatoid arthritis (RA) patient and representative TNFAIP3-positive RA patient (at mRNA level). Right panel shows that the individual plots of mean fluorescence intensity (MFI) showed almost comparable expression between RA (n = 13) and healthy subjects (HS) (n = 10). (b) Expression of TNFAIP9 and TNFAIP3 on sorted CD14+ cells of patients with rheumatoid arthritis (RA, n = 23) and HS (n = 13) by real-time reverse transcription–polymerase chain reaction (RT–PCR). The thick horizontal lines represent the median values. *P < 0·05 by Mann–Whitney U-test.
TNFAIP9 is expressed mainly in CD14brightCD16− monocytes
Clinical and experimental evidence suggests that monocytes are heterogeneous populations consisting, in humans, of at least three subpopulations based on surface expression of CD14 and CD16: classical CD14brightCD16−, non-classical CD14dimCD16+ and intermediate CD14brightCD16+ monocytes 13,14. We investigated TNFAIP9 expression levels in the three subsets using flow cytometry. Monocytes were identified as the HLA-DR+CD14+ population, and the three subpopulations were then subdivided according to CD14/CD16 expression (Fig. 3a). In both the RA and control groups, the mean fluorescence intensity (MFI) of TNFAIP9 was high in the CD14bright cell population, especially CD14brightCD16−, among the three subsets (Fig. 3b). The expression pattern was almost comparable in patients with RA and controls. The percentage of CD14+ monocytes (relative to the total number of PBMC) was similar in RA and controls (Fig. 3c). However, the percentages of CD14bright cells expressing TNFAIP9, CD14brightCD16− and CD14brightCD16+ cells were significantly higher in RA than controls (Fig. 3d–f), in contrast to CD14dimCD16+ cells (Fig. 3g). The percentage of CD14bright cell population could probably explain the high expression level of TNFAIP9 in PBMC of RA.
Fig 3.
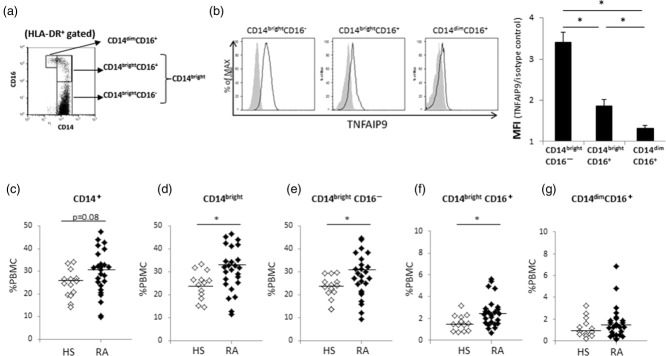
Localization of tumour necrosis factor (TNF)-α-induced protein (TNFAIP)9 in CD14+ cells. (a) Human leucocyte antigen D-related (HLA-DR)+CD14+ monocytes were divided into three subsets according to the CD14 and CD16 levels. (b) Comparison of TNFAIP9 expression in the three subsets of HLA-DR+CD14+ monocytes. Left: fluorescence activated cell sorter (FACS) data of a representative rheumatoid arthritis (RA) patient. Right: mean ± standard deviation (s.d.) values of mean fluorescence intensity (MFI) of TNFAIP9 to isotype control for each subset of HLA-DR+CD14+ monocytes in patients with RA. (c–g) Comparison of data of patients with RA (n = 23) and healthy subjects (HS, n = 13). Percentages of (c) CD14+ cells, (d) CD14bright cells, (e) CD14brightCD16-cells, (f) CD14brightCD16+cells and (g) CD14dimCD16+ cells in peripheral blood mononuclear cells (PBMC). *P < 0·05 by Mann–Whitney U-test.
