

Abstract
Non-essential amino acid L-glutamine (Gln) possesses anti-inflammatory activity via deactivating cytosolic phospholipase A2 (cPLA2). We showed previously that Gln deactivated cPLA2 indirectly via dephosphorylating p38 mitogen-activated protein kinase (MAPK), the major kinase for cPLA2 phosphorylation, through inducing MAPK phosphatase-1 (MKP-1). In this study, we investigated the precise mechanism underlying Gln deactivation of cPLA2. In lipopolysaccharide (LPS)-treated mice, Gln injection resulted in dephosphorylation of phosphorylated cPLA2 (p-cPLA2), which coincided with rapid Gln induction of MKP-1. MKP-1 small interfering RNA (siRNA) abrogated the ability of Gln to induce MKP-1 as well as the dephosphorylation of cPLA2. Co-immunoprecipitation and in-situ proximity ligation assay revealed a physical interaction between MKP-1 and p-cPLA2. In a murine model of allergic asthma, we also demonstrated the physical interaction between MKP-1 and p-cPLA2. Furthermore, Gln suppressed various allergic asthma phenotypes, such as neutrophil and eosinophil recruitments into the airway, airway levels of T helper type 2 (Th2) cytokines [interleukin (IL)-4, IL-5 and IL-13], airway hyperresponsiveness, mucin production and metabolites (leukotriene B4 and platelet-activating factor) through inhibiting cPLA2 in a MKP-1-dependent manner. These data suggest that MKP-1 uses cPLA2, in addition to p38, as a substrate, which further potentiates the anti-inflammatory action of Gln.
Keywords: asthma, cPLA2, glutamine, inflammation, MKP-1
Introduction
Phospholipase A2 (PLA2) is a family of proteins that consists of five subtypes: cytosolic PLA2 (cPLA2), secreted PLA2, calcium-independent PLA2, platelet-activating factor (PAF) acetylhydrolase and lysosomal PLAs 1,2. Among PLA2s, many research studies have focused on cPLA2 due to its preference for hydrolyzing membrane glycerophospholipids at position sn-2 to form arachidonic acid (AA) 3. AA is converted to potent inflammatory lipid mediator eicosanoids, including leukotrienes (LTs), lipoxins, thromboxanes and prostaglandins, through catalytic action of lipoxygenase or cyclooxygenase 4,5. The involvement of cPLA2 in lipid mediator production makes it a potentially important pharmacological target to control inflammation and cancer. Despite its importance, the mechanisms for cPLA2 down-regulation have not yet been determined. Understanding the mechanisms of down-regulation of cPLA2 will provide a novel approach to improve the management of inflammatory diseases.
Non-essential amino acid L-glutamine (Gln), the most abundant amino acid in plasma 6, is an energy substrate for most cells 7,8. Consequently, Gln is important in multiple ways in the nitrogen- and carbon-skeleton exchange of different tissues 9. Human clinical trials with several patient populations have demonstrated that Gln treatment can decrease infectious complications, shorten hospitalization and decrease hospital costs 10. Additionally, extensive research studies using animal models of endotoxin shock, including severe injury models, have demonstrated that Gln supplementation can improve survival, enhance immune and gut barrier function, decrease bacteraemia and inhibit gut mucosal atrophy 11,12.
To understand the molecular mechanism of Gln anti-inflammatory activity, we have shown that Gln can protect animals effectively from endotoxin shock as well as inhibit bronchial allergic asthma by inhibiting cPLA2 phosphorylation 13,14. To decipher the mechanism of Gln inhibition of cPLA2, we demonstrated recently that Gln deactivated p38 and Janus kinase (JNK) mitogen-activated protein kinases (MAPKs) by rapidly inducing MAPK phosphatase (MKP)-1 protein 15, which preferentially dephosphorylated p38 and JNK 16,17, suggesting that Gln inhibited cPLA2 phosphorylation indirectly by blocking the kinase activity of p38, a major upstream pathway for cPLA2 phosphorylation 18.
In this study, we further investigated the precise mechanism of how Gln inhibits cPLA2 phosphorylation. We found a physical interaction between Gln-induced MKP-1 and phosphorylated cPLA2 (p-cPLA2). Our data suggest that MKP-1 can directly dephosphorylate cPLA2, thus further delineating the mechanism underlying Gln deactivation of cPLA2.
Materials and methods
Animals
Specific pathogen-free female BALB/c mice were purchased from Samtako Bio Korea (Osan, Korea) and kept in our animal facility for at least 1 week before use. Mice used at the start of each experiment were at 7–8 weeks of age. All experimental animals used in this study protocol were approved by the Institutional Animal Care and Use Committee of Chonbuk National University Medical School.
Cell culture
MH-S murine alveolar macrophage cells were grown in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1% antibiotics (Invitrogen) at 37°C in a 5% CO2 atmosphere.
Reagents
Lipopolysaccharide (LPS) from Escherichia coli (O127:B8) and L-Gln (biotechnology performance certified, G-8540) were purchased from Sigma-Aldrich (St Louis, MO, USA). LPS (2·5 mg/mouse) was injected intravenously (i.v.) via the lateral tail vein. Gln was dissolved in sterilized distilled water to 3%. A volume of 0·5 ml (750 mg/kg) 12–15 was administered intraperitoneally (i.p.) 10 min before or 10 min after LPS injection.
