

Abstract
Hepatocellular carcinoma (HCC)-related mortality is high because early detection modalities are hampered by inaccuracy, expense and inherent procedural risks. Thus there is an urgent need for minimally invasive, highly specific and sensitive biomarkers that enable early disease detection when therapeutic intervention remains practical. Successful therapeutic intervention is predicated on the ability to detect the cancer early. Similar unmet medical needs abound in most fields of medicine and require novel methodological approaches. Proteomic profiling of body fluids presents a sensitive diagnostic tool for early cancer detection. Here we describe such a strategy of comparative proteomics to identify potential serum-based biomarkers to distinguish high-risk chronic hepatitis C virus infected patients from HCC patients. In order to compensate for the extraordinary dynamic range in serum proteins, enrichment methods that compress the dynamic range without surrendering proteome complexity can help minimize the problems associated with many depletion methods. The enriched serum can be resolved using 2D-difference in-gel electrophoresis and the spots showing statistically significant changes selected for identification by liquid chromatography-tandem mass spectrometry. Subsequent quantitative verification and validation of these candidate biomarkers represent an obligatory and rate-limiting process that is greatly enabled by selected reaction monitoring (SRM). SRM is a tandem mass spectrometry method suitable for identification and quantitation of target peptides within complex mixtures independent on peptide-specific antibodies. Ultimately, multiplexed SRM and dynamic multiple reaction monitoring can be utilized for the simultaneous analysis of a biomarker panel derived from support vector machine learning approaches, which allows monitoring a specific disease state such as early HCC. Overall, this approach yields high probability biomarkers for clinical validation in large patient cohorts and represents a strategy extensible to many diseases.
Keywords: Hepatocellular carcinoma, Biomarkers, Early detection, Selected reaction monitoring, Targeted proteomics
Core tip: The projected rise in hepatocellular carcinoma (HCC) is largely attributed to hepatitis C virus infection with onset of HCC being a latent consequence occurring decades after the original infection. However, other environmental risk factors including alcohol, tobacco, and diet-derived insults that cause liver injury increase the incidence of HCC. The poor prognosis associated with late stage diagnosis renders successful intervention difficult. The methodology described in this review article shows the feasibility of a highly multiplexed manner using multiple reaction monitoring using internal standard peptides to more easily quantify proteins, which narrows the time between discovery and validation in the biomarker pipeline in general.
PROTEOMICS APPROACHES: PATH TO EARLY DIAGNOSIS
This article highlights a proteomics pipeline that is being applied to develop and validate a panel of candidate biomarkers suitable for early hepatocellular carcinoma (HCC) diagnosis. However, the strategies described are also applicable to other objectives including other diseases, drug development, and therapeutic monitoring. Given the time and money involved in bringing a drug to market, the availability of biomarkers capable of identifying potential drug failures early in development is key, but this depends increasingly on advanced proteomic technologies. The characterization of unique protein patterns associated with specific diseases as a discovery strategy to identify candidate biomarkers is one of the most promising areas of clinical proteomics. Cancer, although often classified as a genetic disease, is functionally a proteomic disease. The proteomic tissue microenvironment directly impacts the tumor-host communication system by affecting enzymatic events and the sharing of growth factors[1], so the tumor microenvironment represents a potential source for biomarkers. An example of an early disease biomarker is the prostate-specific antigen (PSA). Today, serum PSA levels are regularly used in the diagnosis of prostate cancer in men. Unfortunately, a reliance on a single protein biomarker is frequently found to be unreliable. Despite decades of effort, single biomarkers generally lack the specificity and sensitivity required for routine clinical use[2]. Due to the heterogeneity that exists from tumor to tumor, biomarker discovery is moving away from the pursuit for an idealized single cancer-specific biomarker in favor of identifying a panel of markers. Disease complexity almost dictates that accurate screening and diagnosis of HCC will require multiple biomarkers. Proteomics affords us the ability to simultaneously interrogate the entire proteome or sub-proteome in order to identify correlations between protein expression (or modifications) and disease progression. In this manner, a panel of biomarkers can be constructed that exhibit the desired sensitivity and specificity necessary for the detection and monitoring of the disease. Recent advances in proteome analysis have focused on the more accessible body fluids including plasma, serum, urine, cerebrospinal fluid, saliva, bronchoalveolar lavage fluid, synovial fluid, nipple aspirate fluid, tear fluid, and amniotic fluid[3]. These body fluids are obtained using minimally invasive procedures, are readily processed, and hence represent clinically tractable cost effective sources of biological material[4].
An effective, clinically useful biomarker should be quantifiable in a readily accessible body fluid such as serum. Since blood comes in contact with almost every tissue, it constitutes a treasure trove of potential biomarkers that provide a systemic picture of the physiological state of the entire body. Every cell in the body contributes to the blood proteome through normal metabolic processes, consequently defined changes characteristic of disease will be reflected in the blood proteome due to perfusion of the affected tissue or organ[5]. Therefore alteration of serum protein profiles can effectively reflect the pathological state of liver injury. It is also ideal to use serum because its sampling is minimally invasive and it is easily prepared with high reproducibility. The disease-related differences in the proteome profile can be attributed to altered protein levels reflecting changes in expression, or dysregulated proteolytic activity affecting protein turnover in diseased cells.
While on the surface this sounds simple to discover a biomarker in serum, the actual process is laborious and time consuming because of the inherent complexity of the human serum proteome, which is composed of thousands of individual proteins. Moreover, the prevalence of serum proteins spans a wide dynamic range-approximately 9 orders of magnitude from pg/mL to mg/mL. These properties represent substantial challenges that often prevent the development and wide-scale utilization of this treasure chest of biological information.
TARGET DISEASE BACKGROUND: HEPATITIS C AND HCC
Liver cancer accounted for 745000 deaths in 2012 alone (World Health Organization. Fact Sheet No 297: 2014). Worldwide, the prevalence of HCC is estimated to affect 180 million people and the incidence continues to rise[6], especially in the United States. It is the fifth most common cancer in men (554000 cases, 7.5% of the total) and the ninth in women (228000 cases, 3.4%)[7]. Chronic infection with hepatitis C virus (HCV) is a major risk factor for the development of HCC with an estimated 130-150 million people having chronic HCV infection (World Health Organization. Fact Sheet No 164:2014). The prognosis for HCC is very poor with an overall 5 year survival rate below 5%, primarily because HCC frequently goes undetected prior to advanced stage disease when therapeutic options are limited. Major risk factors for HCC are infection by the HCV and HBV, alcoholic liver disease, and associated liver cirrhosis. In the developed world however, non-alcoholic fatty liver disease (NAFLD) is increasingly being recognized as a risk factor for HCC without evidence of underlying cirrhosis. Currently, HBV and HCV account for 80%-90% of all HCC worldwide[8-10]. Although HBV remains the most common HCC risk factor worldwide to date, the use of a HBV vaccine in newborns is expected to decrease the HCC incidence associated with HBV infection[8]. In contrast, despite the existence of HCV tests and moderately effective anti-viral therapies, HCV remains a major risk factor for HCC. In fact, the incidence of HCC increased from 2.7 per 100000 to 3.2 per 100000 in 5 years and an estimated 78% of this increase was attributable to latent HCV infections in the general population[11]. In the United States, the incidence of HCC is on the rise stemming from HCV exposures several decades earlier[12], and retrospective studies suggest that once cirrhosis develops, liver disease progresses to either hepatic decompensation (liver failure) or HCC occurs at a rate of 2% to 7% per year[12-16].
