

Abstract
The aging kidney undergoes structural and functional alterations which make it more susceptible to drug-induced acute kidney injury (AKI). Previous studies in our lab have shown that the expression of α(E)-catenin is decreased in aged kidney and loss of α(E)-catenin potentiates AKI-induced apoptosis, but not necrosis, in renal tubular epithelial cells (NRK-52E cells). However, the specific apoptotic pathway underlying the increased AKI-induced cell death is not yet understood. In this study, cells were challenged with nephrotoxicant cisplatin to induce AKI. A ~5.5-fold increase in Fas expression in C2 (stable α(E)-catenin knockdown) relative to NT3 (non-targeted control) cells was seen. Increased caspase-8 and -9 activation was induced by cisplatin in C2 as compared to NT3 cells. In addition, decreased Bcl-2 expression and increased BID cleavage and cytochrome C release were detected in C2 cells after cisplatin challenge. Treating the cells with cisplatin, in combination with a Bcl-2 inhibitor, decreased the viability of NT3 cells to the same level as C2 cells after cisplatin. Furthermore, caspase-3/-7 activation is blocked by Fas, caspase-8, caspase-9 and pan-caspase inhibitors. These inhibitors also completely abolished the difference in viability between NT3 and C2 cells in response to cisplatin. These results demonstrate a Fas-mediated apoptotic signaling pathway that is enhanced by the age-dependent loss of α(E)-catenin in renal tubule epithelial cells.
Keywords: Aging, AKI, α(E)-catenin, Apoptosis, Fas
Introduction
Aging is a major challenge facing scientists and doctors today because of the substantial increase in the human lifespan during the last century [1]. By 2050, it is expected that the number of individuals aged 60 or more will double, accounting for 11%, currently, to 22% of world’s population [2]. Several structural and functional alterations occur in the aging kidney which makes aging a major risk factor for acute kidney injury (AKI) [3]. Clinical studies performed in Spain showed the incidence of AKI is 3.5 times higher in aged patients (≥70 years) compared with those less than 70 years old [4]. In addition, increased medication use in elderly patients can also increase the incidence of AKI since nephrotoxic drugs are the cause for approximately 20% of AKI cases [5]. In our study, cisplatin, a widely used nephrotoxicant-induced AKI model, was used to investigate the pathophysiological mechanism of AKI in aged kidney [6].
α-catenin, which bridges the E-cadherin-β catenin complex and actin cytoskeleton, is essential for maintaining the integrity of the intercellular adherens junction [7]. There are three forms of α-catenin: neural (N), epithelial (E) and testis/heart (T) [8]. There is an increasing recognition that in addition to the well-established role in cell adhesion, α-catenin regulates multiple pathways controlling cell density, polarity, proliferation and apoptosis [9–11]. Previous studies in our lab have shown the expression of α(E)-catenin is dramatically decreased in proximal tubular epithelium cells in aged male Fisher 344 rats [12]. The decreased expression of α(E)-catenin is coupled with increased cisplatin induced apoptosis, rather than necrosis, in a caspase dependent manner [13].
The intrinsic and extrinsic pathways are two major caspase-dependent pathways to induce apoptosis, which are distinguished by the initiating signal [5]. The intrinsic pathway is triggered by cell stress-induced mitochondria outer membrane permeabilization (MOMP), resulting in the release of cytochrome c that activates caspase-9. The extrinsic pathway is initiated by the binding of apoptotic ligand to death receptors leading to the activation of caspase-8. Both intrinsic and extrinsic pathways will ultimately cleave caspase-3/7 which initiates the morphological changes of apoptosis [14]. In this study, the specific apoptotic pathway promoted by decreased α(E)-catenin was identified by using a stable α(E)-catenin knockdown cell line (C2 cells) generated in NRK-52E cells; NT3 cells will be used as the non-targeted control [15, 16]. These results provide the initial evidence that age-dependent loss of α(E)-catenin increases the susceptibility to acute kidney injury by facilitating the Fas-mediated apoptosis pathway in renal tubule epithelial cells.
Results
Target genes involved in apoptosis were assessed by RT2 Profiler PCR Array in NT3 and C2 cells. The gene expression (fold-change) in C2 cells relative to NT3 cells is depicted by the heat map with up-regulation in red and down-regulation in green (Fig. 1). The up-regulated genes include Fas, TNF-α related genes, caspases and pro-apoptotic Bcl-2 family members. The down-regulated genes include Card 10, II10 and Birc3, which are mainly anti-apoptotic [17].
