

Abstract
Lately, the world has faced tremendous progress in the understanding of non-alcoholic fatty liver disease (NAFLD) pathogenesis due to rising obesity rates. Peroxisome proliferator-activated receptors (PPARs) are transcription factors that modulate the expression of genes involved in lipid metabolism, energy homeostasis and inflammation, being altered in diet-induced obesity. Experimental evidences show that PPAR-alpha is the master regulator of hepatic beta-oxidation (mitochondrial and peroxisomal) and microsomal omega-oxidation, being markedly decreased by high-fat (HF) intake. PPAR-beta/delta is crucial to the regulation of forkhead box-containing protein O subfamily-1 expression and, hence, the modulation of enzymes that trigger hepatic gluconeogenesis. In addition, PPAR-beta/delta can activate hepatic stellate cells aiming to the hepatic recovery from chronic insult. On the contrary, PPAR-gamma upregulation by HF diets maximizes NAFLD through the induction of lipogenic factors, which are implicated in the fatty acid synthesis. Excessive dietary sugars also upregulate PPAR-gamma, triggering de novo lipogenesis and the consequent lipid droplets deposition within hepatocytes. Targeting PPARs to treat NAFLD seems a fruitful approach as PPAR-alpha agonist elicits expressive decrease in hepatic steatosis by increasing mitochondrial beta-oxidation, besides reduced lipogenesis. PPAR-beta/delta ameliorates hepatic insulin resistance by decreasing hepatic gluconeogenesis at postprandial stage. Total PPAR-gamma activation can exert noxious effects by stimulating hepatic lipogenesis. However, partial PPAR-gamma activation leads to benefits, mainly mediated by increased adiponectin expression and decreased insulin resistance. Further studies are necessary aiming at translational approaches useful to treat NAFLD in humans worldwide by targeting PPARs.
Keywords: Peroxisome proliferator-activated receptors, Non-alcoholic fatty liver disease, Obesity, Treatment, Insulin resistance, Beta-oxidation, Lipogenesis
Core tip: Multiple pathways disrupted in obesity and non-alcoholic fatty liver disease (NAFLD) are regulated by genes encoded by peroxisome proliferator-activated receptors (PPARs). Thus, PPARs emerged as potential targets to alleviate NAFLD. The use of PPAR-alpha agonist yields increased mitochondrial beta-oxidation coupled with reduced lipogenesis. Both of them are essential to tackle insulin resistance and hepatic steatosis. PPAR-beta/delta agonist is still not available as a medicine, but PPAR-beta/delta agonist elicited expressive reduction in hepatic glucose production in murine models. PPAR-gamma agonist is extensively used, and beneficial effects come from partial activation as total PPAR-gamma activation leads to hepatic lipogenesis, being harmful to the liver.
INTRODUCTION
The current obesity epidemics have resulted in a significant rise in its comorbidities prevalence[1]. Liver is often significantly affected by obesity and, hence, the non-alcoholic fatty liver disease (NAFLD) is regarded as the hepatic manifestation of metabolic syndrome, showing rising prevalence regardless of economic status or age worldwide[2]. Despite being a benign process at first, the continuation of the triggering stimuli can lead to harmful conditions such as non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma[3].
Excessive energy intake concomitant with sedentism are considered essential underpinnings of lipid droplets accumulation[4]. When the metabolism faces obesity, excessive adipose tissue fat pads elicit low-grade inflammation, which is linked to insulin resistance development[5,6]. The resulting hyperinsulinemia yields high lipolysis rate in the white adipose tissue coupled with reduced fatty acid oxidation within the hepatocytes[7]. The balance between fatty acid input and output in the liver is controlled by integrated enzymes that act in the catalysis of hepatic uptake, lipogenesis, oxidation and exportation of fatty acids[8]. Whenever the hepatic fatty acid synthesis and/or uptake surpass the liver oxidative and/or the exportation capacity, lipid droplets accumulate within the hepatic parenchyma, configuring NAFLD[9].
Dietary quality has a paramount importance for the hepatic fatty acid metabolism[10]. Excessive intake of simple carbohydrate such as fructose and sucrose are implicated in high rates of de novo lipogenesis (DNL) in the liver, which is defined as the synthesis of fatty acids from a non-lipid source[11]. In conjunction with a high intake of dietary fats that generates lipotoxicity through the excessive production of ceramides from palmitate and aggravates insulin resistance, the high DNL due to excessive dietary carbohydrate makes a great demand for hepatic oxidation of fatty acids, which exceeds the oxidative capacity of hepatic peroxisomes, mitochondria and microsomes (endoplasmic reticulum). In turn, hepatic lipid metabolic equilibrium is disrupted due to abnormal fat partitioning within hepatocytes[11-13].