Specific cPLA2 inhibitor pyrrophenone (Cayman Chemical Company, Ann Arbor, MI, USA) was administered i.p. at 20 mg/kg 30 min before LPS injection. Triptolide (1 μM) (Calbiochem, Darmstadt, Germany), a MKP-1 inhibitor, was added to the culture 30 min before LPS stimulation. Antibodies against cPLA2 and p-cPLA2 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-MKP-1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunoblotting analysis
Mice were killed by cervical dislocation. Lungs were collected immediately thereafter, washed briefly with cold phosphate-buffered saline (PBS) and dried with blotting paper. Isolated lung tissues were frozen in liquid nitrogen and stored at −70°C until analysis. Small lung specimens were homogenized in PhosphoSafe Extraction Reagent (Novagen, Madison, WI, USA) and subjected to immunoblotting analysis, as described previously 15.
Immunoprecipitation
Lungs and cells were lysed in non-denaturing lysis buffer consisting of 20 mM Tris HCl pH 8, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM ethylendiamine tetraacetic acid (EDTA), protease inhibitor cocktail and phosphatase inhibitor. Equal amounts (100 μg) of cell or tissue extracts were incubated with primary antibodies for 4 h at 4°C in the same total volume of lysis buffer. Protein A/G-conjugated agarose beads (Santa Cruz Biotechnology) were added and incubated overnight. The beads were washed four times with lysis buffer and boiled to separate proteins. The beads were removed by centrifugation. The supernatant was used for Western blot.
RNA interference
Small interfering RNA (siRNA) targeting mouse MKP-1 and non-targeting siRNA controls were purchased from Santa Cruz Biotechnology. In-vivo siRNA transfection was performed using polyethyleneimine (PEI) purchased from Polyplus-tansfection (BP 90018 ; F-67401 Illkirch Cedex, France) according to the manufacturer's instruction. siRNA and PEI dissolved in 5% glucose was mixed in a volume of 200 μl for i.v. or 30 μl for intratracheal (i.t.) injection. The mixture was incubated for 15 min at room temperature and injected 24 h before LPS treatment 12.
In-situ proximity ligation assay (PLA)
Interaction of p-cPLA2 and MKP-1 was detected in situ by PLA technology. Duolink II fluorescence compound was purchased from Olink Bioscience (Uppsala, Sweden). The assay was performed according to the manufacturer's protocol. Briefly, 1 × 104 of MH-S cells were cultured in a covered glass-bottomed dish overnight and treated with LPS (0·1 μg/ml). Cells were washed with PBS for 50 min, fixed with 4% paraformaldehyde for 15 min and treated with cold 100% methanol for 10 min for membrane permeabilization. Samples were blocked with blocking solution for 30 min and incubated with primary antibodies (anti-p-cPLA2 at 1 : 500 or anti-MKP-1 at 1 : 500) at 4°C for 2 h. After washing twice for 5 min, samples were incubated with PLA probes at 37°C in a humidity chamber for 30 min and washed twice for 5 min. Oligonucleotides conjugated with PLA probes were ligated for 30 min and washed twice for 2 min. Ligated oligonucleotides were amplified for 60 min in a preheated humidity chamber and washed. Covered glass-bottomed dishes were mounted with mounting medium and covered with a coverslip. Samples were analysed by confocal microscopy.
Immunization and challenge
Mice were immunized i.p. with 20 μg OVA (grade V; Sigma-Aldrich) plus 1·0 mg aluminium hydroxide adjuvant (Imject® Alum; Pierce, Rockford, IL, USA) on day 0 and ovalbumin (OVA) alone without alum on day 14. The immunized mice were exposed to aerosolized OVA on days 28 and 35. Aerosolization of OVA was performed using a chamber adapted for mice. Animals were exposed to 1% OVA using a model NE-U12 ultrasonic nebulizer at output of 0·8 ml/min (Omron, Tokyo, Japan) for 30 min at first challenge and 10 min at second challenge 19. Gln (750 mg/kg) was administered i.p. immediately after the cessation of the second challenge. Pyrrophenone (20 mg/kg) was administered i.p. 30 min before the second airway challenge.
Bronchoalveolar lavage (BAL)
Mice were anaesthetized and the trachea was cannulated while gently massaging the thorax. Lungs were lavaged with 800 μl of saline. An equal volume of BAL fluid sample was centrifuged. The supernatant was collected and stored at −70° until cytokine assay. The pellet was resuspended in saline and cytospin preparations (5 min, 202 g) of BAL cells were stained with Diff-Quik (International Reagents, Kobe, Japan). Different cell types were enumerated based on morphology and staining profile.
Determination of airway hyperresponsiveness (AHR)
AHR was determined as changes in airway function after challenge by aerosolized methacholine using published procedures 14,19. After methacholine challenge, airway resistance (RL) data were collected continuously. Maximum RL values representing percentage of change from baseline after aerosol saline challenge were used to express changes in airway function.
Histological analysis
Lungs were fixed with 10% formalin. Tissues were embedded in paraffin. To detect mucin, sections (4 μm) were stained with periodic acid-Schiff and counterstained with haematoxylin.
