

Abstract
Citrate, a central component of cellular metabolism, is a widely used anti-coagulant due to its ability to chelate calcium. Adenosine triphosphate (ATP)-citrate lyase, which metabolizes citrate, has been shown to be essential for inflammation, but the ability of exogenous citrate to impact inflammatory signalling cascades remains largely unknown. We hypothesized that citrate would modulate inflammatory responses as both a cellular metabolite and calcium chelator, and tested this hypothesis by determining how clinically relevant levels of citrate modulate monocyte proinflammatory responses to lipopolysaccharide (LPS) in a human acute monocytic leukaemia cell line (THP-1). In normal medium (0·4 mM calcium), citrate inhibited LPS-induced tumour necrosis factor (TNF)-α and interleukin (IL)-8 transcripts, whereas in medium supplemented with calcium (1·4 mM), TNF-α and IL-8 levels increased and appeared independent of calcium chelation. Using an IL-8–luciferase plasmid construct, the same increased response was observed in the activation of the IL-8 promoter region, suggesting transcriptional regulation. Tricarballylic acid, an inhibitor of ATP-citrate lyase, blocked the ability of citrate to augment TNF-α, linking citrate's augmentation effect with its metabolism by ATP-citrate lyase. In the presence of citrate, increased histone acetylation was observed in the TNF-α and IL-8 promoter regions of THP-1 cells. We observed that citrate can both augment and inhibit proinflammatory cytokine production via modulation of inflammatory gene transactivation. These findings suggest that citrate anti-coagulation may alter immune function through complex interactions with the inflammatory response.
Keywords: citrate, epigenetics, inflammation, monocytes
Introduction
Citrate anti-coagulation is central to the storage of blood products for transfusion and to maintain the patency of extracorporeal circuits such as continuous renal replacement therapy (CRRT), used to treat renal failure1–3. Citrate prevents activation of the clotting cascade by chelating free extracellular calcium, which is a necessary co-factor for many steps in the coagulation cascade4. When used with CRRT, regional anti-coagulation with citrate lowers ionized calcium concentrations from ∼1·2 mM in the patient down to ∼0·4 mM in the circuit5. However, plasma citrate concentrations increase from 0·1 mM prior to initiating CRRT to 4–6 mM within the circuit and 0·3–1·0 mM in the patient after initiation of the CRRT circuit5. In order to prevent the development of complications (e.g. citrate ‘lock’), the increased citrate load delivered as a result of CRRT must be metabolized6. None the less, the use of citrate for regional anticoagulation in CRRT results in longer circuit lifetimes, fewer bleeding complications and possibly increased patient survival6–10.
In normal states, citrate is metabolized within the mitochondria as part of the citric acid cycle or by adenosine triphosphate (ATP)-citrate lyase (ACLY) in the cytoplasm for fatty acid synthesis11,12. However, recent studies on immune cells suggest that citrate may play a larger role beyond simply energy production. The mitochondrial citrate carrier, a transport protein that facilitates the efflux of citrate from the mitochondrial matrix into the cytosol, has been shown to promote immune cell activation in response to lipopolysaccharide (LPS)13,14. Once outside the mitochondria, ACLY metabolizes citrate into acetyl-CoA and oxaloacetate, which can be used in the generation of prostaglandins and reactive oxygen species, respectively13–16. Indeed, acetyl-CoA has often been associated with fatty acid synthesis; however, it can also be utilized as an acetyl-donor for histone acetylation15,17,18. An increase in histone acetylation could have a significant impact on the inflammatory response as there is accumulating evidence that this post-translational epigenetic modification alters gene expression patterns19. To date, the ability of metabolic intermediates, and specifically exogenously added metabolites such as citrate, to regulate or alter immune responses has garnered little attention.
Here, we investigate the impact of externally added citrate on the monocyte inflammatory response following LPS-induced immune activation. LPS, a component of Gram-negative bacterial cell walls, is a potent activator of innate immune cells leading to the production of proinflammatory cytokines20. Monocytes, orchestrators of the innate inflammatory response, are dependent upon the opening of cell surface calcium-channels in order to recognize and respond to pathogens and their endotoxins21. LPS binds to Toll-like receptor 4 on the surface of monocytes and macrophages, initiating a calcium-dependent signal transduction cascade leading to nuclear factor-κB (NF-κB) activation22–24. Once translocated to the nucleus, NF-κB is then able to induce the production of various proinflammatory cytokines such as tumour necrosis factor (TNF)-α and interleukin (IL)-825. Given the calcium-dependent nature of monocyte activation induced by LPS, as well as the importance of energy metabolism during inflammation, we sought to characterize the impact of citrate on proinflammatory cytokine responses.
Materials and methods
Chemicals
All chemicals and reagents including sodium citrate and tricarballylic acid (TCA) were obtained from Sigma-Aldrich (St Louis, MO, USA) unless noted otherwise.
Cell culture and experimental setup
Human acute monocytic leukaemia cell line (THP-1) monocytic cells were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA)26. Cells were maintained in RPMI-1640 complete growth medium (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies, Grand Island, NY, USA) and Kanamycin solution from Streptomyces kanamyceticus26. Prior to experiments, THP-1 cells were rested overnight in medium containing 0·5% FBS. On the day of the experiment, cells were resuspended in fresh medium containing 0·5% FBS for 30 min. Experiments were performed in either standard tissue culture medium which contained 0·4 mM or a supplemented calcium medium in which an additional 1 mM CaCl2 was added to bring the total calcium concentration to 1·4 mM. Cells were then pretreated (30 min) with varying concentrations of sodium citrate (1, 6 mM), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid [ethylene glycol tetraacetic acid (EGTA)], 0·05–5 mM), or TCA (1–6 mM) at pH 7·4, prior to stimulation with LPS (Escherichia coli O55:B5, 100 ng/ml).
TNF-α protein measurement
Tissue culture supernatants were collected and TNF-α protein concentration was determined by enzyme-linked immunosorbent assay (ELISA) using a TNF-α human antibody pair (Life Technologies, Carlsbad, CA, USA), according to the manufacturer's instructions.