The absence of randomized clinical trials notwithstanding, there is compelling evidence suggesting that surgical resection, liver transplantation, or ablative therapies significantly improve survival in HCC patients[17,18]. However, few patients with advanced HCC meet the criteria for these therapeutic modalities. Hence, these clinical options are generally only available to individuals fortunate enough to have been diagnosed with early stage HCC, typically where the tumor is less than 3 cm in diameter without vascular involvement[19,20]. Since early HCC tumors are asymptomatic[17,21,22], routine surveillance of high-risk patients such as those with cirrhosis is recommended as a strategy to detect tumors at a time when therapeutic intervention still offers markedly improved survival rates[23]. Surgical resection offers a 5-year survival rate of approximately 35%, increasing to 45% for small tumors (2-5 cm), thereby highlighting the value of early detection[24]. Hence a screening modality that provides the requisite sensitivity and specificity for early HCC detection would be of significant clinical benefit[25].
Current diagnostic tools for early HCC detection are unfortunately insensitive and/or nonspecific. To date, most established serological diagnostic test for HCC is measures serum alpha-fetoprotein (AFP) levels, however the assay is insufficiently sensitive (39% to 65%) or specificity (65% to 94%) to be very reliable[26-28]. For example, HCV patients with necro-inflammation and liver fibrosis may register high serum AFP levels unrelated to HCC. AFP levels are also elevated in hyperthyroidism[29] and pancreatitis[30] limiting its efficacy as a reliable biomarker for HCC. Biopsied and histopathologically tested samples to discriminate early HCC from benign nodules can be difficult even for expert pathologists[31]. The two newer serological biomarkers, DCP and AFP-L3, fared no better than AFP as their elevation was nonspecifically common in patients without HCC and was influenced by race, gender, age, and severity of liver disease. Therefore it was concluded that screening protocols based on AFP, AFP-L3, and DCP are in rveillance[32]. More recently, screening modalities based on markers including Dickkopf-1 and Midkine designed to complement AFP, are being developed to facilitate screening and diagnosing HCC at an earlier stage[33,34]. However, rigorous validation studies are required before their clinical value is established. According to the National Cancer Institute’s Early Detection Research Network guidelines for biomarker development, robust prospective, randomized, controlled, multi-center trials using a large cohort of patients with hepatitis B and hepatitis C infectious liver disease, NAFLD, and alcohol-induced liver disease are required for validation[35]. Currently, imaging with triphasic computed tomography scanning and magnetic resonance imaging with intravenous gadolinium can improve the diagnostic accuracy, but these techniques are time consuming and very expensive, and are not practical for screening the millions of people identified with known risk factors for HCC. Although ultrasound is very sensitive (in the order of 80%) it is extremely operator dependent[36-38] and is not well suited to differentiate between malignant and benign nodules in the cirrhotic liver. Consequently, development of a minimally invasive test using serum-based biomarkers with the necessary sensitivity and specificity will enhance surveillance, widespread screening, and early HCC detection among the millions who are at risk of developing liver cancer.
SAMPLE PREPARATION: FRACTIONATION AND ENRICHMENT
High abundance serum proteins comprise fewer than two dozen proteins, including albumin and the immunoglobulins, which account for approximately 99% of the total serum protein[39]. The presence of these highly abundant proteins masks the ready detection of medium and low abundance proteins that comprise the repertoire of potential biomarkers. This renders identification of the biomarkers extremely challenging. Serum contains 60% - 80 mg/mL protein, but approximately 65% of this is serum albumin, and approximately 15% are γ-globulins[40-42]. Finding a disease-related protein in such a complex mixture is like searching for a needle in a haystack. So it becomes important to compress the serum protein large dynamic range and reduce the few over-represented (i.e., abundant) proteins by depleting highly abundant proteins to allow detection of lower abundant proteins.
Developments in biomarker-based proteomics technologies are dramatically impacted by the recent realization that a high percentage of the diagnostically useful lower molecular weight serum protein entities are bound to higher molecular weight carrier proteins such as albumin[5,43-45]. In fact, these carrier proteins likely serve to amplify and protect lower molecular weight proteins from clearance by the renal system[46,47]. Conventional protocols for biomarker discovery discard the abundant high molecular weight carrier species such as albumin without realizing the valuable cargo they harbor. Albumin is a carrier/transport protein that sequesters numerous other serum components. Consequently, stripping away albumin from a serum sample risks removing potentially important species. This is like “throwing the baby out with the bath water” by failing to capture the information associated with this valuable resource. Researchers believe that the albumin-bound proteomic signature in serum can be used for early detection and staging of HCC[48]. Therefore, in choosing a method for removal of over represented proteins, the chosen strategy should protect against the nonspecific loss of unrelated proteins. A novel methodology is the aptamer-based Proteominer technology (Bio-Rad) designed to preserve the complexity of the serum proteome using a strategy that does not merely deplete carrier proteins. It constitutes a novel sample preparation protocol that narrows the dynamic range of the serum protein profile without losing the complexity of the entire proteome. This is accomplished through the use of a highly diverse hexabead-based library of combinatorial peptide ligands[49,50]. When complex biological samples such as serum are applied to the beads, the high-abundance proteins such as albumin readily saturate the finite high affinity sites on the beads. However, the retention of carrier proteins such as albumin guarantees that albumin-bound entities are retained in the enriched sample. Medium- and low abundance proteins on the other hand are concentrated by binding to the specific aptamers. As a consequence, the dynamic range of protein profile is reduced while preserving the full complexity of the protein sample. Before performing subsequent high-resolution identification strategies, the samples should be desalted (using standard cleanup kits or desalting columns) to remove electrolytes and other impurities present in the sample[50].