The carbohydrate and lipid intake, as well as the adipokines and insulin levels, exert considerable influence upon key transcription factors that can modulate hepatic lipid metabolism[14]. Peroxisome proliferator-activated receptors (PPARs) are at the crossroads of NAFLD pathogenesis, once recent evidences point to the modulation of hepatic beta-oxidation and lipogenesis by different PPAR isoforms[15,16]. Thus, even though weight management and exercise are the most efficient approach to treating NAFLD, adjunctive pharmacological intervention is utterly indicated. PPARs emerge as a target to treat NAFLD by modulating diverse pathways that are impaired by obesity[17,18]. The role that total or partial PPAR alpha, beta/delta and gamma activation play in lipogenesis and hepatic oxidation as well as in carbohydrate metabolism through the gluconeogenesis, and DNL are relevant targets to fasten the treatment of NAFLD and obesity.
PPAR PHYSIOLOGY IN EXPERIMENTAL MODELS NAFLD
Hepatic metabolic pathways can be disturbed differently according to the nutrient that is excessive in the diet[19]. Experimental dietary models of NAFLD are influenced by the dietary scheme duration, diet composition, and animal age, all of which directly affect the spectrum of NAFLD pathogenesis[20].
When there is excessive dietary intake of lipids, hepatic PPAR-alpha expression decreases significantly parallel to an expressive increase of PPAR-gamma[16,21]. Obese mice fed during 16 wk a high-fat (HF) diet made up of 60% of energy as lipids, predominantly saturated fatty acids from lard, exhibited overweight, insulin resistance and 34.57% of volume density of hepatic steatosis concomitant to a proinflammatory adipokine profile and activation of hepatic stellate cells (HSCs)[21]. Hepatic PPAR-alpha expression was substantially reduced[21], agreeing with a reduced number in the numerical density of hepatic mitochondria[21,22]. PPAR-alpha is related to mitochondrial beta-oxidation of fatty acids, which has got carnitine palmitoyl transferase-1 (CPT-1) as a pivotal enzyme that allows the fatty acid to go through the inner mitochondrial membrane and reach the mitochondrial matrix to be metabolized[23]. In the absence of a typical PPAR-alpha expression in the liver, the transcription of its target gene CPT-1 is impaired and excessive fatty acids, which are usually stemmed from lipolysis and delivery to the liver of obese individuals, tend to accumulate in the form of triglycerides[24,25].
A similar dietary scheme (50% of energy as fat for 12 wk) elicited 2.3 fold increase in liver triglycerides, followed by 0.7 fold decrease in PPAR-alpha and a 0.4 fold increase in PPAR-gamma protein expression in the liver. These observations feature a frame that predisposes to NAFLD because PPAR-gamma is linked to lipogenesis and its target gene expression, SREBP-1c, was 0.5 fold increased in HF fed animals[16]. SREBP-1c is implicated in the DNL, induced by high insulin levels. Once activated, SREBP-1c activates others lipogenic genes and leads to the conversion of pyruvate into fatty acids. During this process, there is a great production of malonyl co-A, which inhibits CPT-1 and prevents fatty acids from reaching the mitochondrial matrix to be metabolized through mitochondrial beta-oxidation[26,27]. Alternative pathways, peroxisomal beta-oxidation, and microsomal omega-oxidation are upregulated to try to compensate insufficient mitochondrial oxidative activity. Mitochondrial damage found in NAFLD and increased peroxisomal and microsomal oxidation of fatty acids leads to oxidative stress and the consequent progression to NASH if an adequate intervention is not implemented[28,29]. The pivotal role that oxidative stress plays in NAFLD progression to NASH was verified through positive immunoreactions for oxidized phosphatidylcholine close to activated stellate cells and in apoptotic hepatocytes in samples of human liver autopsy. Moreover, immunostaining intensity correlated positively with the degree of steatosis[30].
When the HF diet (49% of energy as lipids) was administered to dams during 8 wk prior to gestation, gestation, and lactation, similar hepatic alterations were detected in the offspring. Pups from HF dams had overweight and glucose intolerance at 3 mo of age, both of which agree with the 1.4 fold increase in hepatic steatosis rate in these animals. PPAR-alpha gene and protein expression were reduced in the liver of offspring of HF dams parallel to increased gene and protein expression of PPAR-gamma. As a result, the hepatic expression of the PPAR-alpha target gene CPT-1 was decreased, because the expression of the PPAR gamma target genes SREBP-1c was elevated[31]. This pattern of gene and protein expression explains the early NAFLD onset in the offspring of obese dams[32], albeit with a discrete overweight. Besides the compromised mitochondrial beta-oxidation through the reduced CPT-1 activity and the enhanced DNL, favored by overexpression of SREBP-1c, the offspring presented with expressive reduction in fatty acid translocase (FAT)/CD36 expression, which limits long chain fatty acids (more than 20 carbons) oxidation. FAT/CD36 can shorten the fatty acid chain in order to allow CPT-1 to catalyze its transport to the mitochondrial matrix[33]. This represents an additive failure in hepatic lipid metabolism due to maternal obesity.