Quantitative real-time polymerase chain reaction (qPCR)
Total RNAs were extracted from cultured cells using TRIZOL reagent (Life Technologies). Template cDNAs were obtained by reverse transcription (RT) using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Quantification of cDNAs was performed by real-time PCR using primers designed for candidate transcript detection (Life Technologies). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH): forward: 5′-GTGGAA GGACTCATGACCACA-3′, reverse: 5′-GGTTTTTCTAGA CGGCAGGTC-3′; TNF-α: forward: 5′-CCAGGCAGTCAG ATCATCTTC-3′, reverse: 5′-ATGAGGTACAGGCCCTC TGAT-3′; and IL-8: forward: 5′-AGCTCTGTGTGAAGG TGCAG-3′, reverse: 5′-AATTTCTGTGTTGGCGCAGT-3′. Real-time polymerase chain reaction (PCR) experiments were performed in triplicate using SYBR Green chemistry (Life Technologies) and a Mastercycler ep gradient S (Eppendorf, Hamburg, Germany). The quantity in each sample was normalized to the level of reference gene GAPDH transcripts. Real-time PCR data were analysed by the delta-delta CT method27.
Plasmid vectors
The −162/+44 fragment of the full-length human IL-8 promoter, subcloned into luciferase (−162/+44hIL-8/Luc)28, was provided by M. Hershenson (University of Michigan, Ann Arbor, MI, USA). The reporter activities of this plasmid have been shown to be identical to the full-length promoter in response to various inflammatory stimuli28–30.
Luciferase activity
THP-1 cells were transfected transiently with IL-8-luc constructs using the 4D-Nucleofector system (Lonza, Basel, Switzerland) as per the manufacturer's protocol. Following transfection, cells were rested (30 min) in medium (0·5% FBS) and incubated with citrate (30 min) prior to LPS stimulation (4 h). Whole cell lysates were prepared in Reporter Lysis Buffer (Promega, Madison, WI, USA), following the manufacturer's protocol. Luciferase activity was determined using a Luciferase Assay System (Promega) and a Spectramax M3 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
Preparation of histones
Histones were isolated from THP-1 cells using an acid-extraction technique. Briefly, cells (106) were harvested following stimulation and washed twice with ice-cold phosphate buffered saline (PBS) supplemented with 10 mM sodium butyrate. Cells were resuspended in lysis buffer [PBS containing 0·5% Triton X 100 (v/v), 0·1 mM phenylmethysulphonyl fluoride, 10 mM sodium butyrate, and a protease inhibitor tablet (Roche, Basel, Switzerland)]. Cells were lysed on ice for 10 min with gentle stirring and centrifuged at 500 g for 10 min at 4°C. The supernatant was discarded and cells were washed in lysis buffer. The supernatant was again discarded and the pellet was resuspended in 0·2 N HCl overnight at 4°C. Following acid extraction, 1/5 volume of 1 M NaOH was added to neutralize extraction and samples were centrifuged at 500 g for 10 min at 4°C. The supernatant was retained and protein concentration determined by a Bradford Assay (Thermo Scientific, Waltham, MA, USA).
Western blot analysis
Acid extracted histones were separated by sodium dodecyl sulphate reducing polyacrylamide gel electrophoresis (SDS-PAGE). Total histones were determined by Coomassie stain (ThermoFisher Scientific, Waltham, MA, USA) as an extraction control or transferred to polyvinylidene difluoride membranes, as described previously31. The membranes were incubated with primary antibodies for proteins of interest, including anti-acetyl-Histone H3 (06-599; Millipore, Billerica, MA, USA) and anti-acetyl-Histone H4 (06-866; Millipore). Blots were then incubated with horseradish peroxidase-conjugated secondary antibodies (NA934; GE Healthcare Biosciences, Piscataway, NJ, USA) and reacted with chemiluminescence reagents (GE Healthcare Biosciences). Relative band intensity was quantified using ImageJ software32.
Chromatin immunoprecipitation-qPCR
Chromatin immunoprecipitation (ChIP) was performed using the LowCell# ChIP kit (Diagenode, Seraing, Belgium), according to the manufacturer's protocol. After cell lysis, the chromatin was sonicated to an average size of 500 base pairs (bp) and enriched with Protein G-coated magnetic bead (Diagenode)-coupled antibody against acetyl-Histone H3K9 (MAb-185-050; Diagenode), acetyl-Histone H4K8 (pAb-103-050; Diagenode), acetyl-Histone H4K12 (17-10121; Millipore) or mouse immunoglobulin (Ig)G (Diagenode) at 4°C overnight. Following cross-link reversal and DNA isolation, samples were subject to qPCR as described above. The specific sequences from immunoprecipitated and input DNA were detected by PCR primers for promoter regions within the TNF-α and IL-8 gene loci that are important nucleosomal binding regions as described previously33–35. The TNF-α PCR primer lies within the distal upstream promoter region of TNF-α (−195/–345)33,34: forward: 5′-CCC AAAAGAAATGGAGGCAAT-3′, reverse: 5-AAGCATCAAG GATACCCCTCAC-3′. The IL-8 PCR primer lies within the promoter region of IL-835: forward: 5′-CAGAGA CAGCAGAGCACAC-3′, reverse: 5′-ACGGCCAGCTTG GAAGTC-3′. The efficiency of the experimental ChIP for the specified genomic locus was calculated from qPCR data as a percentage of starting material with the same primer set.
Data analysis
All data are presented as mean ± standard deviation (s.d.) unless noted otherwise. Figures contain representative data of experiments performed in triplicate. Statistical analysis was performed with Student's t-test to compare data between two groups. A two-tailed P-value <0·05 was considered statistically significant.