PROTEIN EXPRESSION PROFILING: 2D-DIFFERENCE IN-GEL ELECTROPHORESIS DISCOVERY
Expression profiling is central to proteomics and depends on methods that are able to provide accurately and reproducibly differentiate between the expression profiles in two or more biological samples. Despite recent technological advances in methods for separating and analyzing complex protein mixtures, two-dimensional gel electrophoresis (2D-PAGE) remains a widely used approach in proteomics research[51-55]. 2D-PAGE allows for the separation of thousands of proteins on the basis of both size and charge from a tissue or biological fluid. However, inter-gel variability and the extensive time and labor needed to resolve differences in protein expression have hampered the efficacy of 2D-PAGE. A major technical improvement in 2D-PAGE involves development of the multiplexed fluorescent two-dimensional-difference in-gel electrophoresis (2D-DIGE) method[56] which utilizes direct labeling of the lysine groups on proteins with cyanine (Cy) dyes before isoelectric focusing fractionation in the first dimension. 2D-DIGE is a robust technique proving fruitful in the identification of proteins exhibiting differential expression between samples. It is considered to be one of the most significant advances in quantitative proteomics technology and is crucial to the success of many proteomics initiatives[57,58]. A critical benefit of 2D-DIGE technology is the ability to label 2-3 distinct samples using specific dyes prior to isoelectric focusing of the samples on a single (non-linear) IPG strip[59]. This feature limits spot pattern variability in experimental replicates and therefore the number of gels required to establish confidence in spot pattern differences. Consequently, 2D-DIGE successfully identified statistically significant changes in the serum proteome using only 6 sets of randomly paired patient samples (e.g., 6 HCV and 6 HCC) in 3-5 wk. By labeling the 6 random pairs of HCV and HCC samples with three distinct CyDyes (Cy2, Cy3 and Cy5), we generated 18 HCV and 18 HCC samples for 2D-DIGE and identified 43 significantly differentially expressed proteins. Specifically, 2D-DIGE was used to examine changes in protein abundance between HCV and HCC patient samples by performing Cy3 and Cy5 dye swap experiments under conditions involving a Cy2-labeled internal standard-comprising a pooled preparation of all the patient samples - to normalize between technical replicates[58]. Moreover the substantial sensitivity and broad dynamic range available using these dyes enhances the quantitative accuracy beyond that attainable using silver staining[60]. These advantages renders 2D-DIGE more reliable than 2D-PAGE as a qualitative and quantitative method to interrogate complex proteomes[43], and thus has found utility in proteomics studies examining several human cancers (Figure 1).
Figure 1.
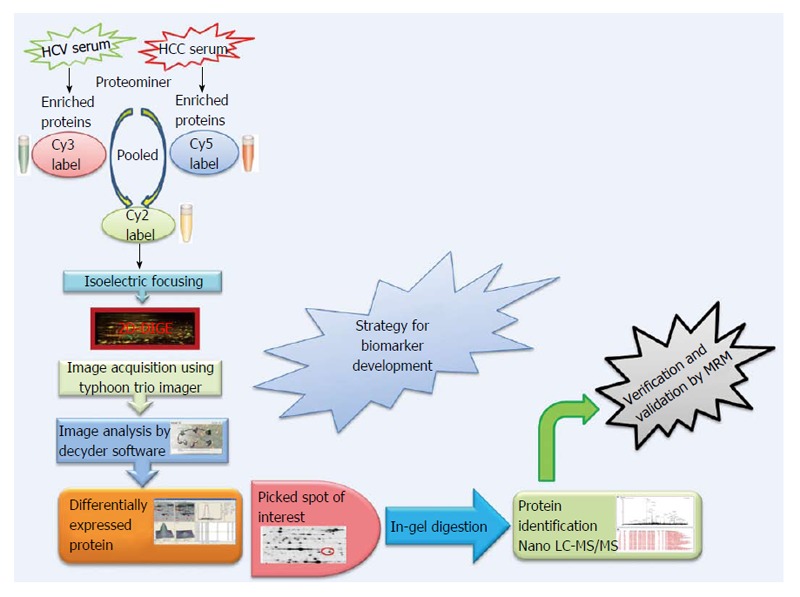
Proteomics strategy for biomarker discovery. HCC: Hepatocellular carcinoma; HCV: Hepatitis C virus; Cy: Cyanine; 2D-DIGE: 2D-difference in-gel electrophoresis; MS: Mass spectrometry; LC: Liquid chromatography.
The CyDye DIGE Fluor minimal dye chemistry relies on a N-hydroxysuccinimidyl ester reactive group that readily forms an amide linkage covalent bond with the epsilon amino group of lysine in proteins. The positive charge of the CyDye substitutes the positive charge in adducted lysine residues at neutral and acidic pH, thus keeping the pI of the protein essentially unchanged. The labeling reaction is designed to be dye that the dye labels approximately 1%-2% of lysine residues. Therefore, each labeled protein typically harbors on average only one dye-labeled residue and is visualized as a single protein spot. The labeling reaction is rapid, taking only 30 min and is quenched by the addition of 1 μL of 10 mmol/L lysine for 10 min. It should be noted that CyDye labeling contributes approximately 500 Da to the mass of the labeled proteins; however, this modest increase in molecular weight does not corrupt the 2D gel pattern since all visualized proteins are labeled. The increased molecular weight conferred by a single CyDye labeling event per protein nor the hydrophobicity of the fluorophore substantially alter gel migration of the labeled proteins. The labeling protocol dictates that the ratio of label to protein be optimized so that less abundant proteins are tagged while keeping the highly abundant proteins in the linear dynamic range for quantitative imaging. 2D-DIGE benefits from the use of a pooled internal standard (i.e., a pooled sample comprising an equal aliquot from each sample in the experiment) labeled with the Cy2 dye. The internal standard is essential for assessing biological and experimental variations and increasing the confidence of the statistical analysis. Sequential scanning of Cy2-, Cy3- and Cy5-labeled proteins (gels) is achieved by the following laser/emission filters: 488/520, 532/580 and 633/670 nm, respectively[61] using a Typhoon Trio Imager (GE Healthcare). Image analysis of the fluorescently labeled proteins requires sequential evaluation of the spot patterns using proprietary (Decyder) software for differential in-gel analysis. In order to reveal changes in protein abundance intra-gel statistical analysis, Cy5/Cy3:Cy2 normalization, and biological variation analysis-which performs inter-gel statistical analysis to provide relative abundance in various groups-are performed. Cy5 or Cy3 samples are normalized against the Cy2 dye-labeled sample (i.e., Cy5:Cy2 and Cy3:Cy2). In the discovery of HCC biomarkers, log abundance ratios were compared between pre-cancerous and cancerous samples from all gels using the chosen statistical analysis (e.g., t-test and ANOVA) using software packages such as DeCyder (GE Healthcare)[62-64]. In addition to being sensitive and quantitative, 2D-DIGE is also compatible with downstream mass spectrometry (MS) protein characterization protocols since most lysine residues in a given protein remain untagged and are accessible for tryptic digestion. Spot detection, the matching and picking of differentially expressed spots of interest among various samples, is done by identifying the spots that reproducibly show expression differences between the cancerous and pre-cancerous samples across biological replicates. Differentially expressed protein spots that satisfy the selection criteria using a statistical significance of P < 0.05 and a threshold of > 1.5-fold change in abundance are selected, and these protein spots are picked from preparative gels involving the 2D-PAGE fractionation of substantially greater amounts of the same protein samples for identification by MALDI-TOF and/or nano-Liquid chromatography (LC)-MS/MS. The combination of 2D-DIGE to confidently detect changes in protein abundance between two samples, with contemporary MS techniques capable of identifying proteins in complex mixtures greatly enhances the biomarker discovery pipeline.
