

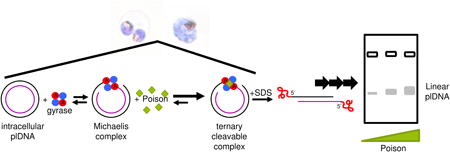
Abstract
Drug-resistant malaria poses a major public health problem throughout the world and the need for new antimalarial drugs is growing. The apicoplast, a chloroplast-like organelle essential for malaria parasite survival and with no counterpart in humans, offers an attractive target for selectively toxic new therapies. The apicoplast genome (plDNA) is a 35 kb circular DNA that is served by gyrase, a prokaryotic type II topoisomerase. Gyrase is poisoned by fluoroquinolone antibacterials that stabilize a catalytically inert ternary complex of enzyme, its plDNA substrate, and inhibitor. We used fluoroquinolones to study the gyrase and plDNA of Plasmodium falciparum. New methods for isolating and separating plDNA reveal four topologically different forms and permit a quantitative exam of perturbations that result from gyrase poisoning. In keeping with its role in DNA replication, gyrase is most abundant in late stages of the parasite lifecycle, but several lines of evidence indicate that even in these cells the enzyme is present in relatively low abundance: about 1 enzyme for every two plDNAs or a ratio of 1 gyrase:70 kb DNA. For a spectrum of quinolones, correlation was generally good between antimalarial activity and gyrase poisoning, the putative molecular mechanism of drug action. However, in P. falciparum there is evidence for off-target toxicity, particularly for ciprofloxacin. These studies highlight the utility of the new methods and of fluoroquinolones as a tool for studying the in situ workings of gyrase and its plDNA substrate.
Keywords: Malaria, plDNA, Gyrase, Topoisomerase, Apicoplast, Fluoroquinolone
Graphical abstract
Introduction
Among the world's top infectious disease killers (along with HIV/AIDS and tuberculosis), malaria infects an estimated 207 million people and kills 630 thousand annually, most of whom are children in Africa under the age of 5 [1-3]. The eukaryotic protozoan parasite that causes malaria belongs to the genus Plasmodium and is transmitted by female anopheline mosquitoes. Many Plasmodium species cause malaria, but P. falciparum, subject of the current work, is most lethal. Malaria is preventable and controllable through a combination of vector control methods, limitation of mosquito and human interaction, and antimalarial drugs. Unfortunately, malaria vaccines remain experimental and many of the available chemotherapies now face widespread resistance. There is a pressing need to identify novel drug targets in the malaria parasite and to develop new antimalarial drugs.
Although a human may simultaneously harbor multiple stages of the lifecycle of P. falciparum, it is the cycling, asexually replicating obligate intraerythrocytic parasites that cause symptomatic clinical disease [1]. After entering a red cell, the initial ring form contains one haploid copy of the nuclear genome. As it evolves into a trophozoite, the parasite enlarges, importing large amounts of glucose, ingesting host cytoplasm and having highly active metabolism. Finally, during schizogony the parasite undergoes multiple rounds of DNA replication and organellar division before cytokinesis. Within a single erythrocyte, a mature schizont may comprise 8-32 progeny, each with a haploid copy of the nuclear genome. The erythrocyte ruptures and releases progeny merozoites, which invade other erythrocytes and reinitiate the cycle. For P. falciparum the erythrocytic cycle lasts ∼48 h.
The malaria parasite has three genomes. Sequence of the 23 Mb, 14 chromosome, nuclear genome of P. falciparum was reported in 2002 [4]. It is exceptionally AT rich, averaging 81% overall and nearly 90% for introns and intergenic regions. At just 6 kb, the mitochondrial genome is the smallest mtDNA known and is arranged in head-to-tail tandem arrays [5]. The third genome is housed in the apicoplast, an organelle intensely studied as a therapeutic target because it has no counterpart in humans [6,7]. The apicoplast is related to chloroplasts and is the site of biosynthetic pathways for isoprenoids, heme, iron-sulfur clusters and fatty acids. Most apicoplast proteins are encoded in the nucleus and imported, but critical components are also provided by its own DNA. The 35-kb circular apicoplast genome (plDNA) has an AT content of ∼87% and is reportedly present in 1 to 15 copies per parasite [8-10]. Both strands of plDNA encode genes required for transcription and translation, and a large inverted repeat accounts for one-third of the genome, coding for 9 duplicated tRNAs and the large- and small-subunit rRNA genes [11,12].
Believed to be of prokaryotic origin, the apicoplast is the target for several antibacterials that have antimalarial action, including doxycycline, clindamycin, azithromycin, and ciprofloxacin. Although parasites treated with these drugs are themselves apparently unaffected, their progeny fail to proliferate even after removal of drug, leading to a phenomenon termed “second cycle effect” [13,14] or, more recently, “delayed cell death” [15].
Topoisomerases are enzymes that change the topology of DNA and are essential for orderly replication and metabolism of nucleic acids and for cell survival [16]. Type II enzymes make a double-stranded break in substrate DNA by tyrosine-mediated cleavage of the phosphodiester backbone of both strands of the helix. The 5′-ends are held in transient covalent linkage to the active site tyrosines, and topology of the DNA is altered by transferring segments of DNA through the double-strand break. The enzyme then religates strands to complete the reaction. Given its three structurally different genomes in distinct subcellular compartments, it is not surprising that P. falciparum encodes multiple type II topoisomerases. One, with homology to mammalian topoisomerase II, is thought to service the nuclear genome. Two additional nuclear genes were found to encode proteins homologous to the A- and B-subunits of DNA gyrase, a prokaryotic type II enzyme capable of negatively supercoiling DNA. Both subunits contain apicoplast-targeting signals and the B subunit has been shown to immunolocalize to the apicoplast [17-19].
The type II topoisomerases are targets for clinically valuable anti-tumor and anti-infective agents (e.g., etoposide, ciprofloxacin). These drugs belong to an important subset of topoisomerase inhibitors, termed poisons, that bind only to a preformed enzyme-DNA Michaelis complex, forming a ternary “cleavable complex,” comprised of DNA, protein and poison [20]. The presence of a poison prevents ligation of the DNA strands, rendering the topoisomerase inert and bound to DNA. In situ, collision of DNA-tracking machinery with fixed cleavable complexes creates the lethal double-stranded breaks that underpin drug efficacy. In the laboratory, addition of a rapid denaturant such as sodium dodecyl sulfate (SDS), leads to protein-DNA adducts with enzyme covalently linked to the 5′-ends of its substrate DNA via phosphotyrosine bonds, a phenomenon that has provided valuable insights into the enzyme's function within the cell [20-22]. Previous studies have demonstrated the sensitivity of malaria parasites and their apicoplast gyrase to various quinolones [23,24].
To further study the effect of quinolones on P. falciparum we developed methods that provide reproducibly accurate parasite counts and that entail minimal manipulation of the parasites or of their plDNA. Using these techniques we treated intraerythrocytic malaria parasites with quinolones, studied the relationship of drug structure with antimalarial activity and with poisoning of apicoplast or nuclear DNA, and asked whether gyrase poisoning correlates with parasite death.
2. Materials and Methods
2.1. Fluoroquinolones and topoisomerase-targeting compounds
Compounds were stored desiccated at -20 °C. Quinolones were dissolved in autoclaved HPLC-grade water or NaOH (both JT Baker, Phillipsburg, NJ) solution; etoposide (Sigma, St. Louis, MO) was dissolved in DMSO (Sigma, St. Louis, MO). Tested were: ciprofloxacin HCl, clinafloxacin (Santa Cruz Biotechnology, Santa Cruz, CA); potassium fleroxacin (Hoffman-La Roche, Basel, Switzerland); gatifloxacin (Selleck Chemicals, Houston, TX); levofloxacin, sodium nalidixate, oxolinic acid, piromidic acid (Sigma, St. Louis, MO); norfloxacin (Merck Sharpe & Dohme, Whitehouse Station, NJ); ofloxacin (Robert Wood Johnson Pharm, NJ); and pefloxacin methanesulfonate (Rhône Poulenc Rorer now Sanofi-Aventis, Bridgewater, NJ).