Results
Citrate's effect on TNF-α protein production following LPS stimulation varies by calcium concentration
To test the ability of citrate to alter proinflammatory cytokine responses, we utilized THP-1 cells to model monocyte cellular responses. In standard tissue culture medium, LPS-induced TNF-α protein production (6 h) was augmented at low citrate concentrations and inhibited at high citrate concentrations relative to controls (Fig. 1a). To test the importance of calcium chelation in the TNF-α response, THP-1 cells were incubated in medium supplemented with additional calcium. In these higher calcium conditions, the LPS-induced TNF-α proinflammatory response is no longer inhibited at high citrate concentrations (Fig. 1b). Instead, high levels of citrate augmented the TNF-α response well above citrate-untreated control culture conditions. Of note, citrate alone, in either normal or supplemented calcium medium, did not alter TNF-α levels in the absence of LPS (data not shown). To determine if the effect of citrate can be explained by extracellular calcium chelation, THP-1 cells were pretreated with the calcium-selective chelator EGTA36,37. As can be seen in Fig. 1c,d, high concentrations of EGTA inhibited TNF-α levels in both standard and supplemental calcium media. In contrast to the effects of citrate, no augmentation effect was seen with EGTA. This pattern of responses to citrate in medium containing varying calcium concentrations indicates that the effect of citrate on the TNF-α response is explained incompletely by calcium chelation alone.
Fig 1.
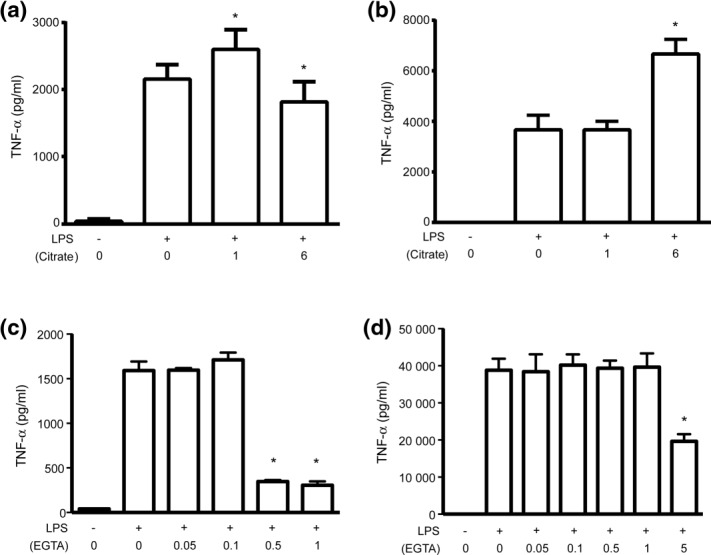
Citrate augments and inhibits tumour necrosis factor (TNF)-α protein production. Human acute monocytic leukaemia cell line (THP-1) cells were incubated (30 min) with varying concentrations of citrate (a,b) or ethylene glycol tetraacetic acid (EGTA) (c,d) prior to stimulation with lipopolysaccharide (LPS) (6 h). TNF-α protein production was determined by enzyme-linked immunosorbent assay (ELISA) in normal medium (0·4 mM calcium, a,c) and in supplemental calcium medium (1·4 mM, b,d) (*<0·05, n.s. = not significant).
Citrate augments and inhibits TNF-α and IL-8 mRNA production in THP-1 cells following LPS stimulation
To determine if citrate is altering the TNF-α proinflammatory response at the level of transcription versus translation, we characterized the levels of TNF-α mRNA transcripts under similar experimental conditions. In standard tissue culture medium, TNF-α mRNA production (2 h) showed a similar pattern to what was seen at the protein level: low concentrations of citrate augmented the TNF-α response while higher citrate concentrations inhibited TNF-α mRNA (Fig. 2a). In medium supplemented with additional calcium, citrate doubled TNF-α mRNA production at moderate and high citrate concentrations, similar to what was seen with TNF-α protein (Fig. 2b). These findings indicated that citrate may be altering TNF-α transcription.
Fig 2.
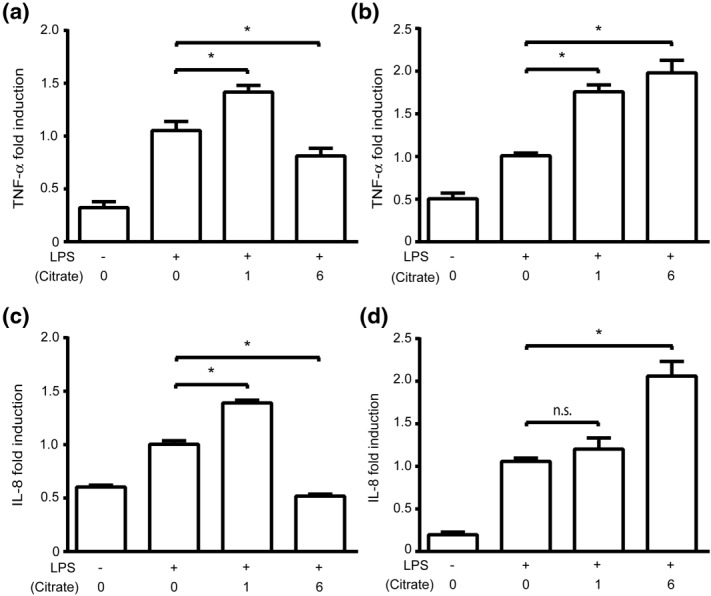
Citrate augments and inhibits proinflammatory cytokine mRNA production. Human acute monocytic leukaemia cell line (THP-1) cells were incubated (30 min) with varying concentrations of citrate prior to stimulation with lipopolysaccharide (LPS) (2 h). mRNA was isolated and subsequently analysed by quantitative polymerase chain reaction (qPCR). (a) Tumour necrosis factor (TNF)-α mRNA production in normal calcium medium (0·4 mM). (b) TNF-α mRNA production in supplemental calcium medium (1·4 mM). (c) IL-8 mRNA production in normal calcium medium (0·4 mM). (d) IL-8 mRNA production in supplemental calcium medium (1·4 mM). Data were normalized to the LPS-stimulated sample with no citrate (*P<0·05, n.s. = not significant).