The many advantages of this approach notwithstanding, there remain significant caveats. For example, proteins with a high percentage of lysine residues are more susceptible to multiple labeling events than proteins encoding few or no lysines. Therefore, it is conceivable that a highly abundant protein with few lysine restudies may be readily detectable by conventional 2D-PAGE but be poorly labeled by the CyDye fluorophores in 2D-DIGE and hence be underestimated. Also, while LC-MS/MS typically requires only 1-5 μg of protein, preparative 2D-gels require substantially more protein (approximately equal to 500 μg) for reliable spot detection, which may become a limiting factor in discovery proteomics. Moreover, despite recent advances in high-resolution mass spectrometers that facilitate quantitative analyzes of thousands of proteins, the technology is still not capable of comprehensively characterizing the entire proteome in complex mixtures such as serum. Thorough assessments of these complex samples require prior fractionations to reduce sample complexity using strategies including multidimensional separation (gel-based and chromatography-based technology). Some of the most common methods used for these complex mixtures are 2D-DIGE, isotope-coded affinity tags, isotope-coded protein labeling, tandem mass tags, isobaric tags for relative and absolute quantitation, stable isotope labeling, and label-free quantification. It is noteworthy that the lower abundance proteins detected by 2D-DIGE are refractory to identification by mass spectrometry due to the detection limits of currently available mass spectrometers.
Proteome analysis is often achieved by the sequential use of 2D-PAGE and MS. However, traditional 2D-PAGE techniques are hamstrung by constraints associated with detection limits of low-abundance proteins in complex samples. These limitations have been addressed by the development of sophisticated front-end separation technologies. LC in combination with tandem LC-MS/MS affords researchers the ability to directly analyze complex mixtures in much greater detail without incurring the detection issues associated with 2D-PAGE[65]. The evolution of proteomics technologies has catalyzed large-scale analyzes of differentially expressed proteins under various experimental conditions, which has greatly enriched our understanding of the global physiological processes that occur at the protein level during cellular signaling events[66]. Bottom-up or shotgun proteomics is a high-throughput strategy capable of characterizing very large numbers of proteins simultaneously. Using LC, hundreds of proteins or peptides can be efficiently separated chromatographically into much simpler protein mixtures if not individual species, prior to identification by MS. By pairing distinct prefractionation technologies with complementary MS capabilities, the researcher can customize the analytical resources to meet their specific experimental needs. For example, Orbitrap mass analyzers are frequently coupled to LC to take full advantage of the MS capabilities. Other common configurations include the quadrupole-TOF and linear ion trap quadrupole-Orbitrap to obtain mass determinations with high accuracy and resolution[67,68].
18O-16O LABELING: VERIFICATION
To increase the odds of success an independent, alternative strategy for biomarker development can be used. For this purpose, enriched or fractionated sera is subjected to differential 18O/16O stable isotope labeling, a quantitative MS-based proteomics technique that separates individual peptides on the basis of a 4 Da m/z change. The ratio of 16O labeled (pre-cancerous) and 18O labeled (cancerous) tryptic digestion products can be analyzed by nano LC-MS/MS to determine quantitative changes in peptide abundance between the samples. 18O/16O labeling can be also used in preliminary experiments of selective reaction monitoring to verify the proteins discovered by 2D-DIGE, and to identify optimal precursor and transition product ions for relative quantitation before doing more expensive absolute quantitation using AQUA peptides. Since the spots in 2D-DIGE gels have more than one protein, this approach also increases the confidence in identification of a protein with an altered expression profile.
Investigators planning to use 18O/16O labeling technique need to be aware that incorporation of 18O atoms into peptides can vary when using trypsin as a catalyst[69]. This issue can be ameliorated under conditions of extensive trypsinization. Also, until recently the availability of suitable computational tools was lacking, but this deficit has been addressed with the development of computational algorithms designed to evaluate 18O/16O labeling spectra[70,71]. It is noteworthy that 18O labeling is far less expensive than the stable labeling techniques, rendering it especially attractive for use in biomarker discovery where large numbers of samples are generally analyzed concurrently[71].
In general, the development and validation of biomarkers for clinical use includes four phases: discovery, quantification, verification, and clinical validation[72,73]. Discovery-phase platforms have to date generated large numbers of candidate cancer biomarkers. However, a comparable system for subsequent quantitative assessment and verification of the myriad candidates is lacking, and constitutes the rate-limiting step in the biomarker pipeline[74]. Clearly, discovery “-omics” experiments only serve to identify candidate biomarkers, which must survive rigorous verification and validation before their clinical utility becomes evident[72,75]. At present, established immunoassay platforms, in particular ELISA, are the paragon for quantitative analysis of protein analytes in sera. However, a reliance on immunological methods to verify candidate biomarkers is impractical given the time, effort and cost required to generate the necessary reagents while their value remains uncertain. These constraints restrict the development of immunoassays to the short list of already highly credentialed candidates. Instead, the large majority of new, unproven candidate biomarkers are best examined using intermediate verification technologies with shorter assay development times, lower assay costs, suitable to multiplexing 10-100 s of candidates, small sample requirements, and a high-throughput capability for analyzing hundreds to thousands of serum or plasma samples with high precision[76,77]. The objective is to winnow down the initial list of candidate biomarkers to a more manageable number worth advancing to traditional candidate-validation.
SELECTIVE REACTION MONITORING: VALIDATION
Targeted or quantitative proteomics has emerged as a new technical approach in proteomics and is an essential step in biomarker development. Among the several types of quantification methods, selected reaction monitoring (SRM) and multiple reaction monitoring (MRM) which enable MS-based absolute quantification (also termed AQUA) are emerging as potential rivals to immunoassays[78]. Where once biomarker discovery workflows were bottlenecked at the verification step, steady improvements over the years have resolved the issues such that MS-based techniques now represent viable strategies for biomarker verification[79,80]. SRM is a powerful tandem mass spectrometry method that can be used to monitor target peptides within a complex protein mixture. The specificity surpasses that of traditional immunoassays that may not identify or distinguish between post-translationally modified peptides. The sensitivity and specificity of the SRM assay to identify and quantify a unique peptide in a complex mixture by triple quadrupole mass spectrometry, lends itself to biomarker discovery and validation. This is especially poignant where affinity-based quantitative assays for proteins are unavailable and generating one is hampered by homology between isotypes. However, even isotypic variants of proteins with high homology can be quantified using SRM[81]. Stable-isotope-dilution MRM mass spectrometry (SID-MRM-MS) is a relatively new technique that enables quantification of a protein using isotopically heavy amino acid labeled peptide internal standards that correspond to the protein of interest. By including three to five diagnostic peptides representing a particular protein, the concentration of a protein of interest can be accurately determined based on the known amount of an internal standard added to the sample. The technique is amenable to multiplexing for the simultaneous quantification of 50 or more proteins, and requires only very small samples. SID-MRM-MS is a high-throughput method and has emerged as a valuable technique for validating multiple potential biomarkers[82]. MRM-MS of peptides using stable isotope-labeled internal standards is increasingly being adopted in the development of quantitative assays for proteins in complex biological matrices. These resultant assays are precise (providing primary amino acid sequence information for the analyte), quantitative, are compatible with tandem MS/MS data, and can be developed very rapidly in comparison to immunoassays. Since hundreds of MRM assays can be incorporated into a single method, the multiplexed nature of the technique allows for parallel monitoring of many targets. This is highly attractive from both a scientific and economic perspective. Furthermore, SRM assay design can target predetermined regions within a protein sequence, which would complement methods designed to enrich targets prior to SRM analysis[83]. Even subtle changes in a protein can be readily measured using the SRM approach. Once a SRM or MRM assay is developed, its utility extends to numerous experimental situations where the target protein(s) are to be measured. This has motivated the development of public repositories containing configured MRM assays[84,85].