Expression of TNFAIP9 and TNFAIP3 in CD14+ cells after TNF-α or LPS stimulation
Monocyte-conditional deficient mice of TNFAIP3 develop spontaneous arthritis 8, and TNFAIP9/TIARP is expressed predominantly in CD11b+ monocytes in mice 4. In the next step, we focused on CD14+ cells and compared the expression of TNFAIP9 and TNFAIP3 in CD14+ cells after TNF-α or LPS stimulation. The expression levels of TNF-α and IL-6 increased immediately after stimulation but decreased gradually thereafter (Fig. 4a,b). The same stimuli resulted in up-regulation of both proteins, but the increase in TNFAIP9 expression occurred after about 6–12 h, while that of TNFAIP3 occurred much earlier than TNFAIP9 (Fig. 4a,b). These findings suggest that both TNFAIP9 and TNFAIP3 can be induced by TNF-α and LPS in human CD14+ monocytes, with an earlier response in TNFAIP3 than TNFAIP9.
Fig 4.

Induction of tumour necrosis factor (TNF)-α-induced proteins (TNFAIP)9 and TNFAIP3 in CD14+ cells by tumour necrosis factor (TNF)-α and lipopolysaccharide (LPS). CD14+ cells of healthy subjects (HS, n = 3) were cultured in the presence or absence of (a) TNF-α (10 ng/ml) or (b) LPS (1 ng/ml) and TNF-α, interleukin (IL)-6, TNFAIP9 and TNFAIP3 mRNA levels were measured before (0 h) and after the indicated time-periods by real-time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Representative data of three independent experiments with similar results. Similar results (data not shown) were obtained using samples from RA patients (n = 2, per test). Values are mean ± standard deviation (s.d.); *P < 0·05 versus control.
Tocilizumab down-regulates TNFAIP9 expression in PBMC
We have reported previously that infliximab, a TNF antagonist, down-regulates the expression of TNFAIP9 in PBMC 15. As both TNFAIP9 and TNFAIP3 in myeloid-conditional deficient mice are IL-6-dependent 5,8, we investigated the effects of tocilizumab on TNFAIP9 and 3 mRNA levels and used abatacept as a control. In this part of the study, 11 RA patients were treated with tocilizumab while another group of 13 RA patients were treated with abatacept for 12 weeks; both were assessed clinically at 12 weeks after completion of therapy using the EULAR response criteria 10. In the tocilizumab-treated group, eight patients were classified as good responders, one as a moderate responder and two as non-responders. In comparison, in patients in the abatacept-treated group, one patient was classified as a good responder, nine as moderate responders and three as non-responders. Further analysis of the tocilizumab-treated group showed a significant decrease in TNFAIP9 expression in PBMC in all 24 RA patients (P = 0·013), especially in the good/moderate responders (P = 0·008). In contrast, the expression of TNFAIP3 in PBMC was increased significantly in the entire population, especially in responders (Fig. 5a). Interestingly, abatacept treatment had no effect on the expression of the two proteins (Fig. 5b).
Fig 5.

Effects of tocilizumab and abatacept treatment on tumour necrosis factor (TNF)-α-induced proteins (TNFAIP)9 and TNFAIP3 expression in peripheral blood mononuclear cells (PBMC) and percentages of CD14+ monocyte subsets. TNFAIP9 and TNFAIP3 mRNA expression levels in PBMC measured by real-time quantitative reverse transcription–polymerase chain reaction (qRT–PCR)before (0wk) and 12 weeks (12wk) after completion of treatment of patients with rheumatoid arthritis with (a) tocilizumab (n = 11) and (b) abatacept (n = 13). Data of individual patients. Patients were classified into responders (good or moderate response) and non-responders, based on their EULAR disease activity score in 28 joints (DAS28) response. (c) Percentages of CD14+ monocytes (left) and CD14bright cells (right) relative to PBMC at baseline and 12 weeks in tocilizumab responders (n = 6). (d) TNFAIP9 mRNA expression levels in CD14+cells isolated from the same six patients shown in (c), at baseline and at 12 weeks. *P < 0·05 by Wilcoxon's signed-rank test.