Enzyme-linked immunosorbent assay (ELISA)
Levels of T helper type 2 (Th2) cytokines interleukin (IL)-4, IL-5 and IL-13 in BAL fluids were measured using ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). The lower limits of detection for the cytokines were as follows: IL-4 (>2 pg/ml), IL-5 (>7 pg/ml) and IL-13 (>1·5 pg/ml). Levels of lipid mediators, leukotriene B4 (LTB4) and PAF in the blood and BAL fluids were also measured using ELISA according to the manufacturer's instructions. The lower limits of detection for lipid mediators were: LTB4 (>13 pg/ml; Cayman Chemical Company) and PAF (>0·78 ng/ml; MyBiosource, San Diego, CA, USA).
Statistical analysis
Data were expressed as mean ± standard deviation (s.d.). Statistical comparisons were performed using one-way analysis of variance (anova) followed by Tukey's test. Statistically significant difference was considered when P-values were less than 0·05.
Results
Gln dephosphorylates p-cPLA2 in a MKP-1-dependent manner
In the course of our studies on Gln deactivation of cPLA2, we have found consistently that Gln could deactivate cPLA2 by dephosphorylating p-cPLA2 13,14,20. In this study, we confirmed this finding in the lungs of LPS-injected mice. Time kinetics of cPLA2 phosphorylation and MKP-1 induction are shown in Fig. 1a. When Gln was administered 10 min after LPS injection, Gln rapidly dephosphorylated the p-cPLA2 within 5 min, concurrent with cPLA2 phosphorylation (Fig. 1b). Gln administration also resulted in an early induction of MKP-1 protein (Fig. 1b), consistent with our previous report 15. MKP-1 siRNA potentiated cPLA2 phosphorylation and suppressed MKP-1 protein induction (Fig. 1c). In mice treated with MKP-1 siRNA, Gln could no longer dephosphorylate cPLA2 (Fig. 1d). To verify the MKP-1/cPLA2 linkage, cPLA2 phosphorylation and MKP-1 expression in response to LPS were evaluated in MH-S murine alveolar macrophage cells. The time kinetics of cPLA2 phosphorylation and MKP-1 induction are shown in Fig. 1e. Addition of Gln resulted in decreased phosphorylation of cPLA2 and early induction of MKP-1 (Fig. 1f). Inhibition of MKP-1 using MKP-1 inhibitor triptolide enhanced the magnitude of cPLA2 phosphorylation but reduced MKP-1 expression (Fig. 1g). Gln-mediated inhibition of cPLA2 phosphorylation was reversed by triptolide (Fig. 1h). Taken together, the observation that Gln deactivated cPLA2 by dephosphorylating it only when cPLA2 was phosphorylated in response to LPS in a MKP-1-dependent manner suggested that Gln could dephosphorylate cPLA2 directly through the induction of MKP-1.
Fig 1.
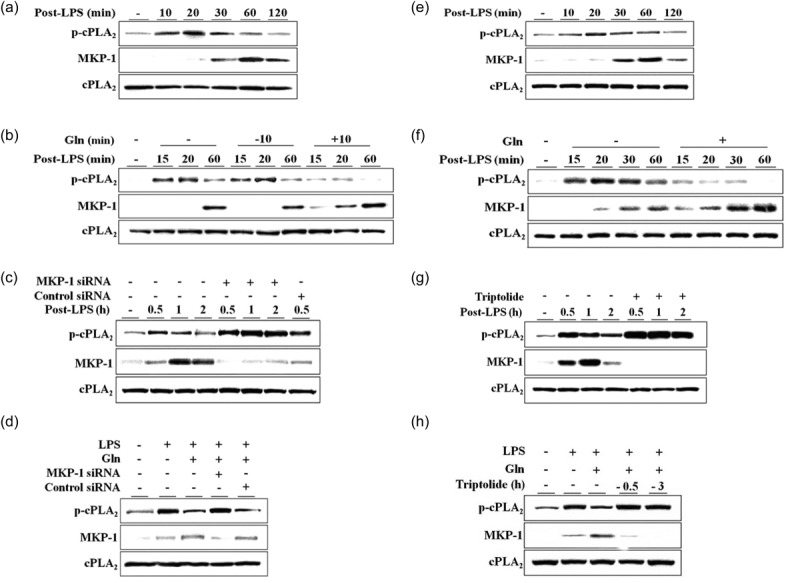
L-glutamine (Gln) dephosphorylates phosphorylated cPLA2 in a mitogen-activated protein kinase phosphatase-1 (MKP-1)-dependent manner. (a) Lungs were removed at the indicated time-points after lipopolysaccharide (LPS) injection [50 μg/mouse, intravenously (i.v.)]; (b) Gln [750 mg/kg, intraperitoneally (i.p.)] was injected 10 min before or 10 min after LPS and mice were killed at the indicated time-points. (c) MKP-1 small interfering RNA (siRNA) or control siRNA (0·4 nM each) was injected i.v. 24 h before LPS; (d) siRNA and Gln were injected 24 h before and 10 min after LPS, respectively; (e) LPS (0·1 μg/ml) was added to the culture medium and cells were harvested at the indicated time-points; (f,h) Gln (20 mM) was added 10 min after LPS; (g,h) triptolide (1 μM) was added 3 h before LPS. (a–d) A representative of three to five independent experiments with three mice/time-point/experiment is shown. (e–g) A representative of two or three independent experiments with three dished/time-point/experiment is shown.