To determine if this response was unique to TNF-α or if it extended to other LPS-induced proinflammatory genes, IL-8 mRNA levels were measured following citrate pretreatment and LPS stimulation. In both normal and calcium-supplemented media, citrate augmented and inhibited the IL-8 mRNA response in a manner that paralleled the TNF-α response (Fig. 2c,d). These data suggest that citrate alters LPS-induced proinflammatory TNF-α and IL-8 cytokine production by augmenting or inhibiting gene transcripts.
Citrate alters transactivation of IL-8 promoter activity
As TNF-α and IL-8 gene expression is largely NF-κB-dependent, THP-1 cells were transfected transiently with a κB-dependent luciferase reporter construct to assess NF-κB transcriptional activation38. In standard tissue culture medium, luciferase expression (4 h) was augmented at low concentrations of citrate and inhibited at high concentrations of citrate compared to stimulated controls (Fig. 3a). These findings are consistent with the response pattern observed in mRNA and protein production. In calcium-supplemented medium, luciferase expression was augmented at high concentrations of citrate (Fig. 3b). Taken together, these data indicate citrate is altering IL-8 cytokine transcription by modulating the transactivation of the IL-8 promoter, in a manner dependent on calcium concentrations.
Fig 3.
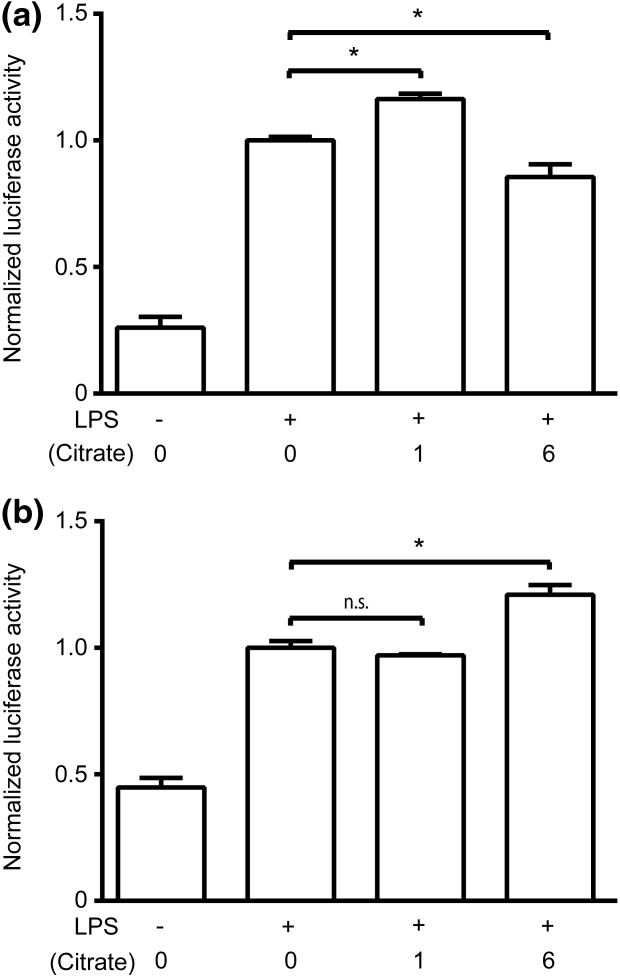
Citrate affects transactivation of the interleukin (IL)-8 promoter. Human acute monocytic leukaemia cell line (THP-1) cells were transiently transfected with a −162/+44 hIL-8 luc plasmid. Cells were stimulated with lipopolysaccharide (LPS) for 4 h following preincubation with varying concentrations of citrate in either (a) normal medium (0·4 mM calcium) or (b) supplemental calcium medium (1·4 mM). Luciferase activity was measured using a Luciferase Assay System (Promega) and a Spectramax M3 spectrophotometer (Molecular Devices). Data were normalized to the LPS-stimulated sample with no citrate (*P<0·05, n.s. = not significant).
TCA, a structural analogue of citrate, inhibits TNF-α mRNA production following LPS stimulation
THP-1 cells were treated with TCA to assess the relative importance of calcium chelation versus flux through ACLY in the ability of citrate to modulate the TNF-α response to LPS. In standard medium, high concentrations of TCA inhibited TNF-α mRNA production (2 h) compared to stimulated controls, a response similar to that in the presence of citrate (Fig. 4a). However, at lower concentrations of TCA (1 mM) in standard medium, there is no augmentation effect as is observed with citrate (Fig. 4a). In stark contrast to treatment with citrate, in supplemented calcium medium high concentrations of TCA inhibited TNF-α mRNA production in a dose–response pattern (Fig. 4b).
Fig 4.
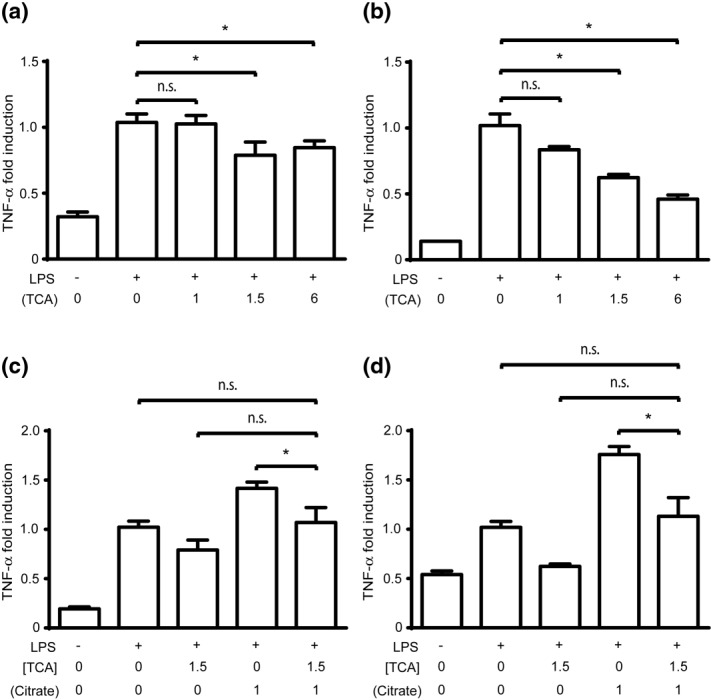
Tricarballylic acid (TCA) inhibits monocyte inflammatory responses. (a,b) Human acute monocytic leukaemia cell line (THP-1) cells were stimulated with lipopolysaccharide (LPS) (2 h) following preincubation with varying concentrations of TCA to determine tumour necrosis factor (TNF)-α mRNA production in (a) normal calcium (0·4 mM) or (b) supplemental calcium (1·4 mM) media. (c,d) THP-1 cells were pretreated (0·5 h) with TCA (1·5 mM) alone, citrate (1 mM) alone, or co-incubated with citrate and TCA prior to LPS stimulation to determine TNF-α mRNA production (2 h) in (c) normal calcium (0·4 mM) or (d) supplemental calcium (1·4 mM) media. Data were normalized to the LPS-stimulated sample with no citrate or TCA (*P<0·05, n.s. = not significant).