2.2. Cultivation and synchronization of P.falciparum
P. falciparum NF54 was maintained in O+ erythrocytes in RPMI, 25 mM HEPES, 27 mM sodium bicarbonate (all Sigma, St. Louis, MO) and 10% heat-inactivated O+ serum (Interstate Blood Bank, Inc., Memphis, TN) in non-vented flasks under 3% O2, 4% CO2, 93% N2. All experiments were done at 37 °C. Human erythrocytes were obtained weekly from healthy donors under an IRB-approved protocol. Parasites were maintained in 1.2% or 2.4% hematocrit. To synchronize, cells were pelleted (1500 × g, 10 min), resuspended in 5% sorbitol (Sigma, St. Louis, MO) (5 min), resedimented, and resuspended in fresh medium [25].
2.3. Parasites, counts, imaging
To determine number of parasitized erythrocytes per mL of culture, 25 μL culture was fixed with 25 μL 10% formalin (3.7% formaldehyde) (Sigma, St. Louis, MO) and stained with 200 μL 0.1% gentian (crystal) violet (Sigma, St. Louis, MO) in 0.8% NaCl (Fisher, Fair Lawn, NJ), 0.02% Na2EDTA (J.T. Baker, Phillipsburg, NJ). Rings, trophozoites and schizonts were counted by hemocytometer. To confirm parasitemia thin smears were fixed with methanol (J.T. Baker, Phillipsburg, NJ), stained with 100 μg/mL acridine orange (Sigma, St. Louis, MO) in 1× PBS (Quality Biologicals Inc., Gaithersburg, MD), visualized (ParaLens Fiber Optic Illuminator, Becton Dickinson), and ∼1000 erythrocytes were examined to determine the percent infected. Thin smears were stained with Giemsa (Accustain, Giemsa Stain, Modified, Sigma St. Louis, MO). Photomicrographs were obtained with a Zeiss Axiphot and Prog Res C14 plus camera.
2.4. Determination of antimalarial activity
A modified microtiter plate-based assay of [3H]hypoxanthine metabolic labeling was used [26]. Briefly, 100 μL P. falciparum at 0.25% parasitemia was incubated for indicated times with 100 μL medium containing solvent or serially diluted drug; each concentration was assayed in quadruplicate. At 40 to 48h, 0.64 μCi [3H]hypoxanthine (10-30 Ci (370GBq-1.11TBq)/mmol; Perkin Elmer, Boston, MA) was added to each well, the plate was incubated (20-24 h), samples from each well were harvested, and tritium counts were measured. All tested compounds achieved 100% efficacy. Curve fitting and EC50 were obtained with the Emax model [27] and DeltaGraph Pro 3.5. Outliers identified by Chauvenet's criterion [28] were discarded from analysis. Each compound was assayed in at least three, and usually four, independent experiments, conducted over several months' time, and by different personnel.
2.5. Purification of apicoplast DNA
Apicoplast DNA was isolated by Qiagen Plasmid Mini Kit (Qiagen Sciences, Germantown, MD). Infected erythrocytes were sedimented, washed twice with and resuspended in PBS with 1% glucose (Fisher, Fair Lawn, NJ) (PBSG), then lysed with 0.05% saponin (Fluka Analytical, Germany). Released parasites were washed twice in PBSG, resuspended in P1 buffer (purchased as such; per Qiagen Sciences this is 50 mM Tris HCl, pH 8.0, 10 mM EDTA, 100 μg/mL RNase A) then processed per manufacturer's instructions. The DNA pellet was resuspended in TE (10 mM Tris HCl, pH 8.0 (Fisher, Fair Lawn, NJ), 1 mM EDTA). Apicoplast DNA markers were prepared from Qiagen-purified plDNA by digestion with EcoR I (New England Biolabs, Ipswich, MA) or exposure to UV irradiation (Fotodyne Transilluminator 3-3000, 120 V; 60 mm from lamp, 30 min).
2.6. Preparation of pJAZZ-OC LSU
A 1.2 kb fragment of plDNA's 2.7 kb large subunit rRNA gene (LSU) (GenBank X61660.1) was cloned into the 12.2 kb linear pJAZZ-OC vector (Big Easy v2.0 Linear Cloning Kit, Lucigen, Middleton, WI). The LSU fragment with NotI (New England Biolabs, Ipswich, MA) ends was amplified by PCR (denature 94 °C; anneal 58 °C; extend 65 °C) from EcoR I-linearized Qiagen-purified plDNA with the following primers: forward 5′GGTGGTGCGGCCGCAGGAACTCGGCAATTTAATCTTG3′; reverse 5′GGTGGTGCGGCCGCATAATTAAATATTCAACTTATTAGGAATTATAC3′. PCR product was digested (NotI, 90 min), gel purified and ligated into NotI-digested pJAZZ-OC. BigEasy TSA cells were electroporated (BTX ElectroSquare Porator ECM 830) per manufacturer's instructions, plated on YT with 30 μg/mL chloramphenicol (Boehringer Mannheim, Basel, Switzerland), 20 μg/mL Xgal (BioExpress, Kaysville, UT) and 1 mM IPTG (Boehringer Mannheim, Basel, Switzerland). Colonies were selected and amplified, plasmid DNA was isolated, and constructs were verified by NotI digestion and sequencing of both strands. The pJAZZ-OC LSU final product, containing only one copy of the 1.2 kb LSU fragment, was dialyzed and its concentration determined by A260, using 1 AU = 50 μg/mL; A260:A280 was 1.84.
2.7. Drug treatment of parasites, isolation and analysis of plDNA by Southern hybridization
An equal number of parasitized erythrocytes (per hemocytometer count of gentian violet-stained cells) was aliquoted into flasks, exposed to drug or solvent, sedimented, resuspended in a small volume of medium with drug or solvent, and transferred to a Thermo-Scientific 20K MWCO dialysis cup immersed in 37 deg;C dialysis buffer (5 mM Tris HCl, pH 8.0, 37.5 mM Na2EDTA, 20 mM NaCl). Parasites were lysed within the cup by an equal volume of 2% SDS (Fisher, Fair Lawn, NJ), 4 mg/mL Proteinase K (Invitrogen/Life Technologies, Grand Island, NY), 10 mM Tris HCl, pH 8.0, 75 mM Na2EDTA, 40 mM NaCl; dialysis buffer was maintained at 37 °C and changed at least twice over ≥2 h. 10 μg/mL RNase A (Sigma, St. Louis, MO) and 20 U/mL RNase T1 (Invitrogen/Life Technologies, Grand Island, NY) (final concentrations) were added (>30 min) and dialysis buffer was changed once. Samples were transferred to microfuge tubes and 100% sucrose (J.T. Baker, Phillipsburg, NJ) loading buffer was added.
DNA was separated by electrophoresis through 0.4% agarose (Denville Sci., Metuchen, NJ) (20 × 25 cm; 1X TAE (40 mM Tris acetate (Sigma, St. Louis, MO), 2 mM EDTA); 1-1.3 V/cm; 40-48 h) at room temperature with circulating buffer. When indicated, gels were cast and run in TAE with ethidium bromide (EtBr) (Sigma, St. Louis, MO). DNA was EtBr-stained for UV visualization, transferred to and UV crosslinked to Hybond XL or N+ membrane (GE Healthcare, Pittsburg, PA) and hybridized [29] with 32P-labeled probe prepared by random primer method from 1.2 kb LSU template DNA (Section 2.6) amplified with forward primer 5′AGGAACTCGGCAATTTAATCTTG and reverse primer 5′ATAATTAAATATTCAACTTATTAGGAATTATAC). Washed membranes were exposed to a PhosphorImager plate that was scanned (Fuji BAS 2500 PhosphorImager) and visualized (Image Gauge version 3.45; Fuji Photo Films Co).