The availability of previous tandem mass spectrometry data provide reliable information as to which fragment ions will yield the greatest signal in an SRM assay using a triple quadrupole mass spectrometer. The concept of monitoring specific peptides from proteins of interest is well established. As the methods exhibit high specificity and sensitivity within a complex mixture, they can be performed in a fraction of the instrument time relative to discovery-based methods. In addition, the capability to multiplex measurements of numerous analytes in parallel highlights the value of this technology in hypothesis-driven proteomics[77,82,86]. Multiplexing also helps with minimizing the amount of sample needed, an important consideration when working with hard to acquire samples.
ASSAY DEVELOPMENT: BIOMARKER VERIFICATION AND VALIDATION
There are a number of criteria to consider when selecting the optimal transitions for MRM analysis[87,88]. Once the proteins of interest found in the discovery phase are selected, the following steps and considerations should be incorporated in the design of an absolute quantification assay using AQUA-peptides (Figure 2).
Figure 2.
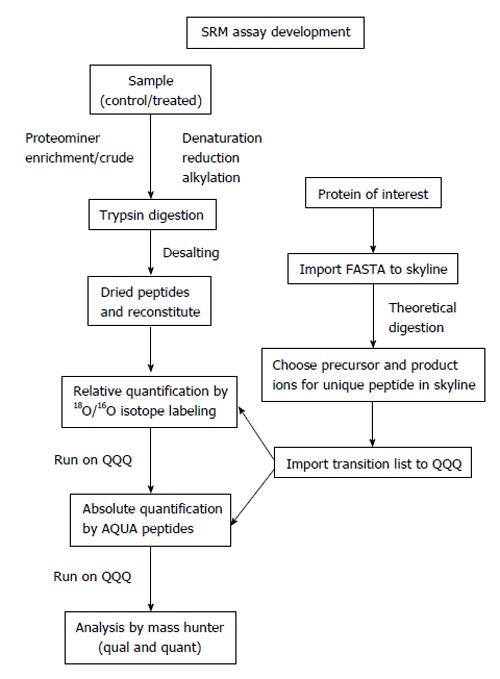
Targeted proteomics work flow for relative and absolute quantification. SRM: Selected reaction monitoring.
Peptide design
First, the peptide sequence for the synthesis of the internal standard is selected. If the absolute amount of a protein is to be quantified, theoretically, any peptide of the protein produced by a proteolytic digestion (e.g., trypsin, chymotrypsin, Glu-C, Lys-C) can be selected. Ideally, two to five peptides are chosen per target protein[82,86,89]. Peptides that deliver strong MS signatures and uniquely identify the target protein - or a specific isoform thereof - have to be identified empirically. Such peptides are termed proteotypic peptides[90] to ensure accurate quantification and exclude artifacts that can originate from unknown modifications of the endogenous protein. In any case, it is essential that the chosen peptide/peptides should be unique to the protein of interest and have a high response in the MS system to afford the greatest sensitivity (“signature peptide”)[91]. Ideally, peptides that have been previously sequenced in a shotgun experiment are chosen to ensure optimal chromatographic behavior, ionization, fragmentation and SRM transitions. Information on sequenced peptides can be found in published proteomics data sets and repositories such as Peptide Atlas[92] (http://www.peptideatlas.org/), Human Proteinpedia[93] (http://www.humanproteinpedia.org/) or PRIDE[94] (http://www.ebi.ac.uk/pride/). These databases can be queried by protein ID, accession number, or peptide sequence. If a peptide is in these databases, the search results return detailed information characterizing the peptide, previously recorded mass spectra of modified and unmodified versions, and information on the samples in which it was identified. To develop methods for targeted SRM analysis on a triple quadrupole mass spectrometer, a key resource exists in academic open source, software called Skyline (http://proteome.gs.washington.edu/software/skyline). The Skyline user interface facilitates the development of mass spectrometer methods and the analysis of data from targeted proteomics experiments performed using SRM. It enables access to MS/MS spectral libraries from a wide variety of sources, to choose SRM filters and verify results based on previously observed ion trap data[95]. The spectrum library can be used to classify fragment ions ranked by intensity, and enable the user to define how many product ions are required to provide a specific and selective measurement given the target sample. The transition lists can be easily exported to triple quadrupole instruments. The Skyline files are in a fast and compact format and are easily shared, even for experiments requiring many sample injections. For a peptide that has not yet been sequenced, an unlabeled peptide can be generated by peptide synthesis and inspected for the ability to function as a reliable AQUA peptide or bioinformatics prediction programs like Skyline can be used to choose potential peptides. In these cases, it is important to take into consideration that certain amino acid sequences or modifications can change the cleavage pattern of the selected protease. For instance, the protease trypsin normally cleaves at the carboxylic side of arginine and lysine. However, if proline is at the amino side of these residues, the bond is resistant to trypsin cleavage. Similarly, if the amino acid on the amino-terminal side is phosphorylated, trypsin cleavage may be inhibited. If the protein contains a series of arginines and lysines, trypsin might cleave after the first arginine or lysine, or after any one following those, creating “missed cleavages” or “ragged ends”. So these theoretical software programs provide a starting point. After using Skyline in preliminary verification, using 18O/16O labeling to check the ratios (relative quantitation) and method development between particular samples is useful because AQUA peptide used for absolute quantitation are very expensive and it is better to be sure which peptides are best suited for absolute quantitation once the method is developed.
Peptide synthesis
AQUA peptides with a peptide sequence corresponding to that generated during digestion of the endogenous protein are synthesized. During peptide synthesis, amino acids containing stable isotopes (18O, 13C, 2H or 15N) are incorporated into the peptide, leading to a peptide with the same chemical and physical characteristics as the endogenous target, but with a defined mass difference. Most commonly, 13C or 15N are used as stable isotopes because they do not lead to chromatographic retention shifts seen in deuterated peptides. Usually, one heavy isotope-labeled leucine, proline, valine, phenylalanine or tyrosine is incorporated into an AQUA peptide leading to a mass shift of 6-8 Da. For tryptic peptides, the C-terminal arginine or lysine is often heavy isotope-labeled such that the resulting y-ion series can be used for monitoring. The peptide is purified, and the exact amount of peptide is determined by amino acid analysis or total nitrogen content. Many commercial vendors synthesize AQUA peptides (including incorporation of a stable isotope-labeled amino acid; e.g., Sigma-Aldrich, Thermo Fisher Scientific or Cell Signaling Technologies).