Citrate is used as a metabolic substrate to augment TNF-α production
Due to the difference in TNF-α responses with citrate and TCA, we hypothesized that citrate may be metabolized by ACLY during monocyte inflammatory responses13,15. To test this hypothesis, THP-1 cells were co-incubated with TCA and citrate prior to LPS stimulation. In both normal and supplemented calcium media, treatment of cells with TCA alone inhibits TNF-α mRNA, and treatment with citrate alone augments TNF-α mRNA levels (Fig. 4c,d). However, co-treatment with citrate and TCA abrogates the ability of citrate to augment LPS-induced TNF-α mRNA. The ability of TCA to prevent augmentation of the inflammatory response, while maintaining an inhibitory effect, implies that citrate's ability to augment inflammatory cytokine production is independent of calcium chelation and may depend instead upon its metabolism through ACLY.
Citrate prolongs histone acetylation following LPS stimulation
Because ACLY metabolizes citrate into acetyl CoA and oxaloacetate, we sought to determine if citrate was increasing the availability of acetyl donors by characterizing the global acetylation of histones H3 and H4 lysine residues. In standard medium, preincubation with low and high concentrations of citrate increased histone H3 and H4 acetylation at baseline compared to control cells (Fig. 5a). In response to LPS stimulation, control cells in standard medium showed an increase in H3 acetylation and a slight decrease in H4 acetylation. Under these same conditions, treatment with citrate increased both H3 and H4 acetylation relative to control (Fig. 5a). In supplemented calcium medium, control cells at baseline prior to LPS stimulation showed high levels of histone acetylation that then decreased following LPS (Fig. 5b). Citrate-treated cells also showed a decrease in histone H3 and H4 acetylation following LPS, although high citrate concentrations limited the decrease in H4 acetylation (Fig. 5b). These data suggest that citrate can increase global levels of histone acetylation.
Fig 5.

Citrate modulates global histone acetylation. Changes in histone H3 and H4 global acetylation were determined by Western blot in acid-extracted lysates of human acute monocytic leukaemia cell line (THP-1) cells. Cells were preincubated with varying concentrations of citrate (0, 1, 6 mM) prior to stimulation with lipopolysaccharide (LPS) (0–90 min) in (a) normal calcium (0·4 mM) or (b) supplemental calcium (1·4 mM) media. Relative band intensity is represented by the numbers below the Western blots with the no citrate time zero condition assigned a relative value of 100.
Citrate increases histone acetylation at inflammatory gene promoters in THP-1 cells
Given the ability of citrate to alter global histone acetylation, we sought to determine if citrate could alter histone acetylation at specific inflammatory gene regions as a means of priming these genes for transcriptional activation. Therefore, we assessed the acetylation marks at three lysine residues on histones H3 and H4 located in the gene promoter regions of TNF-α and IL-8 via ChIP33–35. Experiments were conducted in medium supplemented with calcium and cells were incubated at low citrate concentrations in order to model the steady-state plasma calcium and citrate levels in patients on CRRT5. Histone H3 lysine 9 acetylation, H4 lysine 8 acetylation and H4 lysine 12 acetylation were augmented significantly in THP-1 cells incubated with citrate compared to controls at both the TNF-α and IL-8 promoter regions (Fig. 6a,b, respectively). To determine if these effects were specific to citrate, THP-1 cells were incubated with 1·5 mM TCA instead of citrate. As can be seen in Fig. 6, TCA is also able to increase histone acetylation in the promoter regions of both TNF-α and IL-8.
Fig 6.
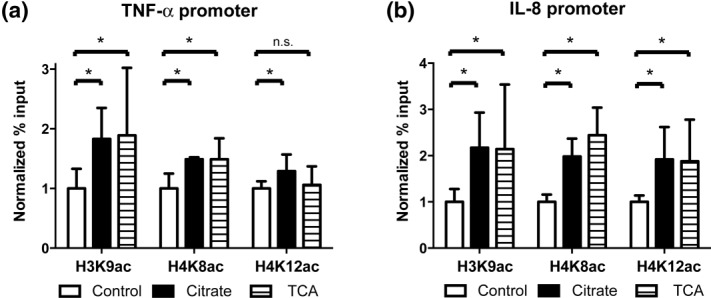
Citrate increases histone lysine acetylation at inflammatory gene promoters. Chromatin immunoprecipitation (ChIP) assays were performed in a human acute monocytic leukaemia cell line (THP-1) with antibodies against histone H3K9ac, H4K8ac and H4K12ac following preincubation with citrate (1 mM) or tricarballylic acid (TCA) (1·5 mM) in supplemental calcium medium (1·4 mM). The efficiency of the experimental ChIP for the specified genomic locus was calculated from quantitative polymerase chain reaction (qPCR) data as a percentage of starting material with the same primer set (% input) and then normalized to control conditions. (a) Tumour necrosis factor (TNF)-α. (b) Interleukin (IL)-8. Error bars represent standard deviation calculated from at least three separate experiments. (*P<0·01, n.s. = not significant).