2.8. Analysis of plDNA-topoisomerase complexes
Parasitized erythrocytes were exposed to drug and lysed with SDS (Section 2.7). While dialyzing, the lysates were incubated with either 4 mg/mL Proteinase K or 4 mg/mL cOmplete EDTA-free protease inhibitor cocktail (37°C, >2 h) (Roche, Nutley, NJ), then digested with RNases. Dialyzed samples were transferred into 1.5 mL microfuge tubes and adjusted to 80 μL with lysis buffer. After addition of 20 μL 325 mM KCl (J.T. Baker, Phillipsburg, NJ), samples were vortexed, incubated (37 °C, 10 min), placed on ice (10 min), and centrifuged (15 min, 4 °C). Supernatants were analyzed for plDNA by Southern hybridization (Section 2.7).
2.9. Precipitation of nuclear DNA-topoisomerase complexes
The KSDS assay, of triplicate samples, was performed as described previously [30]. Briefly, parasites grown in [3H]hypoxanthine (1 μCi/200 μL culture, 18-22 h) were washed, aliquoted, treated with drug or solvent (30 min) and lysed (2% SDS, 10 mM Na2EDTA pH 8.0, 0.8 mg/mL sheared calf-thymus DNA (Invitrogen, Grand Island, NY)). Lysates were incubated with 2 mg/mL Proteinase K or 10 mg/mL protease inhibitor cocktail (37°C, 1 h) then RNase A and RNase T1 (37 °C, 30 min). Samples were incubated (50 °C, 10 min) prior to addition of 100 μL 65 mM KCl, then placed on ice (10 min) and centrifuged (16,000 × g, 4°C, 15 min). Pellets were washed, twice, by being resuspended in 1 mL buffer (10 mM Tris HCl, pH 8.0, 100 mM KCl, 1 mM Na2EDTA 8.0, 0.1 mg/mL calf thymus DNA), incubated (50 °C, 10 min) to dissolve, then placed on ice (10 min) and re-sedimented. Radioactivity in final dissolved pellets was counted by liquid scintillation. For total incorporation of radioactivity into nucleic acids, 50 μL aliquots of lysate, digested or not with RNase, were applied to glass filters, washed with ice cold 5% trichloroacetic acid (J.T. Baker, Phillipsburg, NJ), and counted. Percent DNA covalently linked to protein in RNase-digested lysates was calculated by dividing the radioactivity in KCl-precipitates by that in trichloroacetic acid precipitates.
2.10. Statistical Analyses
Analyses were calculated in Excel for Mac 2011; all values are Mean ± Standard Deviation and p values were obtained from unpaired, two-tailed student's t-test.
3. Results
3.1. Gentian violet stain of malaria parasites and parasite density determination
One objective of this work was to determine the number of plDNA molecules per parasite, and for this purpose we needed an accurate and reproducible method for sampling and analyzing a known number of parasitized erythrocytes. To calculate parasitized erythrocytes per mL of culture, the conventionally used and inexact combination of parasitemia (percent infected erythrocytes, obtained from dried and stained slide preparations) and hematocrit (volume of erythrocytes per mL of culture, assuming 1% hematocrit ≈ 108 erythrocytes per mL) was not satisfactory. Instead we directly measured parasitized erythrocytes per mL of culture by using a method in which cells are stained with gentian violet, visualized and counted by hemocytometer while still suspended in culture medium. The gentian violet method most importantly provided a direct and accurate measure of parasitized erythrocytes per mL of culture, but simultaneously it also afforded a differential count of the lifecycle stages (Fig. 1). Relative to the time-honored Giemsa method, neither acridine orange nor gentian violet counts were statistically significantly different for percent parasitemia, or percent rings, trophozoites or schizonts (p values all >0.05; 5 independent experiments over a period of one month). In this report, gel loading was based on the gentian violet-determined number of infected red cells in the volume of culture taken for analysis (“parasitized erythrocytes/lane”).
Fig. 1.

P. falciparum malaria parasites in human erythrocytes. Thin smears of cells in asynchronous culture were dried on glass slides, fixed with methanol, stained with either (A) Giemsa or (B) acridine orange and examined by light or fluorescence microscopy, respectively. Acridine-stained preparations were excited at 390 nm and emission at 420 nm was captured. (C) Cells in suspension were fixed with formalin, stained with gentian violet and a wet mount was examined by light microscopy. Size marker, 10 μm.
3.2. Characterization of plDNA forms
To optimize analysis of plDNA, numerous methods were tested including saponin release of parasites from erythrocytes, dialysis of lysates in various apparatuses, application of lysates directly into agarose gel wells or via agarose plugs, and widely differing electrophoretic conditions (data not shown). The method that permitted drug exposure and lysis of parasites while still within erythrocytes, that minimized manipulation of large circular plDNA, and that resulted in crisp separation of plDNA forms was SDS lysis of drug-treated parasitized erythrocytes within a Thermo-Scientific dialysis cup, then separation of DNA in the dialysate by conventional electrophoresis in TAE buffer (Section 2.7).
P. falciparum lysates prepared by this method yielded four distinct forms of plDNA: nicked circles, linears, covalently closed circles, and a still-unidentified “form IV” (Fig. 2A, lane 1). Based on quantitation of Southern blots, these comprise 15 ± 4.5, 6.6 ± 1.1, 68 ± 3.8, and 10 ± 1.2 percent of the population, respectively (5 independent experiments; analysis of nicked forms is confounded by the low but overlapping signal from cross-reacting genomic DNA). The most abundant form, covalently closed circles, was assigned based on its relaxation by topoisomerase IB (data not shown) and by the effect of EtBr on its electrophoretic migration, which was retarded and smeared in 1 μg/mL ethidium (Fig. 2B, middle panel, lane 3) but accelerated and compacted in 5 μg/mL ethidium (Fig. 2B, right panel, lane 3). These results are consistent with negatively supercoiled covalently closed plDNA circles that are progressively unwound by ethidium to produce slow-migrating relaxed forms at low concentrations, and compact fully positively supercoiled forms at saturating concentrations of the intercalator. Negative supercoils in covalently closed plDNA circles are in keeping with the in situ co-existence of an apicoplast DNA gyrase.
Fig. 2.

Separation and identification of plDNA forms. (A) Asynchronous cultures of P. falciparum were processed by our method (lane 1) or by Qiagen Kit (lanes 2-4), separated in 0.4% agarose by conventional electrophoresis in TAE buffer, and plDNA was visualized by Southern blotting. Qiagen-purified samples were untreated (lane 2), digested with EcoR I to obtain a linearized plDNA marker (lane 3) or exposed to UV light to obtain a marker for nicked plDNA circles (lane 4). All samples 2 × 107 parasitized erythrocytes/lane. (B) Samples were separated by gel electrophoresis in the presence of zero, 1 or 5 ug/mL EtBr, as indicated. Linear plDNA marker (lanes 1); nicked circle plDNA marker (lanes 2); P. falciparum plDNA prepared by our method (lanes 3, at 1.2 - 2.5 × 107 parasitized erythrocytes/lane). For all panels, the indistinct smear migrating just above nicked molecules is likely non-specific hybridization with genomic DNA. N, nicked circular plDNA; IV, form Four; L, linearized circles; CC, covalently closed circular plDNA. Arrow indicates well.