Mechanism of tocilizumab-induced down-regulation of TNFAIP9
To investigate the reason for the down-regulation of TNFAIP9 in PBMC, we examined changes in CD14+ monocytes population in PBMC. The percentage of CD14+ monocytes was measured before and 12 weeks after the completion of tocilizumab treatment in six patients and all patients were classified as responders. Treatment with tocilizumab decreased significantly the percentage of CD14+ monocytes and CD14bright cells (Fig. 5c).
We also measured TNFAIP9 mRNA expression levels in CD14+ cells isolated from the same six patients at 12 weeks after the completion of tocilizumab treatment. The expression levels in CD14+ cells tended to fall in all patients after treatment (P = 0·075) (Fig. 5d). In contrast, no significant fall was observed in TNFAIP3 mRNA expression levels. These findings suggest that tocilizumab reduces circulating CD14bright monocytes as well as the expression of TNFAIP9 in these cells, resulting in the down-regulation of TNFAIP9 expression in PBMC.
Discussion
In the present study, we analysed the expression of two TNFAIPs, TNFAIP9 and TNFAIP3, in PBMC of RA patients. TNFAIP9 expression was increased significantly while TNFAIP3 expression was decreased in RA compared with the healthy controls. Interestingly, the reverse was observed in splenocytes of mice with GPI-induced arthritis, with decreased expression of TNFAIP9 and increased expression of TNFAIP3 9, suggesting similar localization of both molecules between mice and humans. Previous findings in TNFAIP9/TIARP–/– mice showed that TNFAIP9 regulated arthritis negatively through the suppression of IL-6 production and TNF-α-induced NF-κB signalling in macrophages 5. In patients with RA, TNFAIP9 had been reported to be up-regulated in CD68+ macrophages in synovial tissue 16, while TNFAIP9 protein was highly expressed in synovial fluid 17. In peripheral blood, circulating neutrophils and monocytes are known to express TNFAIP9 16. In the present study, we found significantly elevated TNFAIP9 mRNA expression levels in CD14bright monocytes of patients with RA. In addition, CD14bright monocytes were increased significantly in RA, similar to the results reported by Coulthard et al. 18. CD14brightCD16- monocytes have been reported to produce large amounts of IL-6, IL-8, CCL2 and CCL3, while CD14brightCD16+ monocytes are considered the main producers of TNF-α and IL-1β in response to LPS 19. TNFAIP9 mRNA expression seemed to follow IL-6 mRNA expression (Fig. 4a), and RA patients possessed high amounts of TNFAIP9 in CD14brightCD16- monocytes, suggesting that countersuppression has been noted in these cells in arthritic conditions. Thus, the large percentage of CD14bright monocytes could be related to RA pathogenicity.
Myeloid conditional TNFAIP3-deficient mice also develop arthritis 8. Unlike TNFAIP9, TNFAIP3 is expressed ubiquitously in many types of cells and its basal level is very low, with the exception of thymocytes and peripheral T cells 7,9. Our findings were in agreement with the results of the above studies, and the difference in the distribution of TNFAIP9 and TNFAIP3 is considered to be the reason for the reverse pattern of mRNA expression levels of these two proteins in PBMC. In humans, several studies have described the relationship between single nucleotide polymorphisms (SNPs) of TNFAIP3 gene and autoimmune diseases, such as RA 20,21, systemic lupus erythematosus 22,23, psoriasis 24 and type 1 diabetes 25. However, the expression level of TNFAIP3 in each disease remains to be investigated. In RA, James et al. 26 recently reported high expression of TNFAIP3 in PBMC of RA irrespective of genotype. In contrast, our data showed a low expression of TNFAIP3 in RA compared to controls. The reason for this discrepancy may be due to differences in patient selection. Patients with RA in this study were not treated with biological agents, whereas the above study included some patients who were treated with anti-TNF agent 26, as biologicals could induce up-regulation of TNFAIP3 in PBMC (our preliminary observations). Another group reported recently that lower TNFAIP3 expression was found in RA patients compared to those in HS 27, supporting our data. In recent years, another autoimmune disease, multiple sclerosis (MS), has also been reported to have links to SNPs in the TNFAIP3 gene 28. Gilli et al. demonstrated that the TNFAIP3 transcript was down-regulated in PBMC obtained from MS patients compared to HS, as occurs in RA 29. These findings possibly corroborated the shared hypothesis that TNFAIP3 plays a key role in immunity and a defective expression/activity could be underlying several autoimmune diseases, according to a shared mechanism.