Discussion
Pretreatment of THP-1 cells with citrate resulted in a dichotomous response to LPS that depended upon both citrate and calcium concentrations. When calcium availability is limiting due to sequestration by citrate chelation, as seen within the CRRT circuit, THP-1 monocytes' TNF-α mRNA and protein production is inhibited. This result is expected, given the calcium-dependent nature of this proinflammatory signalling pathway22–24, and is also seen with experiments using the extracellular calcium chelator EGTA (see Fig. 1). However, when calcium availability is not limiting, as can be seen either with low citrate concentrations or with calcium concentrations similar to levels achieved within a patient on CRRT (1–1·4 mM), citrate augmented TNF-α mRNA and protein production following LPS stimulation. Under these same conditions, IL-8 mRNA transcripts followed a similar trend to that of TNF-α mRNA. This same response was also observed with an IL-8-luc reporter construct, indicating that citrate is modulating monocyte proinflammatory TNF-α and IL-8 responses via transactivation of NF-κB and/or activator protein-1 (AP-1). We therefore sought to examine how exogenous citrate may be utilized in the cell to promote an increase in cytokine production. With citrate being an important metabolic substrate, we hypothesized that the metabolism of citrate could contribute to these altered cellular responses.
Citrate metabolism by ATP-citrate lyase
Several factors point to the importance of ACLY in the metabolism of citrate during the inflammatory response. Its activity is increased by the presence of citrate, it is up-regulated following LPS stimulation, and inhibition of ACLY decreases the production of key inflammatory mediators such as nitric oxide, reactive oxygen species and prostaglandin E214,39. We tested the importance of ACLY in our system through the use of TCA, a competitive inhibitor of ACLY that has an inhibition constant of ∼1 mM12. TCA is a structural analogue of citrate that differs due only to the lack of a hydroxyl group at the 2′ carbon. This minor structural change decreases the ability of TCA to chelate calcium by ∼40-fold relative to citrate (citrate has a calcium dissociation constant of 0·4 versus 15 mM for TCA)40. We found that TCA is able to both inhibit LPS-induced TNF-α mRNA transcripts and block the ability of citrate to augment TNF-α. The ability of TCA to inhibit TNF-α at concentrations above the KI for ACLY but below its calcium chelation constant implies that ACLY inhibition may be more important than calcium chelation in the ability of TCA to modulate the TNF-α LPS inflammatory response. These findings support our hypothesis that citrate metabolism by ACLY is essential for its ability to augment LPS-induced proinflammatory responses, and support the importance of ACLY in LPS-triggered inflammation.
ACLY generates both acetyl-CoA and oxaloacetate and therefore it is possible that either metabolite may contribute to augmented LPS inflammatory responses due to exogenously added citrate. During the inflammatory response, oxaloacetate has been shown to be important in the generation of NADPH which is subsequently utilized for reactive oxygen species production13,14. Similarly, acetyl-CoA can be used to generate prostaglandins14. However, both these observations occur after the initial inflammatory insult in accordance with citrate being actively shuttled out of the mitochondria. We therefore sought a mechanism on how external citrate added to the system may contribute to augmented gene activation.
Signalling pathways leading to transcriptional activation have been linked with metabolism41. Protein modifications leading to transcriptional activation, such as acetylation, are generated from metabolites42. In mammalian cells, acetyl-CoA can be generated by ACLY or acetyl-CoA synthetase 1 for use in protein acetylation15. The acetyl-CoA produced by ACLY is derived from the conversion of glucose into citrate which is then cleaved by ACLY to form oxaloacetate and acetyl-CoA43. Cleavage of citrate to acetyl-CoA via ACLY is the preferred pathway for acetyl-CoA production under high nutrient conditions16. It has been shown that ACLY localizes to the nucleus and that nutrient availability alters histone acetylation, suggesting a link between cellular metabolism and histone acetylation via ACLY15. Acetylation of histones allows for protein access to DNA binding sites and increases the transcription of target genes19. We hypothesized that citrate increases the availability of acetyl groups by its conversion into acetyl-CoA by ACLY in the nucleus. Having an increase in acetyl groups at active gene locations may provide a continuous pool of substrate for acetylation, thereby promoting enhanced transcription by facilitating acetylation of histone and non-histone proteins and insulating from metabolic changes17. We showed that citrate prolonged the global acetylation of histones H3 and H4 treated with LPS, thereby potentially linking citrate to enhanced gene transcription via acetyl group formation. Using ChIP analyses, we showed that treatment of THP-1 cells with citrate could induce chromatin remodelling at TNF-α and IL-8 gene promoter regions. However, treatment with TCA was also able to increase histone acetylation at these gene promoter regions. As a competitive inhibitor of ACLY, TCA should decrease the availability of acetyl-CoA. This result is not consistent with the hypothesis that histone acetylation can be increased by simply providing additional acetyl-CoA substrate. Instead, these findings suggest that citrate and TCA may alter histone acetylation via an alternative pathway, such as changes to the activation of histone acetyltransferases.
Clinical implications
The ability of citrate to modulate monocyte inflammatory responses has many potential clinical implications, given citrate's broad usage as an anti-coagulant and its importance in biological pathways. It is known that trauma patients who receive massive blood transfusions (and hence large amounts of citrate) have increased incidence of developing sepsis and systemic inflammatory response syndrome44,45. When used as a regional anti-coagulant for CRRT, citrate has many advantages including increased circuit lifetimes and fewer bleeding complications8,9. However, it has long been recognized that some patients, especially those with liver dysfunction, are at risk of developing increased systemic citrate concentrations46. Recently a prospective study by Link et al. found that CRRT patients with total to ionized calcium ratios >2·4 (reflective of increased citrate concentrations) had a 33·5-fold increased risk of mortality47. We speculate that citrate's ability to modulate inflammatory responses could be playing a central role in increasing the development of SIRS in trauma patients and increasing mortality in CRRT patients with elevated citrate levels.