Qiagen purification yielded an overall lower recovery of plDNA, with selective loss of some forms and the genesis of an unidentified new form, not seen in our method, that migrated just above covalent circles (Fig. 2A, lane 2). EcoR I digestion or UV irradiation of Qiagen-purified plDNA yielded linearized and nicked plDNA markers, respectively, generated at the expense of covalently closed circles (Fig. 2A, lanes 3 and 4). Linearized plDNA migrates between 29 and 32 kb, when compared to the Invitrogen 1kb extension ladder. With or without EtBr during electrophoresis, the bands assigned as nicked or linearized circles comigrated with the nicked or linear markers, supporting their correct identification (Fig. 2B). Despite the many variables that can affect electrophoretic mobility, the nicked, linear and supercoiled covalently closed forms of 35 kb plDNA migrate in the same relative order as do comparable forms for plasmid molecules a fraction of their size.
3.3. Number of plDNA molecules per parasite
Literature reports indicate there are 1 to 15 copies of plDNA/parasite [8-10]. To quantify plDNA content more precisely we prepared an internal standard comprising a single copy fragment of the large subunit rRNA gene (LSU) within the 12.2 kb linear pJAZZ-OC vector. This large vector was selected so the construct would migrate in the vicinity of plDNA. Known concentrations of the construct were electrophoresed alongside samples containing a defined number of twice-synchronized ring form parasites (≥ 98% rings). Rings were chosen because they precede the reported onset of DNA synthesis in the lifecycle [31] and because each ring can reasonably be construed as a single parasite. Parasite samples and serial dilutions of the construct were separated in the same gel, visualized by Southern blotting (Fig. 3A), and signal intensities were quantified (Fig. 3B). A standard curve of pJAZZ-OC LSU was used to interpolate plDNA content in parasite samples. In three replicate experiments, a total of ten parasite samples (4 independent cultures, three of which were sampled in triplicate) were analyzed, and all four plDNA forms were quantified. From these data we estimate there are 18 ± 4.9 plDNA molecules per ring stage parasite. With a 23 Mb nuclear genome, plDNA thus accounts for less than 3% of the parasite's total DNA.
Fig. 3.

Quantification of plDNA per parasite. Cells were synchronized twice with sorbitol (0 and 48 h), early ring forms were collected at 50 h, counted, and the DNA was processed in duplicate, separated by electrophoresis, blotted and probed for plDNA. (A) Lanes 1 and 2, duplicate samples from 1.1 × 107 parasitized erythrocytes. Lanes 3-7, 0.63, 1.27, 2.54, 5.08 or 10.2 ng pJAZZ-OC LSU construct. N, nicked circular plDNA; IV, form Four; L, linearized circles; CC, covalently closed circular plDNA, Construct, pJAZZ-OC LSU. Arrow indicates well. (B) Standard curve of pJAZZ-OC LSU generated from signal intensities in Panel A and known mass amounts determined by UV absorption; R2 = 0.96.
Experiments with synchronous populations enriched for trophozoites or schizonts indicate that trophozoites contain approximately the same number of plDNA molecules as do rings (Fig. 4, compare total mass of plDNA in lanes 3 and 6), and that schizonts have the greatest number (Fig. 4, compare lanes 3 and 9). Correcting for residual rings and trophozoites in the schizont sample, and for parasitized erythrocytes loaded per lane, schizonts appear to have ∼6 times the number of plDNA molecules as do rings or trophozoites (that is, ∼108 copies/parasitized erythrocyte). This is in keeping with plDNA replication occurring in late trophozoites and schizonts [31]. However, since mature schizonts contain 8-32 progeny, the value of 6 is unexpectedly low. This may reflect the presence in the population of very early schizonts (with incomplete DNA replication), or perhaps during sample processing preferential lysis of the more fragile erythrocytes that harbor late schizonts.
Fig. 4.

Ciprofloxacin treatment of synchronized parasite populations. Parasites were synchronized with 5% sorbitol at 0, 48 h and 52 h. Rings were harvested at 55h, then treated for 30 min with solvent, 35 or 350 μM ciprofloxacin, lysed with SDS, digested with proteinase K and further processed for electrophoresis (lanes 3-5; 1.5 × 107 parasitized erythrocytes/lane). Samples taken at 68 or 82 h for trophozoites (lanes 6-8; 1.4 × 107 parasitized erythrocytes/lane) or schizonts (lanes 9-11; 7.1 × 106 parasitized erythrocytes/lane), respectively, were then treated and processed as above. For lanes 4-11, total mass of plDNA relative to that in lane 3 was 0.93, 0.89, 1.1, 1.1, 1.1, 1.7, 1.5, 1.7, respectively, and percent linears in lanes 3-11 were 20, 27, 32, 20, 31, 33, 13, 34, and 47, respectively. Markers are EcoR I-linearized plDNA (lane 1) and UV-nicked plDNA (lane 2). N, nicked circular plDNA; L, linearized circles; CC, covalently closed circular plDNA. Arrow indicates well.
3.4. Characterization of drug-promoted linear forms of plDNA
Having developed a robust method for visualizing the different forms of plDNA, we next assessed the effect of gyrase poisons on plDNA starting with an analysis of the linear molecules. SDS lysis of ciprofloxacin-treated intraerythrocytic P. falciparum generated linearized plDNA molecules (Fig. 5A, compare lanes 3 and 6), attributable to the in situ poisoning of gyrase on its circular plDNA substrate. This is consistent with previous reports using parasites that were released from erythrocytes prior to drug treatment [24]. Since gyrase cleaves both strands of DNA, the expected product is linearized plDNA covalently linked to denatured gyrase A subunit proteins. To confirm this we used the KSDS method in which potassium precipitates SDS, denatured proteins, and DNA that is covalently bound to protein. Protein-free DNA remains in the supernatant. For lysate treated with proteinase K, ciprofloxacin-promoted linears remain in the supernatant (Fig. 5A, lane 8); however for lysate protected by protease inhibitors, linears are lost to the pellet (Fig. 5A, lane 7), indicating that most linears are covalently linked to protein. These forms correspond to the 5′-exonuclease-resistant linears described previously [24]. Linears from cells not treated with ciprofloxacin (lanes 3-5) and the residual linears in the supernatant of ciprofloxacin-treated cells (lane 7) are apparently unaffected by protease digestion or inhibition, suggesting they are not covalently linked to protein and may arise from mechanical damage of plDNA circles or perhaps are a naturally occurring form. On a quantitative basis, the majority of the increase in protein-bound ciprofloxacin-promoted linears can be accounted for by a loss of covalently closed forms (Fig. 5B). This, and the apparent absence of linear fragments smaller than 35 kb, suggest there was only one gyrase situated on the plDNA molecules at the time of poisoning.
Fig. 5.