Fig. 1. Apoptosis gene expression profiling of NT3 and C2 cells.

Apoptosis PCR Array analysis of mRNA from NT3 and C2 cells. A heat map shows the gene expression fold changes of C2 cells relative to NT3 cells with up-regulation in red and down-regulation in green.
Fas and TNF-α are two major death receptors that mediate the extrinsic apoptosis pathway [14]. Real-time PCR revealed the Fas mRNA was elevated 5.5-fold in C2 Cells relative to NT3 cells (Fig. 2A), which is consistent with the PCR Array result (Fig. 1). In addition, increased protein expression of Fas ligand (FasL) was detected in C2 cells but not NT3 cells after cisplatin treatment (Fig. 2B and 2C). The protein expression of Fas was constantly higher in C2 cells than in NT3 cells before and after cisplatin treatment (Fig. 2B and 2D). Confluent cultures of NT3 and C2 cells were challenged with cisplatin in combination with Kp7-6 (FasL/Fas antagonist) for 24h. C2 cells exhibited a significant loss of viability after cisplatin injury as compared with NT3 cells and the susceptibility difference between NT3 and C2 cells to cisplatin was completely abolished by Kp7-6 (Fig. 2F). Although C2 cells showed higher mRNA expression of TNF-α (Fig. 3A), no difference in TNF-α protein expression was observed between NT3 and C2 cells (Fig. 3B and 3C). Furthermore, pentoxifylline, a TNF-α inhibitor, failed to rescue C2 cells from the decreased viability as compared with NT3 cells in response to cisplatin (Fig. 3D and 3E). Taken together, these results indicate Fas, rather than TNF-α, mediates the increased cisplatin-induced apoptosis in C2 cells.
Fig. 2. The role of Fas in cisplatin induced injury in NT3 and C2 cells.

(A) Real time PCR of Fas in NT3 and C2 cells. The result is normalized to NT3 cells (N=3). Immunoblot analysis (B) and quantification of Fas (C) and FasL (D) in whole cell lysates of NT3 and C2 cells (N=3). The β-actin blot serves as the loading control. (E) Cell viability determined by MTT assay in cells treated with indicated concentrations of Kp7-6 alone or in the presence of 150 μM cisplatin for 24 h (N=3). The result is presented as the viability percentage of untreated control in SF media. The asterisks indicate significant differences between NT3 and C2 cells.
Fig. 3. The role of TNF-α in cisplatin induced injury in NT3 and C2 cells.

(A) Real time PCR of TNF-α in NT3 and C2 cells. The result is normalized to NT3 cells (N=3). Immunoblot analysis (B) and quantification of TNF-α (C) in whole cell lysates obtained from cells treated with 150 μM cisplatin for indicated time periods (N=2). The β-actin blot serves as the loading control. (D) Cell viability determined by MTT assay in cells treated with indicated concentrations of pentoxifylline and 150 μM cisplatin for 24 h (N=3). (E) Cell viability determined by MTT assay in cells treated with 150 μM cisplatin alone or in combine with 200 μg/ml pentoxifylline for the indicated time. The results are presented as the viability percentage of untreated control in SF media. The asterisks indicate significant differences between NT3 and C2 cells.
Caspase-8, a member of the cysteine proteases, is directly recruited by the Fas-associated death domain (FADD) [18]. A Caspase-Glo 8 Assay was performed to assess the activity of caspase-8. No significant difference was detected in the basal caspase-8 activity between NT3 and C2 cells. However, C2 cells had increased caspase-8 activation than NT3 cells after cisplatin treatment in a time-dependent manner (Fig. 4A). Interestingly, similar results were observed in a Caspase-Glo 9 Assay (Fig. 4B). Treating the cells with Z-IETD-FMK (caspase-8 inhibitor), Z-LEHD-FMK (caspase-9 inhibitor) and Z-VAD (pan-caspase inhibitor) abolished the difference between NT3 and C2 cells to cisplatin (Fig. 4C). Since caspase-9 is an initiator caspase, which plays a critical role in intrinsic apoptosis pathway [19], these data suggested the involvement of mitochondria in the pathway.
Fig. 4. The role of caspases in cisplatin induced injury in NT3 and C2 cells.

The activities of caspase-8 (A) and caspase-9 (B) in confluent cultures of cells treated with 150 μM cisplatin for indicated time periods were determined by luminescent assay (N=2). (C) Cell viability determined by MTT assay in cells treated with indicated combinations of cisplatin, Z-IETD-FMK, Z-LEHD-FMK and Z-VAD (N=3). The asterisks indicate significant differences between NT3 and C2 cells.