Physical interaction between MKP-1 and p-cPLA2
How could Gln dephosphorylate p-cPLA2 within 5 min? cPLA2 phosphorylation is known to depend upon the kinase activity of p38. In fact, pretreatment of p38 inhibitor SB202190 inhibited cPLA2 phosphorylation significantly 20. Injection of Gln resulted in a rapid dephosphorylation of p38 through MKP-1 induction 15. However, the extent to which Gln-mediated cPLA2 dephosphorylation is p38-dependent remains to be elucidated. Phosphatase activity of MKP-1 could be another possible mechanism underlying Gln dephosphorylation of cPLA2 based on the finding that the activity of Gln was exclusively MKP-1-dependent. To test this hypothesis, we investigated whether or not there was a physical interaction between MKP-1 and cPLA2. Lung lysates from LPS-injected mice were co-immunoprecipitated using anti-MKP-1 and anti-p-cPLA2 antibodies targeting the Ser505 residue. MKP-1 was detected in immunoprecipitates using anti-p-cPLA2 (Fig. 2a). A reciprocal immunoprecipitation and Western blotting experiment using anti-MKP-1 antibody also detected p-cPLA2 (Fig. 2b). A co-immunoprecipitation assay using LPS-treated MH-S cells showed similar results (Fig. 2c,d). However, immunoprecipitates were not detected using anti-MKP-1 or anti-cPLA2 antibodies (Fig. 2e,f). These data support the theory that there was a physical interaction between MKP-1 and p-cPLA2, but not cPLA2 itself. In-situ PLA was performed to confirm further the physical interaction between MKP-1 and p-cPLA2. After MH-S cells were treated with LPS for 50 min, cells were probed with anti-p-cPLA2 and anti-MKP-1 antibodies and conjugated with oligonucleotide PLA probes. When the complementary PLA probes were in close proximity (<40 nm), oligonucleotide strands could be hybridized and amplified via the rolling circle method. As a result of this process, red dots were visualized. When viewed with a fluorescence microscope, the red fluorescence was observed mainly around nucleus (Fig. 2g). However, application of either anti-p-cPLA2 or anti-MKP-1 failed to produce red fluorescence, confirming that there was physical interaction between MKP-1 and p-cPLA2.
Fig 2.

Physical interaction between mitogen-activated protein kinase phosphatase-1 (MKP-1) and phosphorylated cytosolic phospholipase A2 (cPLA2). (a–d) Lung and cell lysates were immunoprecipitated using anti-p-cPLA2 and anti-MKP-1 antibodies. (e,f) Lung and cell lysates were immunoprecipitated using anti-cPLA2 antibody. (g) Cells were probed with anti-phospho-cPLA2 or anti-MKP-1 antibody and conjugated with oligonucleotide PLA probe: nucleus was counterstained with 4',6-diamidino-2-phenylindole (DAPI). A representative of two or three independent experiments with three mice/time-point/experiment is shown in (a–f). A representative of four independent experiments is shown in (g).
Gln inhibits the production of cPLA2 products
We assessed whether Gln inhibition of cPLA2 phosphorylation could lead to the inhibition of cPLA2 products LTB4 and PAF. Serum levels of LTB4 (Fig. 3a) and PAF (Fig. 3b) were increased progressively after LPS injection. Gln administration resulted in the inhibition of both LTB4 (Fig. 3c) and PAF (Fig. 3d). The suppressive activity of Gln was comparable to those of pyrrophenone, the specific cPLA2 inhibitor.
Fig 3.
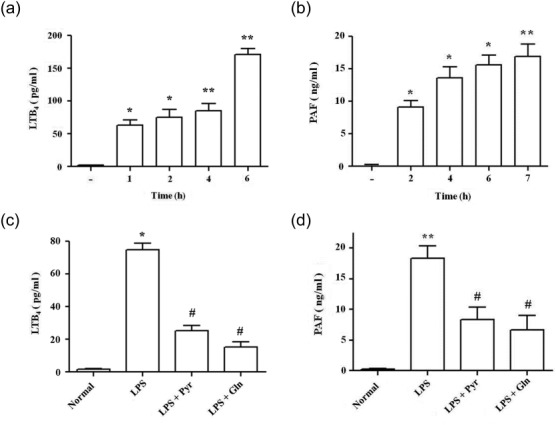
L-glutamine (Gln) and pyrrophenone suppress serum levels of leukotriene B4 (LTB4) and platelet activating factor (PAF). Serum levels of LTB4 (a) and PAF (b) were determined at the indicated time-points. Gln (750 mg/kg, i.p.) and pyrrophenone [20 mg/kg, intraperitoneally (i.p.)] were administered 10 min after and 30 min before LPS injection, respectively. LTB4 (c) and PAF (d) were determined at 1 and 4 h after LPS. Data representing the mean ± standard deviation (s.d.) of three independent experiments with three to five mice/group/experiment are shown. *P < 0·05 compared to saline control; **P < 0·01 compared to saline control; #P < 0·05 compared to the LPS group.