Characterization of plDNA from asynchronous intraerythrocytic parasites treated with topoisomerase poisons. (A) Cultures were incubated with solvent (lanes 3-5) or 700 μM ciprofloxacin (lanes 6-8) for 30 min, lysed with SDS solution containing protease inhibitors (lanes 4, 7) or proteinase K (lanes 3, 5, 6, 8), then digested with RNases. Samples (1.6 × 107 parasitized erythrocytes/lane) were loaded directly (lanes 3, 6) or supernatants were loaded after precipitation of covalent protein-DNA complexes (lanes 4, 5, 7, 8). For lanes 3-8, percent linears were 5.5, 5.2, 4.6, 26, 11, and 27, respectively. Markers are EcoR I-digested plDNA linears (lane 1) and UV-nicked plDNA circles (lane 2). (B) Left panel, cultures were treated with 0, 0.63, 10, 40, 160 or 640 μM ciprofloxacin (lanes 2-7; 9.2 × 106 parasitized erythrocytes/lane) for 30 min and processed for electrophoresis (some sample in lane 2 lost during loading). Right panel, cultures were treated with 0, 4, 80, 200, 500, 1000, 2000 or 4000 μM etoposide (lanes 2-9; 3.3 × 107 parasitized erythrocytes/lane) for 30 min and processed for electrophoresis. Lanes 1, EcoR I-digested plDNA linears. In samples from etoposide-treated cells, increased signal in nicked region likely represents nonspecific binding to cleavable complex-mediated fragmented nuclear DNA. (C) From data as in Panel B, percent linearized plDNA in ciprofloxacin- (□) or etoposide-treated (■) samples as a function of drug concentration. Ciprofloxacin data were from four experiments (EC50 59 μM, R2 0.76), etoposide data were from two (EC50 350 μM, R2 0.87). (D) Parasitized erythrocytes were sampled just before (lane 1) and at 5, 15, and 30 m, and at 2, 6, 24, 48 and 72 h (lanes 2-9, respectively) of exposure to 10 μM ciprofloxacin, then processed for electrophoresis (1.1 × 107 erythrocytes/lane). For lanes 2-9, total plDNA mass was 1.2 ± 0.2 of lane 1 control, and percent linear values were 1.4 ± 0.2 of lane 1 control. (E) Parasite density over time in cultures treated with solvent (◆) or 10 μM ciprofloxacin (□). N, nicked circular plDNA; IV, form Four; L, linearized circles; CC, covalently closed circular plDNA. Arrow indicates well.
3.5. Ciprofloxacin and etoposide dose-dependent cleavable complex formation
We treated asynchronous intraerythrocytic parasites with increasing concentrations of ciprofloxacin or etoposide. Both drugs promoted cleavable complex formation, leading to an increase in 35 kb linearized plDNA (Fig. 5B). Of note, drug-promoted cleavable complexes are evident at clinically relevant concentrations of ciprofloxacin (∼10 μM). Signal intensity of the linear, form IV and covalently closed forms was measured, assuming they comprise the majority of plDNA and are largely free of the non-specific genomic DNA signal that overlaps with nicked circles. Interestingly, for both drugs linearization reaches but does not exceed ∼30% of total plDNA (Fig. 5C), despite an abundance of residual covalently closed forms at highest drug concentrations (Fig. 5B, rightmost lanes).
3.6. Time course of cleavable complex formation with ciprofloxacin
A time course of ciprofloxacin treatment was performed to investigate the kinetics of intracellular gyrase poisoning. At the clinically relevant concentration of 10 μM, poisoning occurs very quickly, as maximum linearization is reached within minutes of exposure to the drug (Fig. 5D). In the presence of ciprofloxacin, linears persist for several days and, strikingly, during this time the various forms and total mass of plDNA remain constant (Fig. 5D, compare lanes 2 through 9). In a separate experiment, we measured parasite density over the same period of time (Fig. 5E). There is a nearly 20-fold increase in parasite density after three days of ciprofloxacin exposure, indicating that although plDNA replication is arrested, cell growth continues in the presence of drug, consistent with the second cycle effect or delayed death phenomenon [15].
3.7. Cleavable complex formation in synchronized parasite populations
Metabolic needs of the ring stage are thought to be small compared to those of trophozoites, in which growth and replication occur. To test whether gyrase activity parallels the demand for RNA and DNA metabolism in more mature parasites, cells were synchronized at 0, 48 and 52 hours with sorbitol, harvested at 55, 68, and 82 hours, and treated with 0, 35 or 350 μM ciprofloxacin (Fig. 4). The 350 μM concentration, near the limit of solubility in our culture system, was included so as to maximize the capture of cleavable complexes. Two effects are evident. As noted previously, there is a ∼6-fold increase in total mass of plDNA in schizonts (from 18 to 108 copies/parasitized erythrocyte), but in addition to this, at both drug concentrations a greater proportion of plDNA is linearized in schizont-infected cells. At 350 μM ciprofloxacin almost half the plDNA in schizonts is linearized, compared to ∼one-third in rings or trophozoites. From this it follows that in schizont-infected cells the number of gyrase molecules has increased from 6 to 54 (9-fold), boosting topoisomerase capacity in these metabolically active late stages.
3.8. Studies of quinolone structure-antimalarial activity
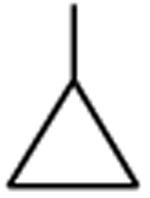
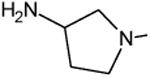
We chose quinolones with varying structures to assess their antimalarial activity. In Table 1 compounds are arranged by EC50 values, in order of decreasing potency over a 75-fold range. Several findings emerge from these cytotoxicity assays. A fluorine substituent at the X6 position was clearly important for antimalarial activity, as all seven of the most potent compounds have this motif, whereas three of the four least potent do not. Despite having a fluorine at X6, fleroxacin was among the least active compounds. Relative to its close analog pefloxacin, it was less active against malaria parasites suggesting an adverse effect of additional fluorines at R1 and/or Y. All three of the most potent compounds contain a cyclopropyl at R1, indicating this group was also favorable for antimalarial action: ciprofloxacin, identical to norfloxacin except at R1, was more potent. Comparison of clinafloxacin, ciprofloxacin and gatifloxacin, and of norfloxacin with pefloxacin, suggested that bulkier substituents at R2 may decrease antimalarial potency. Finally, comparison of pefloxacin with levofloxacin and ofloxacin suggests that a substituent bridging N1 and Y (of nucleus A) decreases antimalarial potency.
Table 1.
Structure and antimalarial activity of selected quinolones.

| ||||||
|---|---|---|---|---|---|---|
| Compound | Structure | EC50a (μM) | ||||
| Nucleus | R1 | X6 | R2 | Y | ||
| Clinafloxacin | A |
|
C-F |
|
C-Cl | 3.2 |
| Ciprofloxacin | A |
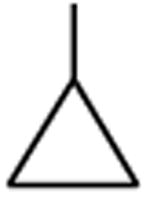
|
C-F |
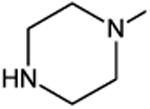
|
C-H | 5.6 |
| Gatifloxacin | A |

|
C-F |
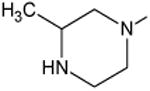
|
C-O-CH3 | 12 |
| Norfloxacin | A | -C2H5 | C-F |
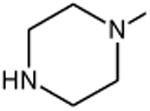
|
C-H | 14 |
| Pefloxacin | A | -C2H5 | C-F |
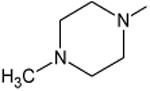
|
C-H | 23 |
| Levofloxacin | B | -(S)CH3 | C-F |
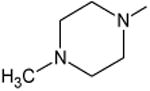
|
-- | 30 |
| Ofloxacin | B | -(R,S)CH3 | C-F |
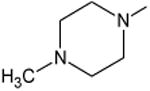
|
-- | 30 |
| Oxolinic Acid | A | -C2H5 | C-O-CH2-O- | C-H | 68 | |
| Piromidic Acid | A | -C2H5 | N |
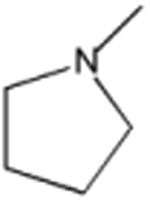
|
N | 80 |
| Fleroxacin | A | -CH2CH2F | C-F |
|
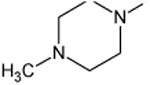
C-F | 90 |
| Nalidixic Acid | A | -C2H5 | C-H | -CH3 | N | 240 |
For each set of quadruplicates the coefficient of variation averaged ≤ 10% with a maximum of 34%. R2 values for the fitted curves were ≥ 0.99. The EC50 of positive control artemisinin, assayed concurrently with the quinolones, was 8.6 ± 1.1 nM (8 determinations).