Members of the Bcl-2 family govern MOMP and have either anti-apoptotic (Bcl-2) or pro-apoptotic (BID) function [20]. Western blot was performed to measure the protein expression of Bcl-2 members (Fig. 5A). Increased BID cleavage was induced by cisplatin in C2 relative to NT3 cells (Fig. 5C). Correspondingly, lower Bcl-2 expression was observed in basal C2 cells. After cisplatin treatment, the expression of Bcl-2 was further decreased in C2 cells, but not in NT3 cells (Fig. 5D). This result was consistent with in vivo data. Aged kidneys have increased BID cleavage (Fig. 5E and 5F) and decreased Bcl-2 expression (Fig. 5E, 5F and 5G) compared to young kidney. Cisplatin induced a further increase of BID cleavage and decrease of Bcl-2 expression in aged kidney. However, the BID cleavage and Bcl-2 expression in young kidney were not influenced by cisplatin. Cytosolic cytochrome c was also measured (Fig. 5A), which revealed that cisplatin induced more cytochrome c release in C2 than NT3 cells (Fig. 5B). Importantly, ABT-199, a Bcl-2 inhibitor, decreased the viability of NT3 cells in a dose-dependent manner and completely abolished the viability difference between NT3 and C2 cells at the dose of 10nM (Fig. 5H). These results demonstrate a key role for the mitochondria in the apoptosis pathway and the decreased viability of C2 cells to cisplatin may be due to decreased anti-apoptotic Bcl-2 expression.
Fig. 5. The role of mitochondria in cisplatin induced injury in NT3 and C2 cells.

Immunoblot analysis (A) and quantification of cytosolic cytochrome c (B), BID (C) and Bcl-2 (D) in whole cell lysates obtained from cells treated with 150 μM cisplatin for indicated time periods (N=2). The β-actin blot serves as the loading control. Immunoblot (E) and quantification of BID (F) and Bcl-2 (G) in kidney tissue lysates obtained from 4- and 24-months-old rats at 72 h after saline or 2.75 mg/kg cisplatin IP injection. The pound indicates the significant difference between saline and cisplatin treated group (N=5). (H) Cell viability determined by MTT assay in cells treated with indicated concentrations of ABT-199 alone or in the presence of 150 μM cisplatin for 24 h (N=3). The asterisks indicate the significant differences between NT3 and C2 cells.
Caspase-3 and caspase-7 are effector caspases that can be activated by either the extrinsic (caspase-8) or intrinsic (caspase-9) pathway leading to apoptosis [21]. Previous studies in our lab showed increased caspase-3/-7 activation in C2 cells after cisplatin treatment [13]. A Caspase-Glo 3/-7 Assay was used to measure the activity of caspase-3/-7 (Fig. 6). Z-IETD-FMK, Z-LEHD-FMK and Z-VAD completely blocked caspase-3/-7 activation induced by cisplatin in NT3 and C2 cells, which indicates that caspase-3/-7 is downstream of both caspase-8 and 9 in the pathway. Interestingly, treating the cells with cisplatin in combination with Kp-6, a FasL/Fas antagonist, selectively decreased the cisplatin-induced caspase-3/-7 activation in C2 cells to the level of NT3 cells. These data demonstrate that loss of α(E)-catenin promotes apoptosis by increasing Fas signaling in renal tubule epithelial cells.
Fig. 6. Caspase-3/7 are the downstream of the Fas mediated apoptosis pathway.
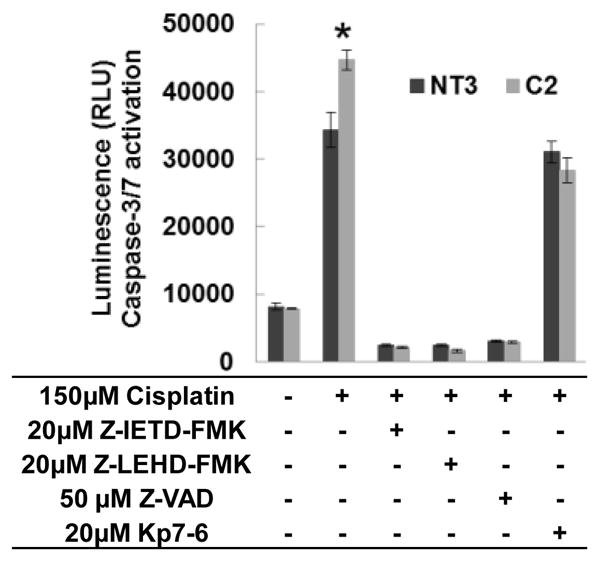
Caspase-3/7 activities in confluent cultures of cells treated with indicated combinations of cisplatin, Z-IETD-FMK, Z-LEHD-FMK, Z-VAD and Kp7-6 for 24 h, were determined by luminescent assay. The asterisks indicate the significant differences between NT3 and C2 cells.