As shown in Fig. 2a, the main difference between TNFAIP9 and TNFAIP3 was the localization of the cells. TNFAIP9 was limited to CD14+ cells, whereas TNFAIP3 was found mainly in CD3+ cells. Navone et al. reported recently that TNFAIP3 mRNA expression on CD14+ monocytes and CD4+ T cells were clearly decreased in MS patients 30. Interestingly, as we show in Fig. 2b, TNFAIP3 in CD14+ cells were equal or tended to be up-regulated in patients with RA compared to HS in spite of the down-regulation of total TNFAIP3 mRNA levels in RA. It is widely accepted that TNF-α blockade is highly beneficial in RA, whereas it can exacerbate MS 31. TNF-α signals rapidly induce TNFAIP3 mRNA as well as TNFAIP9 in CD14+ cells (Fig. 4a), possibly suggesting an important pathological role of the TNF-α signal to CD14+ cells in RA patients. We speculated that the down-regulated expression of TNFAIP3 in CD3+ cells weas the main reason of the down-regulation of TNFAIP3 in PBMC, as in MS 29.
IL-6 plays a pivotal role in arthritis of TNFAIP9- and TNFAIP3-deficient animals 5,8. In the present study, treatment with tocilizumab reduced the expression of TNFAIP9. Circulating CD14bright cells and their TNFAIP9 expression were decreased in patients with RA following treatment with tocilizumab. In this regard, Richez et al. 32 reported that tocilizumab therapy reduced the percentages of circulating monocytes and myeloid dendritic cells in patients with RA. Furthermore, Vaitra et al. 33 reported that tocilizumab reduced the numbers of circulating CD14++CD16− monocytes and CD14+CD16+ monocytes in a patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS). The tocilizumab-induced fall in the number of circulating CD14bright monocytes could be associated with the pathogenicity of RA, probably through reduction of monocyte recruitment into the inflammation site and/or production of proinflammatory cytokines and chemokines. TNFAIP9 regulates both IL-6/signal transducer and activator of transcription-3 (STAT-3) and TNF–NF-κB cascade in mouse macrophages 5. Indeed, in in-vitro experiments with CD14+ cells, TNFAIP9 mRNA expression followed IL-6 mRNA (Fig. 4), suggesting the regulated mechanism by IL-6 signals in human monocytes. It probably reflects the down-regulation of TNFAIP9 in CD14+ cells by tocilizumab (Fig. 5d). Conversely, abatacept did not alter the expression of TNFAIP9 in RA patients. Abatacept [cytotoxic T lymphocyte antigen-4 (CTLA)-4-Ig] suppresses T cell activation and it is considered that it has no direct influence on inflammatory cytokines or the number of circulating CD14+ cell populations. In this regard, we have reported previously that infliximab also reduced the expression of TNFAIP9 in RA patients 15. Considered together, our present and previous results suggest that TNFAIP9 expression could be regulated directly by signals from TNF-α and IL-6 in CD14+ cells in RA patients.