Additionally, hepatic insulin resistance was detected in obese dams offspring through enhanced hepatic expression of glucose 6 phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK)[31]. Both enzymes are crucial to hepatic gluconeogenesis and are activated by glucagon during the fasting period and inhibited by insulin at the postprandial stage. When insulin resistance occurs, insulin, at high levels, loses its capacity to inhibit gluconeogenesis by the downregulating G6Pase and PEPCK expression[34,35]. Hence, high hepatic glucose production aggravates insulin resistance. PPAR-beta/delta induces forkhead box-containing protein O subfamily-1 (FOXO-1), which can regulate G6Pase and PEPCK expression, and might be a viable approach to restoring this pathway[36]. In addition, PPAR-beta/delta is described to induce HSCs proliferation in fibrogenesis activation in vitro and in vivo. HSCs constitutively express PPAR-beta/delta and their activation aim at the hepatic recovery from chronic insult. However, the collagen synthesis by HSCs leads to hepatic fibrosis in the long run, being involved in the progression of simple NAFLD to more dangerous types of liver diseases[37,38].
Excessive intake of sucrose (32% of energy as sucrose) yielded similar effects to liver histology and PPAR expression in C57BL/6 mice. Although animals did not become obese, excessive sucrose supply, elicited 1.3-fold increase in hepatic steatosis coupled with 0.5 fold decrease in PPAR-alpha expression and 0.8 increase in SREBP-1c expression[19]. Likewise, the intake of 34% of energy as fructose impaired hepatic cytoarchitecture and lipid metabolism in the same mouse model. Even in the absence of significant overweight, mice fed a high-fructose diet presented nearly 55% volume density of hepatic steatosis, which can be accounted for by augmented hepatic expression of PPAR-gamma (0.5 fold) and reduced hepatic expression of PPAR-alpha (-0.25 fold)[39]. PPAR-gamma increases the transcription of SREBP-1c, leading to higher lipogenesis, and little PPAR-alpha expression favors high hepatic glucose production. There is the interplay between white adipose tissue and liver as a glucose/glycerol cycle that guarantees energy transport (as glucose) from liver to peripheral tissues[40]. This process relies on PPAR-alpha, being impaired in the fructose-fed animals[39,40]. In addition, high fructose intake augmented the hepatic expression of PEPCK and GLUT2[39], indicating high hepatic glucose output, which aggravates insulin resistance and favors the NAFLD progression to NASH[41].
TARGETING PPARS TO TREAT NAFLD
PPARs encompass a subfamily of a superfamily of nuclear receptors. There are three different isoforms: PPAR-alpha, PPAR-beta/delta, and PPAR-gamma, which are differently expressed in various tissues. They are ligand-dependent transcription factors that regulate the expression of their target genes through specific binding to peroxisome proliferation response elements (PPERs). Each isoform heterodimerize with its retinoid X receptor alpha, beta/delta or gamma and binds to its respective PPRE, forming a structure able to recognize specific DNA sequences (AGGTCA) to activate the transcription of its target genes[42-44].
Briefly, PPAR-alpha is closely linked to the transcription of genes related to hepatic beta-oxidation, such as CPT-1 and is highly expressed in the liver[45,46]. Thus, treatment with PPAR-alpha agonist usually yields body mass loss as this isoform is implicated in lipid metabolism pathways[43]. PPAR-beta/delta is ubiquitously expressed and is crucial to beta-oxidation in skeletal muscle, not in the liver. In the liver, anti-inflammatory properties by the activation of macrophages and the protection against lipotoxicity are reported. The induction of Stearoyl-CoA desaturase 1 by PPAR-beta/delta activation promotes monounsaturated fatty acids formation instead of saturated fatty acids, decreasing oxidative stress, emerging as a promising approach to the tackle insulin resistance[47,48]. PPAR-gamma is expressed at low concentrations in the liver (9%-12% of the expression in the white adipose tissue), being related to adipogenesis and insulin-sensitizing effects through the diversion of fatty acids to adipose tissue storage. Patients with NAFLD exhibit abnormal high expression of PPAR-gamma in the liver, which coincides with overexpression of SREBP-1c and the consequent hepatic lipogenesis[49,50].
Taking into account the above-mentioned PPAR related effects, the use of PPAR agonist to treat NAFLD seems to be a viable strategy. In this regard, the activation of PPAR-alpha by fenofibrate markedly ameliorated the hepatic insulin resistance by the upregulation of enzymes involved with beta-oxidation in fructose-fed mice and the expressive reduction of DNL, albeit with high endoplasmic reticulum stress[51]. In addition, fenofibrate significantly ameliorated microcirculatory perfusion in a HF mice mouse model of NAFLD, besides the upregulation of genes involved in hepatic lipid oxidation[52]. These reported effects comply with a significant decrease in hepatic steatosis percentage after the activation of PPAR-alpha by fenofibrate[51,52]. Activation of PPAR-alpha by fish oil, a nutriceutical, yielded alleviation of hepatic insulin resistance through low G6Pase and PEPCK expression in the liver and reduced steatosis by upregulation of mitochondrial beta-oxidation (high CPT-1 expression) concomitant to reduced lipogenesis (low fatty acid synthase expression)[53].