Discussion
These results provide the first evidence that, in renal tubule epithelial cells, loss of α(E)-catenin increases the susceptibility to acute injury by enhancing a Fas mediated apoptosis pathway.
α-Catenin, previously known as a linking protein between the cadherin-β-catenin complex and actin cytoskeleton, has recently been shown to exert other functions [22]. Our study focused on the role of α-catenin in regulating cell death since renal tubular epithelium cell death is a hallmark of AKI [23]. Increased apoptosis has been reported in α-catenin deleted mammary glands of mice [24]. In addition, increased expression of p53 was coupled to decreased α-catenin expression in both gastric and lung cancer [25]. However, in epidermis or central nervous systems, deletion of α-catenin has been reported to have a protective effect from apoptosis by up regulating NF-κB [26]. α-catenin knockdown in epithelial cancer cells also attenuates DR4/DR5 mediated apoptosis [27]. In a myeloid leukemia cell line, reintroducing α-catenin leads to a decrease in cell proliferation and increase in apoptosis [28]. Hence, the role of α-catenin in cell death may depend on the cellular context. Our laboratory generated a stable α-catenin knockdown cell line (C2 cell) in NRK-52E cells. A previous study showed C2 cells are more susceptible to staurosporine, a commonly used drug to induce apoptosis, but not inorganic mercury which induces necrosis in renal epithelial cells [13]. Increased susceptibility to cisplatin was also detected in C2 cells, and data suggests that the increased susceptibility is due to increased apoptosis rather than necrosis [13].
Apoptosis, or programed cell death, is a fundamental process needed to maintain homeostasis [29]. Two major pathways have been demonstrated to induce apoptosis: the mitochondrial mediated intrinsic pathway and the receptor-dependent extrinsic pathway [30]. Treating the cells with cisplatin induced more caspase-8 and caspase-9 activation in C2 cells, which indicates both the intrinsic and extrinsic pathways are involved (Fig. 4A and 4B). The crosstalk between the extrinsic and intrinsic pathways occurs through caspase-8 [6]. High levels of caspase-8 directly cleaves caspase-3/-7 leading to apoptosis, while low levels of caspase-8 cleaves BID to truncated BID (tBID), which translocates to the mitochondria membrane to activate the intrinsic pathway by inducing MOMP [31, 32]. Our studies showed that Z-LEHD-FMK, the caspase-9 inhibitor, is able to abolish the viability difference between NT3 and C2 similar to Z-IETD-FMK, the caspase-8 inhibitor (Fig. 4C). This demonstrates that the cisplatin induced elevation of caspase-8 activity in C2 cells initiated the mitochondria mediated apoptosis pathway rather than leading to apoptosis directly. Concurrently, increased BID cleavage and cytochrome C release were detected in C2 cells after the cisplatin injury as compared to NT3 cells (Fig. 5A, 5B and 5C). Importantly, the expression of Bcl-2, which is an apoptosis inhibitor, was decreased in C2 cells (Fig. 5D). Meanwhile, increased BID cleavage and decreased Bcl-2 expression were also detected in aged rat kidney. The cisplatin challenge induced further increase of BID cleavage and decrease of Bcl-2 in C2 cells and aged kidney, but not in NT3 cells or young kidney. ABT-199, a Bcl-2 inhibitor, decreased viability of NT3 cells to the extent of C2 cells in response to cisplatin (Fig. 5H). These results demonstrate that the increased susceptibility of C2 cells to cisplatin is due to the increased activity of the caspase-8/mitochondria/caspase-9 pathway, and the decreased expression of Bcl-2, which works as a suppressor in this apoptosis pathway. Apoptosis can also be induced by endoplasmic reticulum (ER) stress [33]. However, this possibility was excluded because caspase-12 expression is not different between young and aged kidney (data not shown).