In contrast to TNFAIP9, tocilizumab increased TNFAIP3 expression in PBMC. Tocilizumab is known to alter myeloid cell linage, but not T cells 32. TNFAIP9 expression in the spleen seems to be the reverse of that of TNFAIP3 in GPI-induced arthritis, and the expression of TNFAIP3 in macrophages seems to be associated closely with that of TNFAIP9 under IL-6 culture conditions 9. Further studies are needed to determine the relationship between the expression of TNFAIP9 and TNFAIP3 in RA and its role in the pathogenesis of RA through TNF-α and IL-6.
Conclusions
To our knowledge, this is the first study that has examined the comparative expression of TNFAIP9 and TNFAIP3 in RA. The results showed up-regulation of TNFAIP9 mRNA expression on CD14bright monocytes and down-regulation of TNFAIP3 mRNA expression in PBMC in patients with RA. TNFAIP9 expression was associated with TNF-α, and tocilizumab reduced TNFAIP9 expression levels on CD14+ monocytes. Further studies to investigate the role of TNFAIP9 expression on CD14bright monocytes may highlight its relation to the pathogenesis of RA.
Acknowledgments
We thank Dr F. G. Issa for the critical reading of the manuscript. This work was supported in part by Health and Labour Sciences Research Grants for research on intractable diseases from the Ministry of Health, Labour and Welfare of Japan, and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology and from the Japan Society for the Promotion of Science.
Author contributions
C. T., I. M. and T. S. wrote the manuscript, C. T. performed all the experiments and I. M. co-ordinated study design and conception. A. I., N. U., Y. T. and Y. K. supported the experiment and data acquisition; Y. W. and I. N. helped to revise it critically for important intellectual content. All authors approved the final version to be published. I. M. had full access to all the data in this study.
Disclosures
The authors declare that they have no competing interests.
References
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–60. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- Al-Shakarchi I, Gullick NJ, Scott DL. Current perspectives on tocilizumab for the treatment of rheumatoid arthritis: a review. Patient Prefer Adherence. 2013;4:653–66. doi: 10.2147/PPA.S41433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Matsumoto I, Tanaka Y, et al. Tumor necrosis factor alpha-induced adipose-related protein expression in experimental arthritis and in rheumatoid arthritis. Arthritis Res Ther. 2009;11:R118. doi: 10.1186/ar2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Matsumoto I, Tanaka Y, et al. Murine tumor necrosis factor α-induced adipose-related protein (tumor necrosis factor α-induced protein 9) deficiency leads to arthritis via interleukin-6 overproduction with enhanced NF-κB, STAT-3 signaling, and dysregulated apoptosis of macrophages. Arthritis Rheum. 2012;64:3877–85. doi: 10.1002/art.34666. [DOI] [PubMed] [Google Scholar]
- Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011;21:22–39. doi: 10.1038/cr.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;29:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matmati M, Jacques P, Maelfait J, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011;14:908–12. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Inoue A, Takai C, et al. Regulatory roles of tumor necrosis factor alpha-induced proteins (TNFAIPs) 3 and 9 in arthritis. Clin Immunol. 2014;153:73–8. doi: 10.1016/j.clim.2014.03.015. [DOI] [PubMed] [Google Scholar]
- van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League against rheumatism criteria. Arthritis Rheum. 1996;39:34–40. doi: 10.1002/art.1780390105. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Tewari M, Wolf FW, Seldin MF, O'Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol. 1995;15:1699–706. [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- Zawada AM, Rogacev KS, Rotter B, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. 2011;118:e50–61. doi: 10.1182/blood-2011-01-326827. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Matsumoto I, Iwanami K, et al. Six-transmembrane epithelial antigen of prostate 4 (STEAP4) is expressed on monocytes/neutrophils, and is regulated by TNF antagonist in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2012;30:99–102. [PubMed] [Google Scholar]