In humans, the evaluation of fenofibrate use to treat NAFLD is difficult as it is usually taken with others drugs. It seems that insulin-sensitizing action of fenofibrate is more important to counter hepatic steatosis than its lipid-lowering property. A recent study showed a significant decrease in hepatic transaminases coupled with a marked decrease of hepatocellular ballooning in humans[54,55].
As far as mice models of NASH are concerned, APOE2 mice fed a western diet showed decreased hepatic macrophage accumulation, which precedes lipid accumulation within hepatocyte, and expressive reduction of lipotoxicity after treatment with fenofibrate. A marked reduction in the expression of proinflammatory genes, great expression of genes implicated in β-oxidation and the suppression of procollagen type 1 expression underlie these findings[56,57].
Total PPAR-gamma activation by rosiglitazone counters insulin resistance, but do not manage to reduce NAFLD in HF mouse models. It can be argued that full activation of PPAR-gamma favors the transcription of lipogenic transcription factors, such as SREBP-1c, and even though animals benefit from anti-inflammatory effects of high adiponectin levels, the upregulation of lipogenesis results in obesity, increased hepatic triglycerides and the maintenance of NAFLD[16,58]. In contrast, mice with NASH benefit from the use of rosiglitazone as it inhibited cell proliferation and diminished collagen expression in hepatic stellate cells in vivo and in vitro[59,60]. In addition, the increase in insulin sensitivity due to enhanced adiponectin transcription and reduced levels of tumor necrosis factor (TNF)-alpha is crucial to the treatment of NASH with rosiglitazone in murine models[61,62].
Humans with NASH also benefit from the regular use of rosiglitazone. The randomized placebo-controlled Fatty Liver Improvement With Rosiglitazone Therapy (FLIRT) trial revealed that 47% of the patients with histologically proved NASH had marked reduction (> 30%) in steatosis score after one year of treatment. Moreover, 38% of the patients achieved normalization of alanine aminotransferase (ALT) values, albeit with no significant improvement of NASH histological features such as hepatocyte ballooning, fibrosis and lobular inflammation/necrosis. In agreement with experimental background, rosiglitazone significantly increased insulin sensitivity and adiponectin levels and the former was correlated with the reduction in the percentage of steatosis in patients of FLIRT trial[63].
When the same set of patients were revisited two years later (FLIRT 2 extension trial), the long-term efficacy of rosiglitazone was attested by the maintenance of reduced ALT levels and reduced HOMA-IR and insulin levels. However, once again, no beneficial effect on liver histology was perceived. It can be argued that even though rosiglitazone exhibited an antisteatogenic effect during the first year and normalized insulin sensitivity and ALT levels, these effects were not enough to tackle NASH features and additional treatments are encouraged[64]. In agreement to the FLIRT trial, the Pioglitazone vs Vitamin E vs Placebo for the Treatment of Nondiabetic Patients with nonalcoholic steatohepatitis trial showed that vitamin E and pioglitazone were able to reduce ALT levels, increase insulin sensitivity, decrease hepatic steatosis and ameliorate lobular inflammation, but without significant improvement in hepatic fibrosis and hepatocyte ballooning. The significant weight gain after pioglitazone treatment was an adverse effect[65].
Recently, the partial activation of PPAR-gamma coupled with the selective activation of PPAR-alpha in the liver by telmisartan (also an AT1 receptor blocker) elicited positive effects to hepatic cytoarchitecture and ultrastructure in mice fed a HF diet[21,66]. Animals showed normal volume density of hepatic steatosis when compared to the untreated group, followed by reduced SREBP-1c expression and insulinemia parallel to greater mitochondrial numerical density revealed by transmission electron microscopy[21]. The same drug was also able to counter steatohepatitis in a murine model through the suppression of macrophage infiltration within hepatocytes, induction of high adiponectin levels and reduction of adipocyte size[66]. In humans, telmisartan showed beneficial effects when compared to losartan (pure AT1 receptor blocker) in the management of NAFLD, highlighting the importance of PPAR activation to treat NAFLD. Telmisartan also yields decreased expression of Nuclear factor κB target genes, such as TNF-alpha and interleukin-6 in diet-induced obese mice, which coupled with increased adiponectin prevent these animals from NASH onset[21,67]. In resemblance with telmisartan, ragaglitazar, a dual PPAR-alpha/PPAR-gamma agonist, tackled hepatic insulin resistance, hepatic steatosis and overweight in a mouse model of metabolic syndrome, whereas total PPAR-gamma agonist rosiglitazone elicited visceral adiposity and hepatomegaly[68].