The receptor Fas, also called CD95, is a member of TNF receptor superfamily [34]. When Fas encounters its ligand FasL, the receptor trimerizes in the cytoplasmic domain and recruits the Fas-associated death domain protein (FADD). The death effector domain on FADD is capable of recruiting, cleaving and activating caspase-8 [35]. FasL is most prevalent in natural killer (NK) cells and T lymphocytes [31]. In the kidney, FasL has been reported to be expressed in tubular epithelial cells, renal endothelial cells, renal fibroblasts and mesangial cells [36]. Evidence has shown that FasL plays a key role in mediating cisplatin-induced AKI [37]. The expression of Fas is significantly reduced in cisplatin-resistant ovarian cancer epithelial cells, and up-regulation of Fas reverses the development of cisplatin resistance [38]. Epigallocatechin-3-gallate, a green tea polyphenol, and amifostine ameliorate cisplatin nephrotoxicity by inhibiting Fas-dependent apoptosis [39, 40]. FasL expression was increased dramatically in cisplatin exposed HeLa cells after centrocyte/centroblast marker 1 stimulation [41]. In our study, increased expression of Fas was detected in C2 cells (Fig. 1). Kp7-6, a Fas/FasL antagonist, abolished the cisplatin induced viability difference between NT3 and C2 cells (Fig. 2E). Most importantly, caspase-3/7 activity was decreased by Kp7-6 in C2 cells, but not in NT3 cells (Fig. 6). These data confirmed that increased Fas mediated apoptosis is the cause of increased susceptibility of C2 cells to acute injury. A possible scheme of the Fas apoptotic signaling pathway is shown in Fig. 7.
Fig. 7. Loss of α(E)-catenin promotes Fas mediated apoptosis pathway.

The expression of α(E)-catenin is decreased in aged kidney. Cisplatin causes further decrease of α(E)-catenin and promotes the Fas mediated apoptosis pathway. The pathway starts with the activation of caspase-8 and the cleavage of BID. The truncated BID (tBID) translocates to the surface of the mitochondria inducing cytochrome c release into the cytosol. The cytosolic cytochrome c activates caspase-9, followed by the activation of downstream caspase-3/-7. Bcl-2 is a negative regulator which inhibits the release of cytochrome c from mitochondria.
In conclusion, this study showed that age dependent loss of α(E)-catenin in renal tubule epithelial cells facilitate the Fas mediated apoptotic signaling pathway in response to cisplatin-induced AKI injury. This result may explain, in part, the increased incidence of AKI in aged kidney and shed light on the development of prospective treatments to AKI in aged patients.
Materials and Methods
Materials
The following chemicals were used in the experiments. cis-Diamineplatinum(II) dichloride (Sigma-Aldrich Cat #P4394); Kp7-6 (CalBiochem Cat #341291); Pentoxifylline (Sigma-Aldrich Cat #P1784); Z-IETD-FMK (CalBiochem Cat #218759); Z-LEHD-FMK (CalBiochem Cat #218761); Z-VAD (OMe)-FMK (CalBiochem Cat #627610); ABT-199 (Selleckchem Cat # S8048).
Animals
Male Fisher 344 rats (4-, and 24-month-old) were obtained from the NIA colony. Animals were randomly assigned to the cisplatin treated group and saline control group with n=5 each. Animals received a single intraperitoneal (IP) injection of 2.75 mg/kg cisplatin, or an equal volume of saline as control. Animals were placed in metabolic cages overnight before harvesting. 72 h after cisplatin injection, rats were anesthetized with ketamine (80–120 mg/kg)/xylazine (5–10 mg/kg) via IP injection. Kidney tissue lysates were obtained to perform western blot. All animal experiments and care were approved by the University of Missouri Animal Care and Use Committee in accordance with the National Institutes of Health (NIH).
Cell Culture
C2 (single cell clone of stable a(E)-catenin knockdown) and NT3 (vector control) cells were generated and cultured as previously described [15, 16]; and used within 20 passages of establishing the clonal cell line for all studies. For these experiments, cells were plated at a density of 5×104 cells/cm2 and cultured in DMEM/F12 (1:1) with L-glutamine and HEPES (Gibco Cat #11039-021) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals Cat #S11150), 5 μg/ml puromycin dihydrochloride (Sigma Cat #P9620) and incubated at 37°C in 5% CO2. Cells were harvested with TrypLE™ Express (Gibco Cat #12604-021) and pelleted at 1500 rpm for 5 min. at room temperature (RT).
RT2 Profiler PCR Array
Total RNA was extracted using the RNeasy mini kit (Qiagen Cat #74104) and subjected to cDNA synthesis using the RT2 Easy First Strand Kits (Qiagen Cat # 330421). Genes associated with apoptosis were assessed by real time RT2 Profiler Rat Apoptosis PCR Array (Qiagen Cat# PARN-012Z) following the kit protocol.