Physical interaction between MKP-1 and p-cPLA2 in asthmatic lungs
We have reported previously a beneficial effect of Gln on allergic asthma in mice 14,20. In this study, we assessed whether cPLA2/MKP-1 linkage was involved in the beneficial effect of Gln. Phosphorylation of cPLA2 was prominent at 10–30 min after the second airway challenge. Expression of MKP-1 was at 30–60 min after the cessation of the second OVA airway challenge (Fig. 4a). MKP-1 siRNA abrogated MKP-1 induction and potentiated cPLA2 phosphorylation in asthmatic lungs (Fig. 4b). Administration of Gln immediately after the cessation of the second airway challenge resulted in the dephosphorylation of cPLA2 and induced early strong induction of MKP-1 protein (Fig. 4c). MKP-1 siRNA reversed Gln inhibition of cPLA2 phosphorylation as well as MKP-1 induction (Fig. 4d). We assessed the interaction between MKP-1 and p-cPLA2 in asthmatic lungs. MKP-1 was detected in immunoprecipitates using anti-p-cPLA2 antibody (Fig. 4e). Immunoprecipitates using anti-MKP-1 antibody showed p-cPLA2 (Fig. 4f). Taken together, these data indicated that MKP-1-deactivated cPLA2, through their physical interaction, also occurred in asthmatic lungs.
Fig 4.
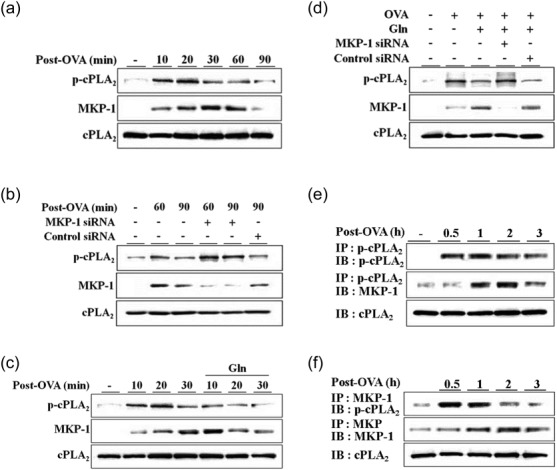
L-glutamine (Gln) deactivates phosphorylated (p)-cytosolic phospholipase A2 (cPLA2) through mitogen-activated protein kinase phosphatase-1 (MKP-1) in asthmatic lungs. After a second ovalbumin (OVA) airway challenge, lungs were removed at the indicated time-points (a–c). Small interfering RNAs (siRNAs) (0·2 nM) were administered intratracheally (i.t.) 24 h before the second challenge (b,d). Gln (750 mg/kg) was administered intraperitoneally (i.p.) immediately after the cessation of the second challenge (c,d). Lungs were removed at 10 min post-challenge (d). Lungs were removed at the indicated time-points. Lung lysates were immunoprecipitated using anti-p-cPLA2 or anti-MKP-1 antibodies (e,f). A representative of three to five independent experiments with three to five mice/time-point/experiment is shown.
Gln suppresses allergic pulmonary inflammation in a MKP-1-dependent manner
We evaluated the role of MKP-1 in the suppression of allergic asthmatic reactions by Gln using MKP-1 siRNA. Gln and pyrrophenone suppressed the recruitment of neutrophils (Fig. 5a) and eosinophils (Fig. 5b), but not macrophages (Fig. 5c), BAL fluid levels of Th2 cytokines (Fig. 5d), AHR (Fig. 5e) or mucus secretion (Fig. 5f). MKP-1 siRNA abrogated all the suppressive effects of Gln. We also assessed LTB4 and PAF in the airways. BAL fluid levels of LTB4 (Fig. 6a) and PAF (Fig. 6b) were increased progressively up to 3 h after challenge, but declined thereafter. Gln and pyrrophenone suppressed the BAL levels of both LTB4 and PAF. The suppressive effect of Gln was reversed by MKP-1 siRNA (Fig. 6c). These data indicate that Gln ameliorated allergic asthma by inhibiting cPLA2 after MKP-1 induction.
Fig 5.

Mitogen-activated protein kinase phosphatase-1 (MKP-1) Small interfering RNA (siRNA) abrogates the suppressive effect of L-glutamine (Gln) in allergic pulmonary inflammation. Small interfering RNAs (siRNA) and Gln were administered as described in Fig. 4. Pyrrophenone (Pyr; 20 mg/kg) was administered intraperitoneally (i.p.) 30 min before the second challenge. Bronchoalveolar lavage (BAL) fluids were collected 12 h (a), 48 h (b) and (c) and 18 h (d) after the second ovalbumin (OVA) challenge. Airway hyperresponsiveness (AHR) was performed at 48 h (e). Lungs were removed at 48 h post-challenge for mucin production (f). Data representing the mean ± standard deviation (s.d.) of three independent experiments with three to five mice/group/experiment are shown. *P < 0·05 compared to saline control; **P < 0·01 compared to saline control; #P < 0·05 compared to the OVA group; ##P < 0·05 compared to the Gln group.
Fig 6.
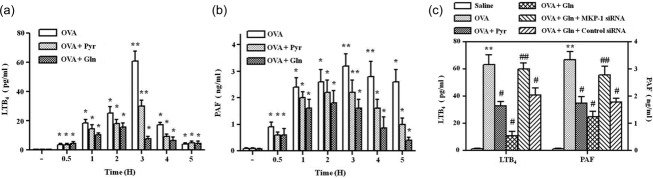
L-glutamine (Gln) and pyrrophenone suppress bronchoalveolar lavage (BAL) levels of leukotriene B4 (LTB4) and platelet activating factor (PAF). BAL fluid levels of LTB4 (a) and PAF (b) were determined at the indicated time-points. Gln [750 mg/kg, intraperitoneally (i.p.)] and pyrrophenone (Pyr; 20 mg/kg, i.p.) were administered immediately after and 30 min before the second airway challenge, respectively. (c) LTB4 and PAF were measured at 3 h. Data representing the mean ± standard deviation (s.d.) of three independent experiments with three to five mice/group/experiment are shown. *P < 0·05 compared to saline control; **P < 0·01 compared to saline control; #P < 0·05 compared to ovalbumin (OVA) challenge group.