Pan-PPAR activation by bezafibrate triggered beneficial effects in offspring from obese dams derived from PPAR-alpha activation (increased CPT-1 expression in the liver); PPAR-beta/delta activation (reduced gluconeogenesis due to low hepatic G6Pase and PEPCK expression caused by downregulation of FOXO-1 gene); and PPAR-gamma activation (high FAT/CD36 liver expression, causing greater hepatic lipid oxidation in conjunction with the high CPT-1 expression)[31]. Bezafibrate and GW501516, a PPAR-delta agonist, inhibited NASH development in mice fed a methionine choline-deficient diet. Both treatments elicited greater expression of genes related to beta-oxidation and lipid transportation in hepatocytes concomitant with reduced levels of genes linked to inflammation[69]. An overview of the effects of PPAR activation upon the pathways involved with NAFLD pathogenesis is shown in Figure 1.
Figure 1.
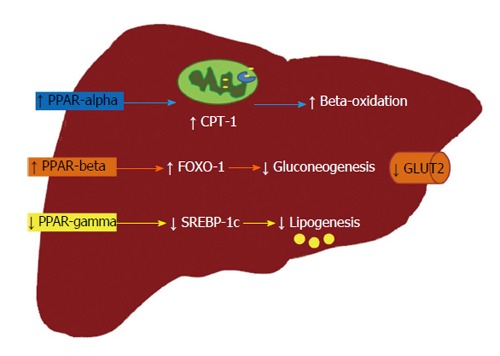
Effects of each peroxisome proliferator-activated receptor isoform in the treatment of non-alcoholic fatty liver disease. PPAR-alpha activation leads to the transcription of CPT-1, a target gene that is crucial to beta-oxidation as it allows the fatty acid to reach the mitochondrial matrix; PPAR-beta/delta activation is involved with FOXO-1 transcription, which reduces the hepatic expression of enzymes involved in gluconeogenesis. Thus, GLUT2 and hepatic glucose production are also significantly reduced; conversely, the partial activation of PPAR-gamma or, even, its reduced expression is linked to diminished lipogenesis. All these events are efficient to tackle NAFLD. PPAR: Peroxisome proliferator-activated receptor; CPT-1: Carnitine palmitoyl transferase-1; FOXO-1: Forkhead box-containing protein O subfamily-1; GLUT2: Glucose transporter 2; SREBP-1c: Sterol regulatory element-binding protein-1c.
PPAR transcriptional activity can be influenced by several kinases as they are phosphoproteins[70]. Mitogen-activated protein kinase (MAPK) activation leads to phosphorylation of PPAR-alpha and PPAR-gamma isoforms. Phosphorylated PPAR-alpha exhibits higher transcription activity, whereas PPAR-gamma shows reduced transcriptional potential after phosphorylation. This knowledge is utterly important when it comes to the attempt to obtain new drugs with huge effectiveness. PPAR-alpha and gamma activate MAPK, while MAPK activation leads to PPAR-alpha and gamma phosphorylation and modulation, configuring an interplay between these two pathways. So, it can be argued that PPAR-alpha beneficial effects are maximized by the interplay with MAPK, which result in favored beta-oxidation in the liver[71]. On the other hand, the reduced transcriptional potential of PPAR-gamma due to MAPK activation might also be beneficial provided that insulin-sensitizing effects of PPAR-gamma are more expressive when there is partial activation of this isoform[72].
CONCLUSION
It is widely understood that PPARs are critically involved in the regulation of hepatic beta-oxidation and lipogenesis pathways, besides influencing hepatic carbohydrate metabolism. These observations prompted the attempt to treat NAFLD by targeting PPARs. Targeting PPAR-alpha has been proved a promising therapeutic approach to control NAFLD through the upregulation of beta-oxidation genes and the inhibition of DNL and gluconeogenesis enzymes. On the other hand, total PPAR-gamma activation shows deleterious effects upon liver histology and physiology based on the increased hepatic lipogenesis. However, partial PPAR-gamma activation as well as dual or pan-PPAR activation shows beneficial effects upon liver structure and functioning. In this way, PPAR modulation by partial activation or selective activation is a promising field of study as it possibilities the reduction of side effects that may stem from total agonism of the receptor. In addition, the role that PPAR-beta/delta has upon liver metabolism remains to be completely unraveled as there is not a PPAR-beta/delta agonist available to the population. Evidences related to this isoform come from experimental studies using selective agonists that are not commercialized or pan-PPAR agonist, which challenges the identification of each isoform properties. Further studies are necessary aiming at translational approaches useful to treat this prevalent metabolic disease in humans worldwide by targeting PPARs.