MTT Assay
Cells were seeded in a 96 well flat bottom tissue culture plate (Sigma Cat #Z707910) at a density of 5×104 cells/cm2. After 24h, culture media was replaced by serum free (SF) media supplemented with desired treatments. Three hours before harvest, 10 μl of 5 mg/ml MTT (Sigma Cat #M2128), dissolved in DPBS (Gibco Cat #14190-144), was added to each well. Upon harvesting, cells were washed with cold DPBS and dissolved by adding 50 μl solubilization solution (10% Triton X-100, 0.1N HCl in isopropanol). The plates were read at 570/690 nm on the Synergy HT Multi-Detection Microplate Reader (BioTek, Winooski, VT). The results are expressed by percent viability as [Abs570-690 treated/Abs570-690 control×100].
Real-Time PCR
1×107 cells were harvested and suspended in 1 ml PBS. RNA was isolated using the NucleoSpin (Clotech Cat #740955) followed by determining the RNA concentration using NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA). cDNA was generated from 2 μg RNA using the High Capacity cDNA Synthesis Kit (Life Technologies Cat #4368814). Quantitative PCR was performed in duplicate using 50 ng cDNA/reaction via Taqman assays with SsoFast™ Probes Supermix with ROX (Bio-Rad Cat #172-5251) and the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA). The following cycling conditions were used: 95°C for 20 sec., then 95°C for 1 sec and 60°C for 20 sec repeated 40 times. Commercially available TaqMan primer sets were used to assess Fas (Rn00685720_m1) (Life technologies Cat #4331182) and TNF-α (Rn01525859_g1) (Life technologies Cat #4453320). Relative quantitation was performed using the Pfaffl method [1] normalized to Casc3.
Caspase Activity Assay
Confluent cultures of NT3 and C2 cells in 96 well plates were challenged with desired treatments. Caspase activity was determined by Caspase-Glo® 3/7 Assay (Promega Cat #G8091), Caspase-Glo® 8 Assay (Promega Cat #G8201) and Caspase-Glo® 9 Assay (Promega Cat #G8211), according to the manufacturer’s instructions.
Western Blot
Subconfluent cells were washed twice with ice-cold DPBS and lysed with lysis buffer (10mM Tris-HCl, 1% SDS) containing Halt™ Protease/Phosphatase inhibitors (Thermo Scientific Cat #78444). Cells were scraped and incubated on a rocker for 15 min at 4°C. Cells were further disrupted by pipetting 15 times and spun at 12,000 g for 15 min at 4°C. Protein concentration was determined by NanoDrop 2000c Spectrophotometer at 280 nm.
The following antibodies were used: Cytochrome c Release Assay Kit (GeneTex Cat #GTX85531), TNF-α (NOVUS Cat #NB600-587), BID (NOVUS Cat #NB100-56106), Bcl-2 (Cell Signaling Cat #2876), Fas (Abcam Cat #15285), FasL (Abcam Cat #82419) and anti-β-actin (Sigma Cat #A2228). Goat-anti-mouse HRP conjugate and Goat-anti-rabbit HRP conjugate (Jackson ImmunoResearch Laboratories, Cat #115035003 and 305035003) were used at 1:20,000 dilutions. Blots were developed using SuperSignal West Femto Chemiluminescent Substrate (Pierce Cat #34095), imaged using the ChemiDoc™ imaging system (Bio-Rad, Hercules, CA), and quantitation performed using the ImageLab 3.0 software (Bio-Rad, Hercules, CA).
Statistics
Results are expressed as mean ± S.E. A two-way analysis of variance (ANOVA) was performed with the exception of Fig. 2A, 2C, 2D and 3A in which a one-way ANOVA was performed, followed by Student’s t-test using the statistical software GraphPad Prism 6 (GraphPad Software, La Jolla, CA). The differences were considered statistically significant when p<0.05.
Acknowledgments
The authors contributed the following to the work: experiments (XW), manuscript preparation (XW, ARP). Research reported in this publication was supported by the National Institute of Aging of the National Institutes of Health under award number RO1AG034154.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Funding: This study was funded by the National Institute of Aging of the National Institutes of Health under award number RO1AG034154
- Conflict of Interest: The authors declare that they have no conflicts of interest.
- All animal experiments and care were approved by the University of Missouri Animal Care and Use Committee in accordance with the National Institutes of Health (NIH).