3.9. Correlation of antimalarial activity with plDNA cleavable complex formation
We tested five quinolones (ciprofloxacin, gatifloxacin, levofloxacin, fleroxacin, nalidixic acid) that spanned the range of antimalarial potency to ask whether antiparasitic activity correlates with ability to promote the in situ formation of cleavable complexes (manifested as linearized plDNA molecules in SDS lysates of treated cells). Parasites were treated with 1, 5.6, 12, 30, 240, or 500 μM selected quinolone, lysed with SDS, and linearized plDNA was quantified from Southern blots (results for two concentrations depicted in Figs. 6A and B). Similar to what was done in Fig. 5C, for each quinolone the extent of plDNA linearization was plotted as a function of log drug concentration to obtain EC50 values. These in turn were graphed versus the EC50s of antimalarial activity from Table 1 (Fig. 6C). For three compounds (gatifloxacin, levofloxacin, and fleroxacin) there was close correlation between antimalarial activity and the linearization of plDNA (R2 = 0.97), in keeping with the notion that antiparasitic action is linked to the intracellular capture of gyrase-plDNA complexes. Of note, the y-intercept is positive, indicating antiparasitic activity in the absence of plDNA linearization and suggesting these compounds may have additional molecular targets. Two compounds did not fall on the line. Nalidixic acid had no detectable gyrase poisoning activity and could not be plotted. More interestingly, ciprofloxacin falls above the line, indicating antimalarial activity greater than expected from plDNA linearization. Although the quinolones and fluoroquinolones preferably target prokaryotic type II topoisomerases (such as gyrase), they do have detectable activity against eukaryotic type II enzymes, which suggested that perhaps the additional antimalarial potency of ciprofloxacin stems from capture of covalent complexes between topoisomerase II and genomic DNA in the nucleus.
Fig. 6.

Correlation of antimalarial activity with plDNA cleavable complex formation. (A) Asynchronous parasites were treated with solvent (lane 3), 5.6 μM ciprofloxacin (lane 4), gatifloxacin (lane 5), levofloxacin (lane 6), fleroxacin (lane 7), or nalidixic acid (lane 8) for 30 min, and processed for plDNA analysis (107 parasitized erythrocytes/lane). (B) Parasites were treated with solvent or 12 μM drug as in (A); 2 × 107 parasitized erythrocytes/lane. (C) Antimalarial activity versus plDNA cleavable complex formation for ciprofloxacin (□), gatifloxacin (●), levofloxacin (▲), and fleroxacin (○). Depicted potencies are the negative log of the cognate EC50s (in molar). EC50 of antimalarial activity is from Table 1. EC50 of cleavable complex formation was obtained from seven experiments, including those in Panels A and B; EC50 and R2 values for plDNA linearization were: gatifloxacin (7.9 μM, 0.80), ciprofloxacin (71 μM, 0.98), levofloxacin (22 μM, 0.92), fleroxacin (260 μM, 0.97). Markers are EcoR I-digested plDNA linears (lanes 1) and UV-nicked plDNA (lanes 2). N, nicked circular plDNA; IV, form Four; L, linearized circles; CC, covalently closed circular plDNA. Arrow indicates well.
3.10. Drug-promoted formation of cleavable complexes with nuclear DNA
To test the ability of fluoroquinolones to capture nuclear DNA in protein-DNA complexes we used KSDS assays. In this method, parasite nucleic acids are metabolically labeled with [3H]hypoxanthine, then cells are treated with compound, lysed with SDS (to capture DNA-topoisomerase covalent adducts), the lysate is digested with RNases, then SDS and proteins are precipitated with cold KCl. Only DNA covalently bound to protein is found in the pellet. Since less than 3% of DNA in P. falciparum is plDNA (Section 3.3), counts in the KSDS pellet are attributable to nuclear DNA-topoisomerase adducts.
In five independent experiments with asynchronous parasites, the RNase-resistant [3H] signal of DNA accounted for 46 ± 7.0% of total incorporation. In no-drug controls, 4.9 ± 1.8% of DNA was pelleted in the KSDS assay, from the capture of naturally occurring DNA-topoisomerase catalytic intermediates that arise in situ in the normal course of nuclear topoisomerase catalysis. Etoposide, a potent poison of eukaryotic type II topoisomerases included as a positive control, promotes the capture of 28 ± 8.3% of nuclear DNA. Interestingly, at high concentrations fluoroquinolones also poison P. falciparum nuclear topoisomerase (Fig. 7), which may contribute to the positive y-axis intercept noted in Fig. 6C. As expected for covalent topoisomerase-DNA complexes, these KSDS precipitates are protease sensitive. The rank order potency of gatifloxacin >levofloxacin >fleroxacin is in keeping with their antimalarial potency (Table 1) and their propensity to poison apicoplast gyrase (Fig. 6C). Ciprofloxacin is again an outlier in this assay, with less than expected poisoning activity vs that of its congeners, indicating that its disproportionately potent antimalarial activity cannot be explained by cleavable complex formation with nuclear DNA. Nalidixic acid does not promote detectable formation of cleavable complexes with nuclear DNA at concentrations up to 10 mM.
Fig. 7.

Cleavable complex formation with nuclear DNA in P. falciparum treated with topoisomerase poisons. Parasites were metabolically labeled with [3H]hypoxanthine, treated with etoposide (■), gatifloxacin (●), levofloxacin (▲), ciprofloxacin (□), fleroxacin (○), or nalidixic acid (∆) at the indicated concentrations for 18-22 h, lysed with SDS and digested with RNases. SDS and protein-bound DNA were precipitated with potassium. The fold increase in nuclear DNA capture as compared to solvent controls (mean of triplicate determinations) was plotted against concentration. Coefficient of variation for each data point was ≤19%.
4. Discussion
To study the effect of topoisomerase poisons on the DNA of malaria parasites we devised methods for quantitating, treating, and lysing intraerythrocytic parasites, with minimal subsequent manipulation of DNA during sample processing, and for separating plDNA forms by conventional electrophoresis. These techniques provide a high yield of plDNA and they preserve the readily distinguishable topological forms of this 35 kb circular organellar genome (Fig. 2A, compare lanes 1 and 2). Our rigorous method for quantifying plDNA provides accurate values for the number of plDNA molecules per parasitized erythrocyte.
Four distinct forms of plDNA are detected from untreated malaria parasites (Fig. 2A, lane 1). The most rapidly migrating and abundant (68%) is negatively supercoiled covalently closed circles. About 7% of plDNA is linearized, likely from endogenous gyrase-plDNA adducts and perhaps also from mechanical breakage. These findings are comparable to previous reports [24,31]. However, as confirmed by comigration with UV-nicked plDNA markers and by insensitivity to EtBr during electrophoresis, the slowest migrating form (15%) comprises noncovalent (likely nicked) circles of plDNA, not covalently closed relaxed circles. These nicked circles could also result from damage but their abundance suggests otherwise. Perhaps they are replication intermediates or newly segregated daughter circles.
What is form IV? The indistinct nature of this newly-identified moiety suggests it is heterogeneous in structure. It may be a replication intermediate, perhaps containing twin D-loops [31]. Alternatively it may be a cruciform. Visualization by electron microscopy has revealed that a small proportion of Plasmodium plDNA contains cruciform structures [31], likely a result of their inverted repeat sequence, that would be expected to travel much more slowly through agarose than do supercoiled covalently closed circles [32].