Peptide validation
After synthesis, the AQUA peptide is analyzed by LC-MS/MS to verify its chromatographic behavior and fragmentation spectra. If they correspond to the previously detected or predicted characteristics, the peptide is ready for use. Quadrupole-based instruments are much better suited to SRM methods by virtue of their ability to generate a continuous ion beam in the SRM transition. In general, trapping instruments are easier to set up and operate, whereas quadrupole-based instruments often require more expertise to optimize fully. However, well-designed targeted SRM methods on a triple quadrupole MS instrument are capable of significantly lower limits of quantification in mixtures of very high complexity, such as whole-cell lysate or unfractionated serum.
Method optimization
An MS spectrum of the peptide (or peptides) is first collected, either by infusion or by LC-MS or by theoretical digestion (using Skyline). Typically, the initial charge-state distribution is interrogated to establish the most sensitive charge state for further monitoring. Note that the actual charge state distribution in a complex mixture may be different than that observed from purified peptides. When an AQUA method is first deployed in a real biological matrix, it is advisable to test multiple charge states to ensure that the most sensitive form of the analyte is ultimately used. A SIM method with a narrow m/z scan range for the charge state with the highest intensity that covers both the AQUA peptide and the endogenous peptide is established from this MS spectrum. For SRM-based methods, the MS/MS spectra of the most intense precursor ions of the AQUA peptide are collected and inspected. Fragment ions at m/z ratios higher than the precursor ion are often more suitable for monitoring because of reduction of noise compared with the lower m/z space. Ion intensity and retention time can be optimized by varying the amount of organic solvent in the peptide loading buffer and in the column equilibration phase of an LC-MS method. In addition, software tools are now available to assist in developing scheduled SRM methods and interpreting their data.
Sample preparation
The biological samples are collected and digested with trypsin. The sample can be directly protease digested, or, to reduce complexity, the sample can be fractionated or enriched before digestion. Measurement of protein abundance by AQUA is indirect and based on the abundance of the resulting peptides; therefore, complete proteolysis is essential, and care should be taken to digest the target mixture with increasing amounts of protease and/or for longer time periods. Before digestion, a denaturation step is important for optimal trypsinization followed by reduction and alkylation. To remove any salts, desalting of sample is also very important.
MS analysis
The peptide mixture containing the endogenous and the AQUA peptide is analyzed on the mass spectrometer by a SIM or a SRM method, and the amount of endogenous peptide is determined. In contrast to a full MS scan, in a SIM experiment only a very narrow mass range is scanned, often by selectively injecting or trapping ions from the narrow scan range to increase the target ion signal-to-noise ratio. In an SRM experiment, a fragment ion or set of fragment ions is monitored. Typically, triplicate measurements for well-designed AQUA experiments produce coefficients of variation between 8%-15%. Using this workflow of proteomics technologies with other novel biostatistical tools, along with the inclusion of clinical factors such as age and gender shown to improve predictive performance[96], will increase the probability of early diagnosis.
These results can be analyzed using machine learning statistical approaches such as a Multivariate Adaptive Regression Splines (MARS) model, which has the ability to search through a large number of candidate predictor variables to determine those most relevant to the classification model. It is a nonparametric regression procedure that seeks to create a classification model based on piecewise linear regressions. MARS are able to reliably track the very complex data structures that are often present in multi-dimensional data[97-100]. In this way, MARS effectively reveals important data patterns and relationships that other models are typically unable to detect.
CONCLUDING REMARKS: THE FUTURE OF CLINICAL PROTEOMICS
Proteomics is a fast maturing discipline that brings the great promise for extending our understanding of the molecular basis of human diseases, and to identify novel biomarkers suitable for patient diagnosis, prognosis and treatment. Hopefully, this improved understanding will inform precision medicine and its application to patient care in the clinical setting. Several recent advances in proteomics have substantially simplified the analysis of serum proteins. Powerful workflows open up new possibilities for biomarker research that may lead to improved clinical assays and faster, more robust drug discovery and development. However there are still some considerations that must be addressed to meet the conditions that satisfy clinical applications. One key issue concerns sample collection, which varies from site to site, but this can be evaluated using the reference sample set from the National Cancer Institute’s Early Detection Research Network, and will serve as a quality control during validation studies. Finally, the power of a panel of biomarkers is exemplified by AFP which requires huge sample numbers compared to statistical approaches. By contrast, quantifiable biomarker panel greatly reduces the requirements for large sample sets during validation[101].
Drugs are launched to market after the lengthy process of development. Despite careful preclinical assessment to identify the most promising candidates, drug development is a lengthy and expensive process. The risk that a drug candidate is withdrawn from testing during clinical trials, especially in the latter stages, is a real concern that comes at considerable cost. There is an enormous impetus within the pharmaceutical industry to adopt new technologies that will hasten drug development and reduce the costs associated with bringing new drugs to market. Biomarkers are emerging as valuable tools in identifying potential drug failures at an early stage that can inform go/no-go decisions. Omics technologies serve an increasingly important role in biomarker discovery and the latter stages of drug development (e.g., target discovery, mechanism of action or predicting toxicity). Recent advances in mass spectrometry including SRM and novel high-resolution capabilities have catalyzed the advent of proteomics and metabolomics as a central component in biomarker discovery, quantification, validation, and clinical verification.
Clinical proteomics is poised to bring important direct “bedside” applications. We foresee a future in which the physician rely upon targeted proteomic analyses during various aspects of disease management. The paradigm shift will directly affect medicine from the development of new therapeutics to clinical practice during the treatment of patients.