- This article does not contain studies with human participants
References
- 1.Stevens PE, Lamb EJ, Levin A. Integrating Guidelines, CKD, Multimorbidity, and Older Adults. Am J Kidney Dis Off J Natl Kidney Found. 2014 doi: 10.1053/j.ajkd.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 2.WHO | Facts about ageing. WHO; 2014. [Accessed 19 Feb 2015]. http://www.who.int/ageing/about/facts/en/ [Google Scholar]
- 3.Wang X, Bonventre JV, Parrish AR. The aging kidney: increased susceptibility to nephrotoxicity. Int J Mol Sci. 2014;15:15358–15376. doi: 10.3390/ijms150915358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pascual J, Orofino L, Liaño F, Marcén R, Naya MT, Orte L, Ortuño J. Incidence and prognosis of acute renal failure in older patients. J Am Geriatr Soc. 1990;38:25–30. doi: 10.1111/j.1532-5415.1990.tb01592.x. [DOI] [PubMed] [Google Scholar]
- 5.Peres LAB, da Cunha AD. Acute nephrotoxicity of cisplatin: molecular mechanisms. J Bras Nefrol. 2013;35:332–340. doi: 10.5935/0101-2800.20130052. [DOI] [PubMed] [Google Scholar]
- 6.Marquez RT, Tsao BW, Faust NF, Xu L. Drug Resistance and Molecular Cancer Therapy: Apoptosis Versus Autophagy. Apoptosis 2013 [Google Scholar]
- 7.Sun Y, Zhang J, Ma L. α-catenin. Cell Cycle Georget Tex. 2014;13:2334–2339. doi: 10.4161/cc.29765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folmsbee SS, Morales-Nebreda L, Van Hengel J, Tyberghein K, Van Roy F, Budinger GR, Bryce PJ, Gottardi CJ. The cardiac protein αT-catenin contributes to chemical-induced asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308:L253–258. doi: 10.1152/ajplung.00331.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell. 2001;104:605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 10.Herr KJ, Tsang YN, Ong JWE, Li Q, Yap LL, Yu W, et al. Loss of α-catenin elicits a cholestatic response and impairs liver regeneration. Sci Rep. 2014;4:6835. doi: 10.1038/srep06835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, et al. Alpha-Catenins Control Cardiomyocyte Proliferation by Regulating Yap Activity. Circ Res. 2015;116:70–79. doi: 10.1161/CIRCRESAHA.116.304472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung K-Y, Dean D, Jiang J, Gaylor S, Griffith WH, Burghardt RC, Parrish AR. Loss of N-cadherin and alpha-catenin in the proximal tubules of aging male Fischer 344 rats. Mech Ageing Dev. 2004;125:445–453. doi: 10.1016/j.mad.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Grunz-Borgmann EA, Parrish AR. Loss of α(E)-catenin potentiates cisplatin-induced nephrotoxicity via increasing apoptosis in renal tubular epithelial cells. Toxicol Sci Off J Soc Toxicol. 2014;141:254–262. doi: 10.1093/toxsci/kfu130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated Cell Death in AKI. J Am Soc Nephrol JASN. 2014;25:2689–2701. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nichols LA, Grunz-Borgmann EA, Wang X, Parrish AR. A role for the age-dependent loss of α(E)-catenin in regulation of N-cadherin expression and cell migration. Physiol Rep. 2014 doi: 10.14814/phy2.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nichols LA, Slusarz A, Grunz-Borgmann EA, Parrish AR. α(E)-catenin regulates BMP-7 expression and migration in renal epithelial cells. Am J Nephrol. 2014;39:409–417. doi: 10.1159/000362250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beach JA, Nary LJ, Hovanessian R, Medh RD. Correlation of glucocorticoid-mediated E4BP4 upregulation with altered expression of pro- and anti-apoptotic genes in CEM human lymphoblastic leukemia cells. Biochem Biophys Res Commun. 2014;451:382–388. doi: 10.1016/j.bbrc.2014.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruidering M, Evan GI. Caspase-8 in apoptosis: the beginning of “the end”? IUBMB Life. 2000;50:85–90. doi: 10.1080/713803693. [DOI] [PubMed] [Google Scholar]
- 19.Yano T. Cellular and molecular mechanisms in drug-induced nephrotoxicity and its prevention. Nihon Yakurigaku Zasshi Folia Pharmacol Jpn. 2013;142:172–177. doi: 10.1254/fpj.142.172. [DOI] [PubMed] [Google Scholar]