Footnotes
P- Reviewer: Hoare M, Ikura Y, la Tijera MFH, Lisotti A
S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: The author discloses any conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 28, 2015
First decision: February 7, 2015
Article in press: April 2, 2015
References
- 1.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 2.Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol. 2004;2:1048–1058. doi: 10.1016/s1542-3565(04)00440-9. [DOI] [PubMed] [Google Scholar]
- 3.De Minicis S, Day C, Svegliati-Baroni G. From NAFLD to NASH and HCC: pathogenetic mechanisms and therapeutic insights. Curr Pharm Des. 2013;19:5239–5249. [PubMed] [Google Scholar]
- 4.Fan JG, Cao HX. Role of diet and nutritional management in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2013;28 Suppl 4:81–87. doi: 10.1111/jgh.12244. [DOI] [PubMed] [Google Scholar]
- 5.Ahima RS. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006;14 Suppl 5:242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- 6.Cerf ME. High fat diet modulation of glucose sensing in the beta-cell. Med Sci Monit. 2007;13:RA12–RA17. [PubMed] [Google Scholar]
- 7.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 8.den Boer M, Voshol PJ, Kuipers F, Havekes LM, Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Arterioscler Thromb Vasc Biol. 2004;24:644–649. doi: 10.1161/01.ATV.0000116217.57583.6e. [DOI] [PubMed] [Google Scholar]
- 9.Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. 2007;65:S57–S63. doi: 10.1111/j.1753-4887.2007.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 10.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Moore JB, Gunn PJ, Fielding BA. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients. 2014;6:5679–5703. doi: 10.3390/nu6125679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crescenzo R, Bianco F, Falcone I, Coppola P, Liverini G, Iossa S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur J Nutr. 2013;52:537–545. doi: 10.1007/s00394-012-0356-y. [DOI] [PubMed] [Google Scholar]
- 13.Mori T, Kondo H, Hase T, Murase T. Dietary phospholipids ameliorate fructose-induced hepatic lipid and metabolic abnormalities in rats. J Nutr. 2011;141:2003–2009. doi: 10.3945/jn.111.143602. [DOI] [PubMed] [Google Scholar]
- 14.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Hardwick JP, Osei-Hyiaman D, Wiland H, Abdelmegeed MA, Song BJ. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res. 2009;2009:952734. doi: 10.1155/2009/952734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraulob JC, Souza-Mello V, Aguila MB, Mandarim-de-Lacerda CA. Beneficial effects of rosuvastatin on insulin resistance, adiposity, inflammatory markers and non-alcoholic fatty liver disease in mice fed on a high-fat diet. Clin Sci (Lond) 2012;123:259–270. doi: 10.1042/CS20110373. [DOI] [PubMed] [Google Scholar]
- 17.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28 Suppl 1:68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 18.Mansour M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog Mol Biol Transl Sci. 2014;121:217–266. doi: 10.1016/B978-0-12-800101-1.00007-7. [DOI] [PubMed] [Google Scholar]
- 19.Oliveira LS, Santos DA, Barbosa-da-Silva S, Mandarim-de-Lacerda CA, Aguila MB. The inflammatory profile and liver damage of a sucrose-rich diet in mice. J Nutr Biochem. 2014;25:193–200. doi: 10.1016/j.jnutbio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14:21240–21257. doi: 10.3390/ijms141121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Souza-Mello V, Gregório BM, Cardoso-de-Lemos FS, de Carvalho L, Aguila MB, Mandarim-de-Lacerda CA. Comparative effects of telmisartan, sitagliptin and metformin alone or in combination on obesity, insulin resistance, and liver and pancreas remodelling in C57BL/6 mice fed on a very high-fat diet. Clin Sci (Lond) 2010;119:239–250. doi: 10.1042/CS20100061. [DOI] [PubMed] [Google Scholar]
- 22.Neto-Ferreira R, Rocha VN, Souza-Mello V, Mandarim-de-Lacerda CA, de Carvalho JJ. Pleiotropic effects of rosuvastatin on the glucose metabolism and the subcutaneous and visceral adipose tissue behavior in C57Bl/6 mice. Diabetol Metab Syndr. 2013;5:32. doi: 10.1186/1758-5996-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serviddio G, Giudetti AM, Bellanti F, Priore P, Rollo T, Tamborra R, Siculella L, Vendemiale G, Altomare E, Gnoni GV. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS One. 2011;6:e24084. doi: 10.1371/journal.pone.0024084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1998;273:23786–23792. doi: 10.1074/jbc.273.37.23786. [DOI] [PubMed] [Google Scholar]
- 25.Yu GS, Lu YC, Gulick T. Co-regulation of tissue-specific alternative human carnitine palmitoyltransferase Ibeta gene promoters by fatty acid enzyme substrate. J Biol Chem. 1998;273:32901–32909. doi: 10.1074/jbc.273.49.32901. [DOI] [PubMed] [Google Scholar]