Footnotes
P- Reviewer: Coling D, Gam LH, Li Z, Provenzano JC S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: The authors declare that they have no competing interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 15, 2014
First decision: September 28, 2014
Article in press: March 9, 2015
References
- 1.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 2.Petricoin EF, Belluco C, Araujo RP, Liotta LA. The blood peptidome: a higher dimension of information content for cancer biomarker discovery. Nat Rev Cancer. 2006;6:961–967. doi: 10.1038/nrc2011. [DOI] [PubMed] [Google Scholar]
- 3.Hu S, Loo JA, Wong DT. Human body fluid proteome analysis. Proteomics. 2006;6:6326–6353. doi: 10.1002/pmic.200600284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veenstra TD, Conrads TP, Hood BL, Avellino AM, Ellenbogen RG, Morrison RS. Biomarkers: mining the biofluid proteome. Mol Cell Proteomics. 2005;4:409–418. doi: 10.1074/mcp.M500006-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Liotta LA, Ferrari M, Petricoin E. Clinical proteomics: written in blood. Nature. 2003;425:905. doi: 10.1038/425905a. [DOI] [PubMed] [Google Scholar]
- 6.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldwell S, Park SH. The epidemiology of hepatocellular cancer: from the perspectives of public health problem to tumor biology. J Gastroenterol. 2009;44 Suppl 19:96–101. doi: 10.1007/s00535-008-2258-6. [DOI] [PubMed] [Google Scholar]
- 8.Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42 Suppl 3:S206–S214. doi: 10.1016/S1590-8658(10)60507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho WC, Cheng CH. Oncoproteomics: current trends and future perspectives. Expert Rev Proteomics. 2007;4:401–410. doi: 10.1586/14789450.4.3.401. [DOI] [PubMed] [Google Scholar]
- 10.El-Aneed A, Banoub J. Proteomics in the diagnosis of hepatocellular carcinoma: focus on high risk hepatitis B and C patients. Anticancer Res. 2006;26:3293–3300. [PubMed] [Google Scholar]
- 11.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 12.El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74–S83. doi: 10.1053/jhep.2002.36807. [DOI] [PubMed] [Google Scholar]
- 13.Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology. 2002;35:519–524. doi: 10.1053/jhep.2002.32089. [DOI] [PubMed] [Google Scholar]
- 14.Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, Nevens F, Solinas A, Mura D, Brouwer JT, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterology. 1997;112:463–472. doi: 10.1053/gast.1997.v112.pm9024300. [DOI] [PubMed] [Google Scholar]
- 15.Seeff LB, Buskell-Bales Z, Wright EC, Durako SJ, Alter HJ, Iber FL, Hollinger FB, Gitnick G, Knodell RG, Perrillo RP. Long-term mortality after transfusion-associated non-A, non-B hepatitis. The National Heart, Lung, and Blood Institute Study Group. N Engl J Med. 1992;327:1906–1911. doi: 10.1056/NEJM199212313272703. [DOI] [PubMed] [Google Scholar]
- 16.Tong MJ, el-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusion-associated hepatitis C. N Engl J Med. 1995;332:1463–1466. doi: 10.1056/NEJM199506013322202. [DOI] [PubMed] [Google Scholar]
- 17.Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, Christensen E, Pagliaro L, Colombo M, Rodés J. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421–430. doi: 10.1016/s0168-8278(01)00130-1. [DOI] [PubMed] [Google Scholar]
- 18.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 19.Gao JJ, Song PP, Tamura S, Hasegawa K, Sugawara Y, Kokudo N, Uchida K, Orii R, Qi FH, Dong JH, et al. Standardization of perioperative management on hepato-biliary-pancreatic surgery. Drug Discov Ther. 2012;6:108–111. [PubMed] [Google Scholar]
- 20.Belghiti J, Fuks D. Liver resection and transplantation in hepatocellular carcinoma. Liver Cancer. 2012;1:71–82. doi: 10.1159/000342403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawlik TM, Abdalla EK, Vauthey JN. Liver transplantation for hepatocellular carcinoma: need for a new patient selection strategy. Ann Surg. 2004;240:923–924; author reply 924-925. doi: 10.1097/01.sla.0000143896.65218.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedrich-Rust M, Ong MF, Herrmann E, Dries V, Samaras P, Zeuzem S, Sarrazin C. Real-time elastography for noninvasive assessment of liver fibrosis in chronic viral hepatitis. AJR Am J Roentgenol. 2007;188:758–764. doi: 10.2214/AJR.06.0322. [DOI] [PubMed] [Google Scholar]
- 23.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 24.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 25.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 26.Gebo KA, Chander G, Jenckes MW, Ghanem KG, Herlong HF, Torbenson MS, El-Kamary SS, Bass EB. Screening tests for hepatocellular carcinoma in patients with chronic hepatitis C: a systematic review. Hepatology. 2002;36:S84–S92. doi: 10.1053/jhep.2002.36817. [DOI] [PubMed] [Google Scholar]
- 27.Giannini EG, Marenco S, Borgonovo G, Savarino V, Farinati F, Del Poggio P, Rapaccini GL, Anna Di Nolfo M, Benvegnù L, Zoli M, et al. Alpha-fetoprotein has no prognostic role in small hepatocellular carcinoma identified during surveillance in compensated cirrhosis. Hepatology. 2012;56:1371–1379. doi: 10.1002/hep.25814. [DOI] [PubMed] [Google Scholar]
- 28.Daniele B, Bencivenga A, Megna AS, Tinessa V. Alpha-fetoprotein and ultrasonography screening for hepatocellular carcinoma. Gastroenterology. 2004;127:S108–S112. doi: 10.1053/j.gastro.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 29.Bayati N, Silverman AL, Gordon SC. Serum alpha-fetoprotein levels and liver histology in patients with chronic hepatitis C. Am J Gastroenterol. 1998;93:2452–2456. doi: 10.1111/j.1572-0241.1998.00703.x. [DOI] [PubMed] [Google Scholar]
- 30.Hiraoka A, Nakahara H, Kawasaki H, Shimizu Y, Hidaka S, Imai Y, Utsunomiya H, Tatsukawa H, Tazuya N, Yamago H, et al. Huge pancreatic acinar cell carcinoma with high levels of AFP and fucosylated AFP (AFP-L3) Intern Med. 2012;51:1341–1349. doi: 10.2169/internalmedicine.51.6536. [DOI] [PubMed] [Google Scholar]
- 31.Roncalli M, Terracciano L, Di Tommaso L, David E, Colombo M. Liver precancerous lesions and hepatocellular carcinoma: the histology report. Dig Liver Dis. 2011;43 Suppl 4:S361–S372. doi: 10.1016/S1590-8658(11)60592-6. [DOI] [PubMed] [Google Scholar]
- 32.Sterling RK, Wright EC, Morgan TR, Seeff LB, Hoefs JC, Di Bisceglie AM, Dienstag JL, Lok AS. Frequency of elevated hepatocellular carcinoma (HCC) biomarkers in patients with advanced hepatitis C. Am J Gastroenterol. 2012;107:64–74. doi: 10.1038/ajg.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu WW, Guo JJ, Guo L, Jia HL, Zhu M, Zhang JB, Loffredo CA, Forgues M, Huang H, Xing XJ, et al. Evaluation of midkine as a diagnostic serum biomarker in hepatocellular carcinoma. Clin Cancer Res. 2013;19:3944–3954. doi: 10.1158/1078-0432.CCR-12-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forner A, Bruix J. Biomarkers for early diagnosis of hepatocellular carcinoma. Lancet Oncol. 2012;13:750–751. doi: 10.1016/S1470-2045(12)70271-1. [DOI] [PubMed] [Google Scholar]