Outside its use as an anti-coagulant, high concentrations of citric acid are found in citrus fruits and citrate salts are used to alkalinize the urine in the treatment of kidney stones48,49. It is tempting to speculate if oral and dietary sources of citrate could alter inflammatory responses. Studies of the renal handling of citrate demonstrate that the increase in urinary citrate does not result directly from an increase in plasma citrate levels. Instead, the kidney increases urinary citrate excretion in response to a metabolic alkalosis, which can be induced by bicarbonate infusion or from the metabolism of citrate into bicarbonate50. These studies imply that, when taken orally, citrate is subject to extensive metabolism by the liver and that oral intake of citrate does not result in elevated plasma levels of citrate. In contrast, when citrate is used as an anti-coagulant, it is infused directly into the systemic circulation. Even then, elevated citrate levels are seen typically only in patients who receive massive blood product transfusions, have liver dysfunction or receive continuous citrate infusions with CRRT46,51,52. As such, we feel it is unlikely that citrate in food or other sources could produce elevated plasma levels of citrate such that it would impact monocyte inflammatory responses.
Lastly, the proinflammatory state caused by diabetes has been attributed to the effects of hyperglycaemia on modulating immune cell responses53,54. Monocytes in diabetic conditions show an altered phenotype leading to an increase in proinflammatory cytokine production55,56. In a hyperglycaemic state, monocytes show an increase in NF-κB localization, histone acetylation and histone acetyltransferase activity localized at inflammatory genes leading to increased cytokine production57,58. However, links between hyperglycaemia, increased protein acetylation and the inflammatory signal cascade have not been defined explicitly. Metabolism of glucose into citrate may provide a link in understanding the augmented inflammatory state seen in patients with diabetes.
Acknowledgments
The authors thank Dr M. Hershenson (University of Michigan) for the gifts of plasmid vectors. This work was supported by a grant from the Robert and Genevieve Berhalter Research Fund from the University of Michigan Department of Pediatrics. N. B. B. is supported by a Career Development Award from the National Institutes of Health (K08 DK093785). T. T. C. is supported by a Career Development Award from the National Institutes of Health (K08 HD062142).
Author contributions
M. J. A., K. L. M., J. J. P., P. L. C. and D. T. S. performed experiments; M. J. A., T. T. C., T. P. S. and N. B. B. designed the research and analysed and interpreted the data; M. J. A. and N. B. B. wrote the manuscript.
Disclosures
The authors declare no competing financial interests.
REFERENCES
- Garcia DA, Baglin TP, Weitz JI, Samama MM American College of Chest Physicians. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th edn. American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e24S–43S. doi: 10.1378/chest.11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao GS, Kim HT, Daley H, et al. Validation of short-term handling and storage conditions for marrow and peripheral blood stem cell products. Transfusion. 2011;51:137–47. doi: 10.1111/j.1537-2995.2010.02758.x. [DOI] [PubMed] [Google Scholar]
- Tolwani A, Wille KM. Advances in continuous renal replacement therapy: citrate anticoagulation update. Blood Purif. 2012;34:88–93. doi: 10.1159/000342378. [DOI] [PubMed] [Google Scholar]
- Engstad CS, Gutteberg TJ, Osterud B. Modulation of blood cell activation by four commonly used anticoagulants. Thromb Haemost. 1997;77:690–6. [PubMed] [Google Scholar]
- Chadha V, Garg U, Warady BA, Alon US. Citrate clearance in children receiving continuous venovenous renal replacement therapy. Pediatr Nephrol. 2002;17:819–24. doi: 10.1007/s00467-002-0963-6. [DOI] [PubMed] [Google Scholar]
- Mariano F, Bergamo D, Gangemi EN, Hollo Z, Stella M, Triolo G. Citrate anticoagulation for continuous renal replacement therapy in critically ill patients: success and limits. Int J Nephrol. 2011;2011:748320. doi: 10.4061/2011/748320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore PM, Jacobs R, Joannes-Boyau O, et al. Septic AKI in ICU patients. Diagnosis, pathophysiology, and treatment type, dosing, and timing: a comprehensive review of recent and future developments. Ann Intens Care. 2011;1:32. doi: 10.1186/2110-5820-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudemans-van Straaten HM, Kellum JA, Bellomo R. Clinical review: anticoagulation for continuous renal replacement therapy – heparin or citrate? Crit Care. 2011;15:202. doi: 10.1186/cc9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudemans-van Straaten HM, Ostermann M. Bench-to-bedside review: citrate for continuous renal replacement therapy, from science to practice. Crit Care. 2012;16:249. doi: 10.1186/cc11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MY, Hsu YH, Bai CH, Lin YF, Wu CH, Tam KW. Regional citrate versus heparin anticoagulation for continuous renal replacement therapy: a meta-analysis of randomized controlled trials. Am J Kidney Dis. 2012;59:810–8. doi: 10.1053/j.ajkd.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Akram M. Citric acid cycle and role of its intermediates in metabolism. 2014. Cell Biochem Biophys ; 68:475–8. [DOI] [PubMed]
- Watson JA, Fang M, Lowenstein JM. Tricarballylate and hydroxycitrate: substrate and inhibitor of ATP: citrate oxaloacetate lyase. Arch Biochem Biophys. 1969;135:209–17. doi: 10.1016/0003-9861(69)90532-3. [DOI] [PubMed] [Google Scholar]
- Infantino V, Convertini P, Cucci L, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. 2011;438:433–6. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. 2013;440:105–11. doi: 10.1016/j.bbrc.2013.09.037. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270–6. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–8. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–4. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Rossol M, Heine H, Meusch U, et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31:379–446. doi: 10.1615/critrevimmunol.v31.i5.20. [DOI] [PubMed] [Google Scholar]
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- Chen BC, Hsieh SL, Lin WW. Involvement of protein kinases in the potentiation of lipopolysaccharide-induced inflammatory mediator formation by thapsigargin in peritoneal macrophages. J Leukoc Biol. 2001;69:280–8. [PubMed] [Google Scholar]