Although ciprofloxacin is substantially more potent than etoposide in promoting plDNA cleavable complex formation, both drugs reach a similar maximum of 30% of plDNA trapped in cleavable complexes (Fig. 5C). This suggests several important conclusions. First, since ciprofloxacin and etoposide generate a similar maximum, despite having differential activity against gyrase or eukaryotic topoisomerase II (respectively), gyrase appears to be the only type II enzyme in the apicoplast. Second, at maximal drug effect the depletion of covalently closed plDNA circles (gyrase substrate) can be accounted for by the appearance of full length protein-bound linears (Fig. 5B), implying that only one active gyrase is associated with a plDNA circle. Since processing replication forks generate positive supercoils that, in prokaryotic systems, can only be released by gyrase, this in turn suggests only one moving replication fork per circle. Perhaps the twin D-loop processes unidirectionally, or replication in these cells is preferentially by rolling circle mechanism. Alternatively, if the apicoplast contains a eukaryotic type I topoisomerase, this enzyme too could relax the positive supercoils created ahead of DNA-tracking synthetic machinery. Third, our findings indicate that in schizonts there is only one active gyrase per approximately 70 kb of plDNA, since approximately one-half of plDNA molecules are linearized as a result of etoposide or ciprofloxacin treatment. This ratio is much lower than those reported in transformed chicken erythroblasts (1 enzyme/5 kb), for the mitochondrial kDNA of trypanosomes (1 enzyme/8 kb), or in rapidly dividing Drosophila embryos (1 enzyme/25 kb) [33-35]. If schizonts have ∼108 copies of plDNA (six times the 18 copies in rings), these replicating forms appear to have only 54 active gyrase molecules/infected erythrocyte.
Treatment of synchronized parasites shows that plDNA metabolism is most vulnerable to gyrase poisoning in the schizont stage (Fig. 4). This is in keeping with plDNA synthesis beginning during the late trophozoite stage and continuing through schizogony [31]. Gyrase may also participate in decatenation of daughter plDNA circles at the end of replication. In bacteria this process is usually catalyzed by topoisomerase IV; however, there is no current evidence of a topoisomerase IV in P. falciparum, so gyrase likely performs both functions. Gyrase has the ability to decatenate DNA, despite its preferred role in vivo of releasing positive supercoils ahead of a replication fork [36].
Fluoroquinolones are among the antibacterials that target apicoplast metabolism and lead to a distinctively delayed death of malaria parasites [15]. Treated cells themselves survive but progressive loss of apicoplast function proves lethal to the next generation of parasites. This phenomenon is quite different from the effect fluoroquinolones have on bacteria [37]. Rapid bactericidal action is attributable to the collision of processing replication machinery with stationary drug-promoted gyrase-DNA complexes, leading to lethal double-strand breaks in the genome. The resulting SOS response contributes further to cytotoxicity. In contrast, despite several days of constant drug pressure, in malaria parasites the pool of plDNA remains remarkably static: it does not appreciably increase, change distribution among forms, fragment, or diminish (Fig. 5D). This suggests several things. First, and in keeping with our quantitative results above, replication fork collisions with gyrase-plDNA complexes are infrequent. Second, the apicoplast appears to lack SOS response machinery comparable to that responsible for finalizing double-strand breaks and the consequent rapid bactericidal activity. Simply inhibiting gyrase catalysis, without creating double-stranded beaks (effecting static but not cidal activity), is consistent with the delayed death effect of quinolones on malaria parasites.
A number of quinolones were assayed for their in vitro antimalarial activity (Table 1). In general the structure-activity profile parallels that for bacteria [38]. Addition of fluorine at C6 increases potency, and a cyclopropyl group at N1 triples activity over N1 ethyl analogs. However, racemic ofloxacin and its L-isomer levofloxacin have the same EC50 against malaria parasites, whereas levofloxacin is generally 2-fold more potent than ofloxacin against most bacteria [39]. Based on the observed structure-activity relationships, we would expect that the antimalarial activity of clinafloxacin (the most potent compound assayed) could be brought into the submicromolar range by removing the amino group of the R2 pyrrolidine (less bulky substituents at this position have greater activity) and removing the chlorine at C8.
A subset of five quinolones, spanning the 75-fold range of antimalarial potency, were chosen to assess the relationship between antiparasitic activity and the putative molecular mechanism of action (gyrase poisoning). In general the correlation was excellent, with one notable exception: ciprofloxacin's antiparasitic activity was disproportionately greater than expected for its potency as a gyrase poison (Fig. 6C). Activity at high concentrations against nuclear topoisomerase II, though observed, does not explain this discrepancy. Does ciprofloxacin have additional targets, accounting for its unexpectedly high antimalarial activity? Partially purified P. falciparum mitochondrial topoisomerase II is reportedly insensitive to fluoroquinolones, requiring minimum inhibitory concentrations of 1 mM or more [40], making this an unlikely explanation and suggesting that in malaria parasites ciprofloxacin at clinically relevant concentrations has off-target effects that contribute to its antiparasitic efficacy.
We have characterized several forms of plDNA in P. falciparum, used fluoroquinolones as a tool to better understand the role of gyrase in plDNA metabolism, and have found evidence supporting gyrase poisoning as a major basis for the antimalarial activity of fluoroquinolones. This drug class has had variable success in treating human infection, leading to cures in some study populations [41] but partial or complete failure in others [42-44]. Our structure-activity studies suggest simple modifications that may improve efficacy and permit this valuable class of anti-infectives to find a meaningful role in malaria treatment, perhaps as a component of combination therapy.
Acknowledgments
This research was supported by the Johns Hopkins Malaria Research Institute, R01AI095453, and the Medical Scientist Training Grant T32GM07039. Funding sources had no involvement in study design; data collection, analysis or interpretation; writing of report or decision to submit the article for publication.
We thank Paul Englund, Robert Jensen and Sean Prigge for their thoughtful discussions and suggestions throughout the course of this study. We are grateful to Rahul Bakshi and Jane Scocca for their assistance with methodology, and to Rahul for carefully reading the manuscript and generously preparing the final figures. Photomicrographs were obtained with the assistance of Norm Barker and Kirsten Meyer.
Footnotes
Conflict of Interests: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sonya C. Tang Girdwood, Email: Sonya.TangGirdwood@cchmc.org.
Elizabeth Nenortas, Email: enenort1@jhmi.edu.