- Tanaka Y, Matsumoto I, Iwanami K, et al. Six-transmembrane epithelial antigen of prostate 4 (STEAP4) is a tumor necrosis factor alpha-induced protein that regulates IL-6, IL-8, and cell proliferation in synovium from patients with rheumatoid arthritis. Mod Rheumatol. 2012;22:128–36. doi: 10.1007/s10165-011-0475-y. [DOI] [PubMed] [Google Scholar]
- Noh R, Park SG, Ju JH, et al. Comparative proteomic analyses of synovial fluids and serums from rheumatoid arthritis patients. J Microbiol Biotechnol. 2014;24:119–26. doi: 10.4014/jmb.1307.07046. [DOI] [PubMed] [Google Scholar]
- Coulthard LR, Geiler J, Mathews RJ, et al. Differential effects of infliximab on absolute circulating blood leucocyte counts of innate immune cells in early and late rheumatoid arthritis patients. Clin Exp Immunol. 2012;170:36–46. doi: 10.1111/j.1365-2249.2012.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco G, Hinks A, Eyre S, et al. Combined effects of three independent SNPs greatly increase the risk estimate for RA at 6q23. J. Hum Mol Genet. 2009;15:2693–9. doi: 10.1093/hmg/ddp193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieguez-Gonzalez R, Calaza M, Perez-Pampin E, et al. Analysis of TNFAIP3, a feedback inhibitor of nuclear factor-κB and the neighbor intergenic 6q23 region in rheumatoid arthritis susceptibility. Arthritis Res Ther. 2009;11:R42. doi: 10.1186/ar2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–4. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrianto I, Wen F, Templeton A, et al. Association between a functional variant downstream of TNFAIP3 and systemic lupus erythematosus. Nat Genet. 2011;43:253–8. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011;39:1086–91. doi: 10.1042/BST0391086. [DOI] [PubMed] [Google Scholar]
- Fung EY, Smyth DJ, Howson JM, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009;10:188–91. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- Maxwell JR, Gowers IR, Kuet KP, Barton A, Worthington J, Wilson AG. Expression of the autoimmunity associated TNFAIP3 is increased in rheumatoid arthritis but does not differ according to genotype at 6q23. Rheumatology (Oxf) 2012;51:1514–5. doi: 10.1093/rheumatology/kes134. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu L, Liao Z, et al. Alternative expression pattern of MALT1-A20-NF-κB in patients with rheumatoid arthritis. J Immunol Res. 2014;2014:492872. doi: 10.1155/2014/492872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–60. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilli F, Lindberg RL, Valentino P, et al. Learning from nature: pregnancy changes the expression of inflammation-related genes in patients with multiple sclerosis. PLOS ONE. 2010;5:e8962. doi: 10.1371/journal.pone.0008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navone ND, Perga S, Martire S, Berchialla P, Malucchi S, Bertolotto A. Monocytes and CD4+ T cells contribution to the under-expression of NR4A2 and TNFAIP3 genes in patients with multiple sclerosis. J Neuroimmunol. 2014;272:99–102. doi: 10.1016/j.jneuroim.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Steinman L, Merrill JT, McInnes IB, Peakman M. Optimization of current and future therapy for autoimmune diseases. Nat Med. 2012;18:59–65. doi: 10.1038/nm.2625. [DOI] [PubMed] [Google Scholar]
- Richez C, Barnetche T, Khoryati L, et al. Tocilizumab treatment decreases circulating myeloid dendritic cells and monocytes, 2 components of the myeloid lineage. J Rheumatol. 2012;39:1192–7. doi: 10.3899/jrheum.111439. [DOI] [PubMed] [Google Scholar]
- Vaitla PM, Radford PM, Tighe PJ, et al. Role of interleukin-6 in a patient with tumor necrosis factor receptor-associated periodic syndrome: assessment of outcomes following treatment with the anti-interleukin-6 receptor monoclonal antibody tocilizumab. Arthritis Rheum. 2011;63:1151–5. doi: 10.1002/art.30215. [DOI] [PubMed] [Google Scholar]