- Letari O, Nicosia S, Chiavaroli C, Vacher P, Schlegel W. Activation by bacterial lipopolysaccharide causes changes in the cytosolic free calcium concentration in single peritoneal macrophages. J Immunol. 1991;147:980–3. [PubMed] [Google Scholar]
- Zhou X, Yang W, Li J. Ca2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol Chem. 2006;281:31337–47. doi: 10.1074/jbc.M602739200. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26:171–6. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Garofalo R, Sabry M, Jamaluddin M, et al. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol. 1996;70:8773–81. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell TT, Hinkovska-Galcheva V, Sun L, et al. Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8 production in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L849–56. doi: 10.1152/ajplung.90516.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Johnson XD, Iazvovskaia S, Tan A, Lin A, Hershenson MB. Signaling intermediates required for NF-kappa B activation and IL-8 expression in CF bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L307–15. doi: 10.1152/ajplung.00086.2002. [DOI] [PubMed] [Google Scholar]
- Blatt NB, Bednarski JJ, Warner RE, et al. Benzodiazepine-induced superoxide signals B cell apoptosis: mechanistic insight and potential therapeutic utility. J Clin Invest. 2002;110:1123–32. doi: 10.1172/JCI16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim NA, Sanford A, Sullivan KE. Histone acetylation and chromatin conformation are regulated separately at the TNF-alpha promoter in monocytes and macrophages. J Leukoc Biol. 2003;73:862–71. doi: 10.1189/jlb.1202618. [DOI] [PubMed] [Google Scholar]
- Sullivan KE, Reddy AB, Dietzmann K, et al. Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol. 2007;27:5147–60. doi: 10.1128/MCB.02429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrisano T, Pero R, Peluso S, et al. LPS-induced IL-8 activation in human intestinal epithelial cells is accompanied by specific histone H3 acetylation and methylation changes. BMC Microbiol. 2010;10:172. doi: 10.1186/1471-2180-10-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Liesegang GW, Berger RL, Czerlinski G, Podolsky RJ. A stopped-flow investigation of calcium ion binding by ethylene glycol bis(beta-aminoethyl ether)-N,N'-tetraacetic acid. Anal Biochem. 1984;143:188–95. doi: 10.1016/0003-2697(84)90575-x. [DOI] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–55. [PubMed] [Google Scholar]
- Houston B, Nimmo HG. Purification and some kinetic properties of rat liver ATP citrate lyase. Biochem J. 1984;224:437–43. doi: 10.1042/bj2240437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert J, Lindenbaum A. Complexes of calcium with citric acid and tricarballylic acids measured by ion exchange. Nature. 1950;166:913–4. doi: 10.1038/166913b0. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15:813–26. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Vander Heiden MG. Metabolism strikes back: metabolic flux regulates cell signaling. Genes Dev. 2010;24:2717–22. doi: 10.1101/gad.2010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale E, Zhu J, Chan L, Shulman I, Harwood R, Demetriades D. Blood transfusion in critically injured patients: a prospective study. Injury. 2006;37:455–65. doi: 10.1016/j.injury.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Sihler KC, Napolitano LM. Complications of massive transfusion. Chest. 2010;137:209–20. doi: 10.1378/chest.09-0252. [DOI] [PubMed] [Google Scholar]
- Meier-Kriesche HU, Gitomer J, Finkel K, DuBose T. Increased total to ionized calcium ratio during continuous venovenous hemodialysis with regional citrate anticoagulation. Crit Care Med. 2001;29:748–52. doi: 10.1097/00003246-200104000-00010. [DOI] [PubMed] [Google Scholar]
- Link A, Klingele M, Speer T, et al. Total-to-ionized calcium ratio predicts mortality in continuous renal replacement therapy with citrate anticoagulation in critically ill patients. Crit Care. 2012;16:R97. doi: 10.1186/cc11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penniston KL, Nakada SY, Holmes RP, Assimos DG. Quantitative assessment of citric acid in lemon juice, lime juice, and commercially-available fruit juice products. J Endourol Mar. 2008;22:567–70. doi: 10.1089/end.2007.0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattle D, Hess B. Preventive treatment of nephrolithiasis with alkali citrate – a critical review. Urol Res. 2005;33:73–9. doi: 10.1007/s00240-005-0464-8. [DOI] [PubMed] [Google Scholar]
- Simpson DP. Citrate excretion: a window on renal metabolism. Am J Physiol. 1983;244:F223–34. doi: 10.1152/ajprenal.1983.244.3.F223. [DOI] [PubMed] [Google Scholar]
- Diaz J, Acosta F, Parrilla P, et al. Correlation among ionized calcium, citrate, and total calcium levels during hepatic transplantation. Clin Biochem. 1995;28:315–7. doi: 10.1016/0009-9120(94)00094-c. [DOI] [PubMed] [Google Scholar]
- Kramer L, Bauer E, Joukhadar C, et al. Citrate pharmacokinetics and metabolism in cirrhotic and noncirrhotic critically ill patients. Crit Care Med. 2003;31:2450–5. doi: 10.1097/01.CCM.0000084871.76568.E6. [DOI] [PubMed] [Google Scholar]
- Pugliese G, Tilton RG, Williamson JR. Glucose-induced metabolic imbalances in the pathogenesis of diabetic vascular disease. Diabetes Metab Rev. 1991;7:35–59. doi: 10.1002/dmr.5610070106. [DOI] [PubMed] [Google Scholar]
- Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70:138–51. [PubMed] [Google Scholar]
- Jain SK, Kannan K, Lim G, Matthews-Greer J, McVie R, Bocchini JA. Elevated blood interleukin-6 levels in hyperketonemic type 1 diabetic patients and secretion by acetoacetate-treated cultured U937 monocytes. Diabetes Care. 2003;26:2139–43. doi: 10.2337/diacare.26.7.2139. Jr. [DOI] [PubMed] [Google Scholar]
- Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–64. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- Guha M, Bai W, Nadler JL, Natarajan R. Molecular mechanisms of tumor necrosis factor alpha gene expression in monocytic cells via hyperglycemia-induced oxidant stress-dependent and -independent pathways. J Biol Chem. 2000;275:17728–39. doi: 10.1074/jbc.275.23.17728. [DOI] [PubMed] [Google Scholar]
- Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279:18091–7. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]