- 26.Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie. 2005;87:81–86. doi: 10.1016/j.biochi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Chien D, Dean D, Saha AK, Flatt JP, Ruderman NB. Malonyl-CoA content and fatty acid oxidation in rat muscle and liver in vivo. Am J Physiol Endocrinol Metab. 2000;279:E259–E265. doi: 10.1152/ajpendo.2000.279.2.E259. [DOI] [PubMed] [Google Scholar]
- 28.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 29.Macdonald GA, Prins JB. Peroxisomal fatty acid metabolism, peroxisomal proliferator-activated receptors and non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2004;19:1335–1337. doi: 10.1111/j.1440-1746.2004.03562.x. [DOI] [PubMed] [Google Scholar]
- 30.Ikura Y, Ohsawa M, Suekane T, Fukushima H, Itabe H, Jomura H, Nishiguchi S, Inoue T, Naruko T, Ehara S, et al. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: impact on disease progression. Hepatology. 2006;43:506–514. doi: 10.1002/hep.21070. [DOI] [PubMed] [Google Scholar]
- 31.Magliano DC, Bargut TC, de Carvalho SN, Aguila MB, Mandarim-de-Lacerda CA, Souza-Mello V. Peroxisome proliferator-activated receptors-alpha and gamma are targets to treat offspring from maternal diet-induced obesity in mice. PLoS One. 2013;8:e64258. doi: 10.1371/journal.pone.0064258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bringhenti I, Ornellas F, Martins MA, Mandarim-de-Lacerda CA, Aguila MB. Early hepatic insult in the offspring of obese maternal mice. Nutr Res. 2015;35:136–145. doi: 10.1016/j.nutres.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJ, Glatz JF, Bonen A. A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J Biol Chem. 2004;279:36235–36241. doi: 10.1074/jbc.M400566200. [DOI] [PubMed] [Google Scholar]
- 34.Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S, McKay R, Monia B, Shutter JR, Lindberg RA, et al. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes. 2006;55:2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 35.Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, Roviaro G, Fargion S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes. 2008;57:1355–1362. doi: 10.2337/db07-0714. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 2014;53:124–144. doi: 10.1016/j.plipres.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, Heirman C, Quartier E, Schuit F, Wahli W, et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- 38.Tsukamoto H, She H, Hazra S, Cheng J, Wang J. Fat paradox of steatohepatitis. J Gastroenterol Hepatol. 2008;23 Suppl 1:S104–S107. doi: 10.1111/j.1440-1746.2007.05294.x. [DOI] [PubMed] [Google Scholar]
- 39.Schultz A, Neil D, Aguila MB, Mandarim-de-Lacerda CA. Hepatic adverse effects of fructose consumption independent of overweight/obesity. Int J Mol Sci. 2013;14:21873–21886. doi: 10.3390/ijms141121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Xiao G, Trujillo C, Chang V, Blanco L, Joseph SB, Bassilian S, Saad MF, Tontonoz P, Lee WN, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J Biol Chem. 2002;277:50237–50244. doi: 10.1074/jbc.M201208200. [DOI] [PubMed] [Google Scholar]
- 41.Day CP. NASH-related liver failure: one hit too many? Am J Gastroenterol. 2002;97:1872–1874. doi: 10.1111/j.1572-0241.2002.05952.x. [DOI] [PubMed] [Google Scholar]
- 42.Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–148. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- 43.Harrington WW, S Britt C, G Wilson J, O Milliken N, G Binz J, C Lobe D, R Oliver W, C Lewis M, M Ignar D. The Effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan Agonists on Body Weight, Body Mass, and Serum Lipid Profiles in Diet-Induced Obese AKR/J Mice. PPAR Res. 2007;2007:97125. doi: 10.1155/2007/97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 45.Tenenbaum A, Motro M, Fisman EZ. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol. 2005;4:14. doi: 10.1186/1475-2840-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brady PS, Marine KA, Brady LJ, Ramsay RR. Co-ordinate induction of hepatic mitochondrial and peroxisomal carnitine acyltransferase synthesis by diet and drugs. Biochem J. 1989;260:93–100. doi: 10.1042/bj2600093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, Gangl MR, Gorgun C, Balschi JA, Ntambi JM, et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J Biol Chem. 2011;286:1237–1247. doi: 10.1074/jbc.M110.138115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts LD, Hassall DG, Winegar DA, Haselden JN, Nicholls AW, Griffin JL. Increased hepatic oxidative metabolism distinguishes the action of Peroxisome proliferator-activated receptor delta from Peroxisome proliferator-activated receptor gamma in the ob/ob mouse. Genome Med. 2009;1:115. doi: 10.1186/gm115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettinelli P, Del Pozo T, Araya J, Rodrigo R, Araya AV, Smok G, Csendes A, Gutierrez L, Rojas J, Korn O, et al. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim Biophys Acta. 2009;1792:1080–1086. doi: 10.1016/j.bbadis.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 50.Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96:1424–1430. doi: 10.1210/jc.2010-2129. [DOI] [PubMed] [Google Scholar]