Discussion
Our data demonstrate a physical interaction between Gln-induced MKP-1 and p-cPLA2, suggesting that Gln could deactivate cPLA2 directly via early induction of MKP-1. Our findings not only provided the underlying mechanism of Gln's anti-inflammatory activity, but also delineated a possible mechanism underlying the down-regulation of cPLA2. Gln deactivates p38 MAPK by rapidly inducing MKP-1 protein 15, which dephosphorylates p38 and JNK preferentially 16,17. Both MKP-1 induction and MKP-1 mediated p38 dephosphorylation within 5 min following Gln administration, suggesting that Gln dephosphorylated cPLA2 indirectly through MKP-1-mediated p38 dephosphorylation. However, Gln dephosphorylated p-cPLA2 rapidly within 5 min, as in the case of p38 in LPS-injected mice 13,14,20, suggesting that phosphatase is involved. Gln injection resulted in a rapid induction of MKP-1 within 5 min, which coincided with Gln-induced dephosphorylation of cPLA2 (Fig. 1b). MKP-1 siRNA abrogated the ability of Gln to induce dephosphorylation of cPLA2, suggesting that MKP-1 was involved in the dephosphorylation of cPLA2 by Gln. Our time-kinetic study revealed that Gln failed to deactivate cPLA2, consistent with our previous report 13. As shown in this and other studies 15,20, Gln dephosphorylation of cPLA2 is an exclusively MKP-1-dependent phenomenon. MKP-1 has been reported to be a labile protein that is normally degraded via the ubiquitin/proteasome pathway. Its phosphorylation reduces its ubiquitination and degradation 21,22 and phosphorylation of MKP-1 is mediated by transient activation of extracellular-regulated kinase (ERK), which enhances MKP-1 stabilization 23. Therefore, in the absence of ERK activation by an exogenous stimulus, Gln cannot up-regulate MKP-1 induction. This is the probable reason why Gln failed to deactivate cPLA2 when it was administered before LPS injection.
If MKP-1 could dephosphorylate cPLA2, these two molecules should interact physically with each other. Co-immunoprecipitation and in-situ PLA experiments revealed a physical interaction between MKP-1 and p-cPLA2, identifying cPLA2 as a novel MKP-1-interacting protein. In-situ PLA revealed red fluorescence in nucleus and the immediate vicinity. MKP-1 is a nuclear protein 24. Activated cPLA2 translocates from the cytosol to cell membranes, such as those of endoplasmic reticulum, Golgi apparatus and perinuclear region 5. Thus, MKP-1 may interact with cPLA2 during its translocation to the perinuclear membrane. These data suggest that Gln could deactivate cPLA2 directly by MKP-1-dependent cPLA2 dephosphorylation. Taken together, these data suggest that two mechanisms are involved in the Gln deactivation of cPLA2. The first mechanism is by indirect cPLA2 dephosphorylation through MKP-1-mediated p38 inactivation; the other mechanism is by direct physical interaction between Gln-induced MKP-1 and p-cPLA2.
In this study, we used anti-p-cPLA2 antibody to recognize phosphorylated Ser505. When MKP-1 was induced by Gln, the antibody could no longer detect p-cPLA2, indicating that MKP-1 could bind to at least the Ser505 residue of cPLA2. Further studies are required to address the possible involvement of phosphorylation site besides Ser505. MKP-1, an archetypal member of the MKP family, plays a pivotal role in the negative control of p38 and JNK. p38 has a role in the production of inflammatory molecules through both transcription-dependent mechanism and post-transcriptional regulation 25–29. MKP-1 functions as a critical negative regulator of inflammation. In a septic shock model, MKP-1-deficient mice produced greater amounts of proinflammatory cytokines and chemokines, with increased incidence and severity of multi-organ failure as well as higher mortality 30–32. These studies identified MKP-1 as an important target for the treatment of inflammatory diseases. Thus, developing novel ligands to up-regulate MKP-1 would be a therapeutically attractive anti-inflammatory strategy.