References
- 1.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014;383:723–35. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Fact sheet on the world malaria report 2013. 2013 http://www.who.int/malaria/media/world_malaria_report_2013/en/
- 3.Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, et al. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–31. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 4.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mather MW, Vaidya AB. Mitochondria in malaria and related parasites: ancient, diverse and streamlined. J Bioenerg Biomembr. 2008;40:425–33. doi: 10.1007/s10863-008-9176-4. [DOI] [PubMed] [Google Scholar]
- 6.Fichera ME, Roos DS. A plastid organelle as a drug target in apicomplexan parasites. Nature. 1997;390:407–9. doi: 10.1038/37132. [DOI] [PubMed] [Google Scholar]
- 7.Kalanon M, McFadden GI. Malaria, Plasmodium falciparum and its apicoplast. Biochem Soc Trans. 2010;38:775–82. doi: 10.1042/BST0380775. [DOI] [PubMed] [Google Scholar]
- 8.Williamson DJ, Preiser PR, Wilson RJM. Organelle DNAs: The bit players in malaria parasite DNA replication. Parasitol Today. 1996;12:357–62. doi: 10.1016/0169-4758(96)10053-3. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki M, Kikuchi T, Kita K, Kojima S, Kuroiwa T. Large amounts of apicoplast nucleoid DNA and its segregation in Toxoplasma gondii. Protoplasma. 2001;318:180–91. doi: 10.1007/BF01306607. [DOI] [PubMed] [Google Scholar]
- 10.Wilson RJ. Progress with parasite plastids. J Mol Biol. 2002;319:257–74. doi: 10.1016/S0022-2836(02)00303-0. [DOI] [PubMed] [Google Scholar]
- 11.Wilson RJ, Denny PW, Preiser PR, Rangachari K, Roberts K, Roy A, et al. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J Mol Biol. 1996;261:155–72. doi: 10.1006/jmbi.1996.0449. [DOI] [PubMed] [Google Scholar]
- 12.Fleige T, Soldati-Favre D. Targeting the transcriptional and translational machinery of the endosymbiotic organelle in apicomplexans. Curr Drug Targets. 2008;9:948–56. doi: 10.2174/138945008786786073. [DOI] [PubMed] [Google Scholar]
- 13.Divo AA, Geary TG, Jensen JB. Oxygen- and time-dependent effects of antibiotics and selected mitochondrial inhibitors on Plasmodium falciparum in culture. Antimicrob Agents Chemother. 1985;27:21–7. doi: 10.1128/aac.27.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seaberg LS, Parquette AR, Gluzman IY, Phillips GW, Brodasky TF, Krogstad DJ. Clindamycin activity against chloroquine-resistant Plasmodium falciparum. J Infect Dis. 1984:904–11. doi: 10.1093/infdis/150.6.904. [DOI] [PubMed] [Google Scholar]
- 15.Dahl EL, Rosenthal PJ. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob Agents Chemother. 2007;51:3485–90. doi: 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–4. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 17.Khor V, Yowell C, Dame JB, Rowe TC. Expression and characterization of the ATP-binding domain of a malarial Plasmodium vivax gene homologous to the B-subunit of the bacterial topoisomerase DNA gyrase. Mol Biochem Parasitol. 2005;140:107–1. doi: 10.1016/j.molbiopara.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Raghu Ram EV, Kumar A, Biswas S, Kumar A, Chaubey S, Siddiqi MI, et al. Nuclear gyrB encodes a functional subunit of the Plasmodium falciparum gyrase that is involved in apicoplast DNA replication. Mol Biochem Parasitol. 2007;154:30–9. doi: 10.1016/j.molbiopara.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Dar MA, Sharma A, Mondal N, Dhar SK. Molecular cloning of apicoplast-targeted Plasmodium falciparum DNA gyrase genes: unique intrinsic ATPase activity and ATP-independent dimerization of PfGyrB subunit. Eukaryot Cell. 2007;6:398–412. doi: 10.1128/EC.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–75. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 21.Froelich-Ammon SJ, Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J Biol Chem. 1995;270:21429–32. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 22.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishna S, Davis TME, Chan PCY, Wells RA, Robinson KJH. Ciprofloxacin and malaria. Lancet. 1988;1:1231–2. doi: 10.1016/s0140-6736(88)92056-9. [DOI] [PubMed] [Google Scholar]
- 24.Weissig V, Vetro-Widenhouse TS, Rowe TC. Topoisomerase II inhibitors induce cleavage of nuclear and 35-kb plastid DNAs in the malarial parasite Plasmodium falciparum. DNA Cell Biol. 1997;16:1483–92. doi: 10.1089/dna.1997.16.1483. [DOI] [PubMed] [Google Scholar]
- 25.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–20. [PubMed] [Google Scholar]
- 26.Posner GH, Gonzalez L, Cumming JN, Klinedinst D, Shapiro TA. Synthesis and antimalarial activity of heteroatom-containing bicyclic endoperoxides. Tetrahedron. 1997;53:37–50. [Google Scholar]
- 27.Holford NH, Sheiner LB. Pharmacokinetic and pharmacodynamic modeling in vivo. Crit Rev Bioeng. 1981;5:273–322. [PubMed] [Google Scholar]
- 28.Taylor JR. Rejection of data. In: McGuire A, editor. An introduction to error analysis: the study of uncertainties in physical measurements. Sausalito, CA: University Science Books; 1997. pp. 165–72. [Google Scholar]
- 29.Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–5. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bodley AL, Shapiro TA. Molecular and cytotoxic effects of camptothecin, a topoisomerase I inhibitor, on trypanosomes and Leishmania. Proc Natl Acad Sci USA. 1995;92:3726–30. doi: 10.1073/pnas.92.9.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williamson DH, Preiser PR, Moore PW, McCready S, Strath M, Wilson RJ. The plastid DNA of the malaria parasite Plasmodium falciparum is replicated by two mechanisms. Mol Microbiol. 2002;45:533–42. doi: 10.1046/j.1365-2958.2002.03033.x. [DOI] [PubMed] [Google Scholar]
- 32.Courey AJ. Analysis of altered DNA structures: cruciform DNA. Methods Mol Biol. 1999;94:29–40. doi: 10.1385/1-59259-259-7:29. [DOI] [PubMed] [Google Scholar]
- 33.Heck MM, Earnshaw WC. Topoisomerase II: A specific marker for cell proliferation. J Cell Biol. 1986;103:2569–81. doi: 10.1083/jcb.103.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro TA, Klein VA, Englund PT. Drug-promoted cleavage of kinetoplast DNA minicircles. Evidence for type II topoisomerase activity in trypanosome mitochondria. J Biol Chem. 1989;264:4173–8. [PubMed] [Google Scholar]
- 35.Fairman R, Brutlag DL. Expression of the Drosophila type II topoisomerase is developmentally regulated. Biochemistry. 1988;27:560–5. doi: 10.1021/bi00402a009. [DOI] [PubMed] [Google Scholar]
- 36.Tretter EM, Berger JM. Mechanisms for defining supercoiling set point of DNA gyrase orthologs: II. The shape of the GyrA subunit C-terminal domain (CTD) is not a sole determinant for controlling supercoiling efficiency. J Biol Chem. 2012;287:18645–54. doi: 10.1074/jbc.M112.345736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drlica K, Hooper DC. Mechanisms of quinolone action. In: Hooper P, Rubinstein E, editors. Quinolone Antimicrobial Agents. ASM Press; 2003. pp. 19–40. [Google Scholar]
- 38.Domagala JM, Hagan SE. Structure-activity relationships of the quinolone antibacterials in the new millennium: some things change and some do not. In: Hooper P, Rubinstein E, editors. Quinolone Antimicrobial Agents. ASM Press; 2003. pp. 1–18. [Google Scholar]
- 39.Wimer SM, Schoonover L, Garrison MW. Levofloxacin: a therapeutic review. Clin Ther. 1998;20:1049–70. doi: 10.1016/s0149-2918(98)80104-5. [DOI] [PubMed] [Google Scholar]
- 40.Chavalitshewinkoon-Petmitr P, Worasing R, Wilairat P. Partial purification of mitochondrial DNA topoisomerase II from Plasmodium falciparum and its sensitivity to inhibitors. Southeast Asian J Trop Med Public Health. 2001;32:733–8. [PubMed] [Google Scholar]
- 41.Sarma PS. Norfloxacin: a new drug in the treatment of falciparum malaria. Ann Intern Med. 1989;111:336–7. doi: 10.7326/0003-4819-111-4-336. [DOI] [PubMed] [Google Scholar]
- 42.Deloron P, Lepers JP, Raharimalala L, Dubois B, Coulanges P, Pocidalo JJ. Pefloxacin for falciparum malaria: only modest success. Ann Intern Med. 1991;114:874–5. doi: 10.7326/0003-4819-114-10-874. [DOI] [PubMed] [Google Scholar]
- 43.Stromberg A, Bjorkman A. Ciprofloxacin does not achieve radical cure of Plasmodium falciparum infection in Sierra Leone. Trans R Soc Trop Med Hyg. 1992;86:373. doi: 10.1016/0035-9203(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 44.Tripathi KD, Sharma AK, Valecha N, Kulpati DD. Curative efficacy of norfloxacin in falciparum malaria. Indian J Med Res. 1993;97:176–8. [PubMed] [Google Scholar]