- 51.Chan SM, Sun RQ, Zeng XY, Choong ZH, Wang H, Watt MJ, Ye JM. Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes. 2013;62:2095–2105. doi: 10.2337/db12-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo K, Sugioka T, Tsukada K, Aizawa M, Takizawa M, Shimizu K, Morimoto M, Suematsu M, Goda N. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, improves hepatic microcirculatory patency and oxygen availability in a high-fat-diet-induced fatty liver in mice. Adv Exp Med Biol. 2010;662:77–82. doi: 10.1007/978-1-4419-1241-1_10. [DOI] [PubMed] [Google Scholar]
- 53.Bargut TC, Frantz ED, Mandarim-de-Lacerda CA, Aguila MB. Effects of a diet rich in n-3 polyunsaturated fatty acids on hepatic lipogenesis and beta-oxidation in mice. Lipids. 2014;49:431–444. doi: 10.1007/s11745-014-3892-9. [DOI] [PubMed] [Google Scholar]
- 54.Fernández-Miranda C, Pérez-Carreras M, Colina F, López-Alonso G, Vargas C, Solís-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–205. doi: 10.1016/j.dld.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Kostapanos MS, Kei A, Elisaf MS. Current role of fenofibrate in the prevention and management of non-alcoholic fatty liver disease. World J Hepatol. 2013;5:470–478. doi: 10.4254/wjh.v5.i9.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lalloyer F, Wouters K, Baron M, Caron S, Vallez E, Vanhoutte J, Baugé E, Shiri-Sverdlov R, Hofker M, Staels B, et al. Peroxisome proliferator-activated receptor-alpha gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arterioscler Thromb Vasc Biol. 2011;31:1573–1579. doi: 10.1161/ATVBAHA.110.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, Staels B, Maeda N, van Bilsen M, Hofker MH. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol. 2006;44:732–741. doi: 10.1016/j.jhep.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 58.Fernandes-Santos C, Carneiro RE, de Souza Mendonca L, Aguila MB, Mandarim-de-Lacerda CA. Pan-PPAR agonist beneficial effects in overweight mice fed a high-fat high-sucrose diet. Nutrition. 2009;25:818–827. doi: 10.1016/j.nut.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 59.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Motomura K, Anania FA, Willson TM, Tsukamoto H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 60.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 61.Nakayama H, Otabe S, Yuan X, Ueno T, Hirota N, Fukutani T, Wada N, Hashinaga T, Yamada K. Effects of adiponectin transgenic expression in liver of nonalcoholic steatohepatitis model mice. Metabolism. 2009;58:901–908. doi: 10.1016/j.metabol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 62.Phillips SA, Kung JT. Mechanisms of adiponectin regulation and use as a pharmacological target. Curr Opin Pharmacol. 2010;10:676–683. doi: 10.1016/j.coph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, Podevin P, Lacorte JM, Bernhardt C, Bruckert E, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100–110. doi: 10.1053/j.gastro.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 64.Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, Hartmann-Heurtier A, Bruckert E, Poynard T. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 65.Violi F, Cangemi R. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;363:1185–1186; author reply 1186. doi: 10.1056/NEJMc1006581. [DOI] [PubMed] [Google Scholar]
- 66.Kudo H, Yata Y, Takahara T, Kawai K, Nakayama Y, Kanayama M, Oya T, Morita S, Sasahara M, Mann DA, et al. Telmisartan attenuates progression of steatohepatitis in mice: role of hepatic macrophage infiltration and effects on adipose tissue. Liver Int. 2009;29:988–996. doi: 10.1111/j.1478-3231.2009.02006.x. [DOI] [PubMed] [Google Scholar]
- 67.Penna-de-Carvalho A, Graus-Nunes F, Rabelo-Andrade J, Mandarim-de-Lacerda CA, Souza-Mello V. Enhanced pan-peroxisome proliferator-activated receptor gene and protein expression in adipose tissue of diet-induced obese mice treated with telmisartan. Exp Physiol. 2014;99:1663–1678. doi: 10.1113/expphysiol.2014.081596. [DOI] [PubMed] [Google Scholar]
- 68.Ye JM, Iglesias MA, Watson DG, Ellis B, Wood L, Jensen PB, Sørensen RV, Larsen PJ, Cooney GJ, Wassermann K, et al. PPARalpha /gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab. 2003;284:E531–E540. doi: 10.1152/ajpendo.00299.2002. [DOI] [PubMed] [Google Scholar]
- 69.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 70.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner OS, Dewar BJ, Earp HS, Samet JM, Graves LM. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. J Biol Chem. 2003;278:46261–46269. doi: 10.1074/jbc.M307827200. [DOI] [PubMed] [Google Scholar]
- 72.Miles PD, Barak Y, He W, Evans RM, Olefsky JM. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J Clin Invest. 2000;105:287–292. doi: 10.1172/JCI8538. [DOI] [PMC free article] [PubMed] [Google Scholar]