- 20.Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and Molecular Targeting Therapy in Cancer. BioMed Res Int. 2014 doi: 10.1155/2014/150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 22.Maiden SL, Hardin J. The secret life of α-catenin: moonlighting in morphogenesis. J Cell Biol. 2011;195:543–552. doi: 10.1083/jcb.201103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagawa S, Omura T, Yonezawa A, Yano I, Nakagawa T, Matsubara K. Extracellular nucleotides from dying cells act as molecular signals to promote wound repair in renal tubular injury. Am J Physiol Renal Physiol. 2014;307:F1404–F1411. doi: 10.1152/ajprenal.00196.2014. [DOI] [PubMed] [Google Scholar]
- 24.Nemade RV, Bierie B, Nozawa M, Bry C, Smith GH, Vasioukhin V, et al. Biogenesis and function of mouse mammary epithelium depends on the presence of functional alpha-catenin. Mech Dev. 2004;121:91–99. doi: 10.1016/j.mod.2003.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nozawa N, Hashimoto S, Nakashima Y, Matsuo Y, Koga T, Sugio K, et al. Immunohistochemical alpha- and beta-catenin and E-cadherin expression and their clinicopathological significance in human lung adenocarcinoma. Pathol Res Pract. 2006;202:639–650. doi: 10.1016/j.prp.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Kobielak A, Fuchs E. Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A. 2006;103:2322–2327. doi: 10.1073/pnas.0510422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu M, Marsters S, Ye X, Luis E, Gonzalez L, Ashkenazi A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell. 2014;54:987–998. doi: 10.1016/j.molcel.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 28.Benjamin JM, Nelson WJ. Bench to bedside and back again: molecular mechanisms of alpha-catenin function and roles in tumorigenesis. Semin Cancer Biol. 2008;18:53–64. doi: 10.1016/j.semcancer.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schleich K, Krammer PH, Lavrik IN. The chains of death. Cell Cycle. 2013;12:193–194. doi: 10.4161/cc.23464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–542. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- 31.Maher S, Toomey D, Condron C, Bouchier-Hayes D. Activation-induced cell death: the controversial role of Fas and Fas ligand in immune privilege and tumour counterattack. Immunol Cell Biol. 2002;80:131–137. doi: 10.1046/j.1440-1711.2002.01068.x. [DOI] [PubMed] [Google Scholar]
- 32.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 33.Gao Z, Liu G, Hu Z, Li X, Yang X, Jiang B, Li X. Grape seed proanthocyanidin extract protects from cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Mol Med Rep. 2014;9:801–807. doi: 10.3892/mmr.2014.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fouqué A, Debure L, Legembre P. The CD95/CD95L signaling pathway: a role in carcinogenesis. Biochim Biophys Acta. 2014;1846:130–141. doi: 10.1016/j.bbcan.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 36.Lorz C, Ortiz A, Justo P, González-Cuadrado S, Duque N, Gómez-Guerrero C, Egido J. Proapoptotic Fas ligand is expressed by normal kidney tubular epithelium and injured glomeruli. J Am Soc Nephrol. 2000;11:1266–1277. doi: 10.1681/ASN.V1171266. [DOI] [PubMed] [Google Scholar]
- 37.Linkermann A, Himmerkus N, Rölver L, Keyser KA, Steen P, Bräsen JH, et al. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int. 2011;79:169–178. doi: 10.1038/ki.2010.317. [DOI] [PubMed] [Google Scholar]
- 38.Yang F, Long W, Xuechuan H, Xueqin L, Hongyun M, Yonghui D. Upregulation of Fas in epithelial ovarian cancer reverses the development of resistance to Cisplatin. BMB Rep. 2015;48:30–5. doi: 10.5483/BMBRep.2015.48.1.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou P, Song J, Jiang B, Pei F, Chen B, Yang X, Liu G, Hu Z. Epigallocatechin-3-gallate protects against cisplatin nephrotoxicity by inhibiting the apoptosis in mouse. Int J Clin Exp Pathol. 2014;7:4607–4616. [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y, Liu Y, Xu L, Guo M. Protective effect of amifostine on cisplatin-induced nephrotoxicity and its mechanism. Zhonghua Zhong Liu Za Zhi. 2006;28:8–12. [PubMed] [Google Scholar]
- 41.Park GB, Kim D, Yoon HS, Kim YS, Lee HK, Kim KT, et al. Antibody ligation of CM1 on cisplatin-exposed HeLa cells induces apoptosis through reactive oxygen species-dependent Fas ligand expression. Int J Oncol. 2014;44:2016–2024. doi: 10.3892/ijo.2014.2361. [DOI] [PubMed] [Google Scholar]