It is important to discuss the mechanism underlying Gln suppression of neutrophilic infiltration into the airway and Th2-dependent asthmatic phenotypes. Many investigators have reported an association between cPLA2 metabolites and Th2 response in asthmatic reactions 33–36. Importantly, LTs play a key role in dendritic cell influx into airways in asthma 37–39 which, in turn, results in inhibition of recruitment of T cells and eosinophils into asthmatic airways and late AHR. Regarding neutrophil infiltration, we reported previously that airway neutrophilia in the asthmatic lungs is mediated by LTB4 rather than CXC chemokines such as keratinocyte-derived chemokine and macrophage inflammatory protein (MIP)-2 20, which are principally chemotactic for neutrophils. Taken together, cPLA2 metabolites play an important role in induction of airway neutrophilia and Th2-dependent asthmatic phenotypes. As a result, Gln can suppress these parameters. In contrast to neutrophils and eosinophils, macrophage infiltration was not suppressed by Gln. This may be due to the fact that macrophage migration is not influenced by cPLA2 metabolites; therefore, the monocytes/macrophages chemokines, C-C chemokines, such as monocyte chemotactic protein-1 and MIP-1, are not inhibited by Gln. In fact, the C-C chemokines are not suppressed by Gln in asthmatic lungs (data not shown). cPLA2 has high selectivity in liberating arachidonic acid that is metabolized subsequently by a panel of downstream enzymes for eicosanoid production. Absence of cPLA2 in mice resulted in their resistance to a variety of inflammatory diseases 40–44, indicating the potential contribution of cPLA2 to inflammation. Therefore, cPLA2 has attracted considerable attention as a potential therapeutic target for the control of inflammation and cancer. Inhibition of cPLA2 as a pharmacological strategy has been proposed for the treatment of asthma 45, inflammatory skin diseases 46,47 and arthritis 48,49. Despite promising preclinical data, the use of cPLA2 inhibitors as a therapeutic approach in inflammatory diseases has not yet been confirmed in clinical trials, due probably to toxicity 50 or poor bioavailability 51. In this regard, our findings showing that Gln exerted a potent anti-inflammatory action by deactivating both p38 and cPLA2, probably the most potent enzymes involved in inflammatory reactions, might make Gln a useful therapeutic agent for many inflammatory diseases that involve cPLA2 in their pathogenesis.
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2008-0062279) and NRF-2014R1A2A2A01004207 and by research funds of Chonbuk National University in 2010.
Disclosures
The authors declare that there are no conflicts of interest.
References
- Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–92. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Leslie CC. Regulation of arachidonic acid availability for eicosanoid production. Biochem Cell Biol. 2004;82:1–17. doi: 10.1139/o03-080. [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Dhalen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–6. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- Niknami M, Patel M, Witting PK, Dong Q. Molecules in focus: cytosolic phospholipase A2-alpha. Int J Biochem Cell Biol. 2009;41:994–7. doi: 10.1016/j.biocel.2008.07.017. [DOI] [PubMed] [Google Scholar]
- Cynober LA. Plasma amino acid levels with a note on membrane transport: characteristics, regulation, and metabolic significance. Nutrition. 2002;18:761–6. doi: 10.1016/s0899-9007(02)00780-3. [DOI] [PubMed] [Google Scholar]
- Fox RE, Hopkins IB, Cabacungan ET, Tildon JT. The role of glutamine and other alternate substrates as energy sources in the fetal rat lung type II cell. Pediatr Res. 1996;40:135–41. doi: 10.1203/00006450-199607000-00023. [DOI] [PubMed] [Google Scholar]
- Encarnacion S, Calderon J, Gelbard AS, Cooper AJ, Mora J. Glutamine biosynthesis and the utilization of succinate and glutamine by Rhizobium etli and Sinorhizobium meliloti. Microbiology. 1998;144:2629–38. doi: 10.1099/00221287-144-9-2629. [DOI] [PubMed] [Google Scholar]
- Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. 1983;63:547–605. doi: 10.1152/physrev.1983.63.2.547. [DOI] [PubMed] [Google Scholar]
- Griffiths RD, Jones C, Palmer TE. Six-month outcome of critically ill patients given glutamine supplemented parenteral nutrition. Nutrition. 1997;13:295–302. [PubMed] [Google Scholar]
- Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological, and clinical importance. Q J Exp Physiol. 1985;70:473–89. doi: 10.1113/expphysiol.1985.sp002935. [DOI] [PubMed] [Google Scholar]
- Wischmeyer PE, Kahana M, Wolfson R, Ren H, Musch MM, Chang EB. Glutamine reduces cytokine release, organ damage, and mortality in a rat model of endotoxemia. Shock. 2001;16:398–402. doi: 10.1097/00024382-200116050-00014. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim GY, Kim JH, et al. Glutamine inhibits lipopolysaccharide-induced cytoplasmic phospholipase A2 activation and protects against endotoxin shock in mouse. Shock. 2006;25:290–4. doi: 10.1097/01.shk.0000194041.18699.6f. [DOI] [PubMed] [Google Scholar]
- Ko HM, Kang NI, Kim YS, et al. Glutamine preferentially inhibits T-helper type 2 cell-mediated airway inflammation and late airway hyperresponsiveness through the inhibition of cytosolic phospholipase A(2) activity in a murine asthma model. Clin Exp Allergy. 2008;38:357–64. doi: 10.1111/j.1365-2222.2007.02900.x. [DOI] [PubMed] [Google Scholar]
- Ko HM, Oh SH, Bang HS, et al. Glutamine protects mice from lethal endotoxic shock via a rapid induction of MAPK phosphatase-1. J Immunol. 2009;182:7957–62. doi: 10.4049/jimmunol.0900043. [DOI] [PubMed] [Google Scholar]
- Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272:16917–23. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- Hammer M, Mages J, Dietrich H, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–11. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Choi IW,Sun-Kim, Kim YS, et al. TNF-α induces the late-phase airway hyperresponsiveness and airway inflammation through cytosolic phospholipase A2 activation. J Allergy Clin Immunol. 2005;116:537–43. doi: 10.1016/j.jaci.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Lee CH, Kim HK, Kim JM, et al. Glutamine suppresses airway neutrophilia by blocking cytosolic phospholipase A(2) via an induction of MAPK phosphatase-1. J Immunol. 2012;189:5139–46. doi: 10.4049/jimmunol.1201599. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Small GW, Shi YY. Evidence that inhibition of p44/42 mitogen-activated protein kinase signaling is a factor in proteasome inhibitor-mediated apoptosis. J Biol Chem. 2002;277:27864–71. doi: 10.1074/jbc.M201519200. [DOI] [PubMed] [Google Scholar]
- Brondello JM, Pouyssegur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999;286:2514–17. doi: 10.1126/science.286.5449.2514. [DOI] [PubMed] [Google Scholar]
- Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16:769–79. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–26. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–8. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- Kracht M, Saklatvala J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine. 2002;20:91–106. doi: 10.1006/cyto.2002.0895. [DOI] [PubMed] [Google Scholar]
- Schieven GL. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem. 2005;5:921–8. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4:372–7. doi: 10.1016/j.coph.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang X, Nelin LD, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–40. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M, Mages J, Dietrich H, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA. 2006;103:2274–9. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myou S, Sano H, Fujimura M, et al. Blockade of eosinophil migration and airway hyperresponsiveness by cPLA2-inhibition. Nat Immunol. 2001;2:145–9. doi: 10.1038/84244. [DOI] [PubMed] [Google Scholar]
- Fischer AR, McFadden CA, Frantz R, et al. Effect of chronic 5-lipoxygenase inhibition on airway hyperresponsiveness in asthmatic subjects. Am J Respir Crit Care Med. 1995;152:1203–7. doi: 10.1164/ajrccm.152.4.7551371. [DOI] [PubMed] [Google Scholar]
- Irvin CG, Tu YP, Sheller JR, Funk CD. 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Physiol. 1997;272:L1053–8. doi: 10.1152/ajplung.1997.272.6.L1053. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Lewis JDB, Albert RK, et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. J Exp Med. 1996;184:1483–94. doi: 10.1084/jem.184.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J. 1999;14:12–8. doi: 10.1034/j.1399-3003.1999.14a04.x. [DOI] [PubMed] [Google Scholar]
- Parameswaran K, Liang H, Fanat A, Watson R, Snider DP, O'Byrne PM. Role for cysteinyl leukotrienes in allergen-induced change in circulating dendritic cell number in asthma. J Allergy Clin Immunol. 2004;114:73–9. doi: 10.1016/j.jaci.2004.03.054. [DOI] [PubMed] [Google Scholar]
- Del Prete A, Shao WH, Mitola S, Santoro G, Sozzani S, Haribabu B. Regulation of dendritic cell migration and adaptive immune response by leukotriene B4 receptors: a role for LTB4 in up-regulation of CCR7 expression and function. Blood. 2007;15:626–31. doi: 10.1182/blood-2006-02-003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–5. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- Hegen M, Sun L, Uozumi N, et al. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J Exp Med. 2003;197:1297–1302. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura C, Inada M, Matsumoto C, et al. An essential role of cytosolic phospholipase A2alpha in prostaglandin E2-mediated bone resorption associated with inflammation. J Exp Med. 2003;197:1303–10. doi: 10.1084/jem.20030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase T, Uozumi N, Ishii S, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–6. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- Marusic S, Leach MW, Pelker JW, et al. Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis. J Exp Med. 2005;202:841–51. doi: 10.1084/jem.20050665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen KA, Legault H, Hang C, et al. In vitro allergen challenge of peripheral blood induces differential gene expression in mononuclear cells of asthmatic patients: inhibition of cytosolic phospholipase A2alpha overcomes the asthma-associated response. Clin Exp Allergy. 2008;38:1590–605. doi: 10.1111/j.1365-2222.2008.03059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Davern LB, Stanley PL, et al. BMS- 229724 is a tight-binding inhibitor of cytosolic phospholipase A2 that acts at the lipid/water interface and possesses anti-inflammatory activity in skin inflammation models. J Pharmacol Exp Ther. 2001;298:376–85. [PubMed] [Google Scholar]
- Yamamoto M, Haruna T, Imura K, et al. Inhibitory effect of a potent and selective cytosolic phospholipase A2alpha inhibitor RSC-3388 on skin inflammation in mice. Pharmacology. 2008;81:301–11. doi: 10.1159/000117816. [DOI] [PubMed] [Google Scholar]
- Hegen M, Sun L, Uozumi N, et al. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J Exp Med. 2003;197:1297–302. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichel L, Berger S, Hadad N, et al. Reduction of cPLA2alpha overexpression: an efficient anti-inflammatory therapy for collagen-induced arthritis. Eur J Immunol. 2008;38:2905–15. doi: 10.1002/eji.200838545. [DOI] [PubMed] [Google Scholar]
- Risse D, Elfringhoff AS, Lehr M. Determination of the cell lytic properties of amphiphilic inhibitors of the cytosolic phospholipase A2 against human platelets by measuring the liberation of serotonin with high-performance liquid chromatography and fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;769:185–90. doi: 10.1016/s1570-0232(02)00013-2. [DOI] [PubMed] [Google Scholar]
- Lee KL, Behnke ML, Foley MA, et al. Benzenesulfonamide indole inhibitors of cytosolic phospholipase A2alpha: optimization of in vitro potency and rat pharmacokinetics for oral efficacy. Bioorg Med Chem. 2008;16:1345–58. doi: 10.1016/j.bmc.2007.10.060. [DOI] [PubMed] [Google Scholar]