- 35.Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, Winget M, Yasui Y. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93:1054–1061. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- 36.El-Serag HB. Surveillance for hepatocellular carcinoma: long way to achieve effectiveness. Dig Dis Sci. 2012;57:3050–3051. doi: 10.1007/s10620-012-2413-z. [DOI] [PubMed] [Google Scholar]
- 37.Aghoram R, Cai P, Dickinson JA. Alpha-foetoprotein and/or liver ultrasonography for screening of hepatocellular carcinoma in patients with chronic hepatitis B. Cochrane Database Syst Rev. 2012;9:CD002799. doi: 10.1002/14651858.CD002799.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amarapurkar D, Han KH, Chan HL, Ueno Y. Application of surveillance programs for hepatocellular carcinoma in the Asia-Pacific Region. J Gastroenterol Hepatol. 2009;24:955–961. doi: 10.1111/j.1440-1746.2009.05805.x. [DOI] [PubMed] [Google Scholar]
- 39.Jaffe JD, Keshishian H, Chang B, Addona TA, Gillette MA, Carr SA. Accurate inclusion mass screening: a bridge from unbiased discovery to targeted assay development for biomarker verification. Mol Cell Proteomics. 2008;7:1952–1962. doi: 10.1074/mcp.M800218-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs JM, Adkins JN, Qian WJ, Liu T, Shen Y, Camp DG, Smith RD. Utilizing human blood plasma for proteomic biomarker discovery. J Proteome Res. 2005;4:1073–1085. doi: 10.1021/pr0500657. [DOI] [PubMed] [Google Scholar]
- 41.Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG, Fagan R, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3:311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Shen Y, Kim J, Strittmatter EF, Jacobs JM, Camp DG, Fang R, Tolié N, Moore RJ, Smith RD. Characterization of the human blood plasma proteome. Proteomics. 2005;5:4034–4045. doi: 10.1002/pmic.200401246. [DOI] [PubMed] [Google Scholar]
- 43.Zhou G, Li H, DeCamp D, Chen S, Shu H, Gong Y, Flaig M, Gillespie JW, Hu N, Taylor PR, et al. 2D differential in-gel electrophoresis for the identification of esophageal scans cell cancer-specific protein markers. Mol Cell Proteomics. 2002;1:117–124. doi: 10.1074/mcp.m100015-mcp200. [DOI] [PubMed] [Google Scholar]
- 44.Lau AT, He QY, Chiu JF. Proteomic technology and its biomedical applications. Shengwu Huaxue Yu Shengwu Wuli Xuebao (Shanghai) 2003;35:965–975. [PubMed] [Google Scholar]
- 45.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 46.Mehta AI, Ross S, Lowenthal MS, Fusaro V, Fishman DA, Petricoin EF, Liotta LA. Biomarker amplification by serum carrier protein binding. Dis Markers. 2003;19:1–10. doi: 10.1155/2003/104879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liotta LA, Petricoin EF. Serum peptidome for cancer detection: spinning biologic trash into diagnostic gold. J Clin Invest. 2006;116:26–30. doi: 10.1172/JCI27467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camaggi CM, Zavatto E, Gramantieri L, Camaggi V, Strocchi E, Righini R, Merina L, Chieco P, Bolondi L. Serum albumin-bound proteomic signature for early detection and staging of hepatocarcinoma: sample variability and data classification. Clin Chem Lab Med. 2010;48:1319–1326. doi: 10.1515/CCLM.2010.248. [DOI] [PubMed] [Google Scholar]
- 49.Millioni R, Tolin S, Puricelli L, Sbrignadello S, Fadini GP, Tessari P, Arrigoni G. High abundance proteins depletion vs low abundance proteins enrichment: comparison of methods to reduce the plasma proteome complexity. PLoS One. 2011;6:e19603. doi: 10.1371/journal.pone.0019603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mustafa MG, Petersen JR, Ju H, Cicalese L, Snyder N, Haidacher SJ, Denner L, Elferink C. Biomarker discovery for early detection of hepatocellular carcinoma in hepatitis C-infected patients. Mol Cell Proteomics. 2013;12:3640–3652. doi: 10.1074/mcp.M113.031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wirth PJ, Hoang TN, Benjamin T. Micropreparative immobilized pH gradient two-dimensional electrophoresis in combination with protein microsequencing for the analysis of human liver proteins. Electrophoresis. 1995;16:1946–1960. doi: 10.1002/elps.11501601321. [DOI] [PubMed] [Google Scholar]
- 52.Seow TK, Ong SE, Liang RC, Ren EC, Chan L, Ou K, Chung MC. Two-dimensional electrophoresis map of the human hepatocellular carcinoma cell line, HCC-M, and identification of the separated proteins by mass spectrometry. Electrophoresis. 2000;21:1787–1813. doi: 10.1002/(SICI)1522-2683(20000501)21:9<1787::AID-ELPS1787>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 53.Park KS, Kim H, Kim NG, Cho SY, Choi KH, Seong JK, Paik YK. Proteomic analysis and molecular characterization of tissue ferritin light chain in hepatocellular carcinoma. Hepatology. 2002;35:1459–1466. doi: 10.1053/jhep.2002.33204. [DOI] [PubMed] [Google Scholar]
- 54.Lim SO, Park SJ, Kim W, Park SG, Kim HJ, Kim YI, Sohn TS, Noh JH, Jung G. Proteome analysis of hepatocellular carcinoma. Biochem Biophys Res Commun. 2002;291:1031–1037. doi: 10.1006/bbrc.2002.6547. [DOI] [PubMed] [Google Scholar]
- 55.Pleguezuelo M, Lopez-Sanchez LM, Rodriguez-Ariza A, Montero JL, Briceno J, Ciria R, Muntane J, de la Mata M. Proteomic analysis for developing new biomarkers of hepatocellular carcinoma. World J Hepatol. 2010;2:127–135. doi: 10.4254/wjh.v2.i3.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unlü M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 57.Gharbi S, Gaffney P, Yang A, Zvelebil MJ, Cramer R, Waterfield MD, Timms JF. Evaluation of two-dimensional differential gel electrophoresis for proteomic expression analysis of a model breast cancer cell system. Mol Cell Proteomics. 2002;1:91–98. doi: 10.1074/mcp.t100007-mcp200. [DOI] [PubMed] [Google Scholar]
- 58.Van den Bergh G, Clerens S, Cnops L, Vandesande F, Arckens L. Fluorescent two-dimensional difference gel electrophoresis and mass spectrometry identify age-related protein expression differences for the primary visual cortex of kitten and adult cat. J Neurochem. 2003;85:193–205. doi: 10.1046/j.1471-4159.2003.01668.x. [DOI] [PubMed] [Google Scholar]
- 59.Lin CP, Chen YW, Liu WH, Chou HC, Chang YP, Lin ST, Li JM, Jian SF, Lee YR, Chan HL. Proteomic identification of plasma biomarkers in uterine leiomyoma. Mol Biosyst. 2012;8:1136–1145. doi: 10.1039/c2mb05453a. [DOI] [PubMed] [Google Scholar]
- 60.Knowles MR, Cervino S, Skynner HA, Hunt SP, de Felipe C, Salim K, Meneses-Lorente G, McAllister G, Guest PC. Multiplex proteomic analysis by two-dimensional differential in-gel electrophoresis. Proteomics. 2003;3:1162–1171. doi: 10.1002/pmic.200300437. [DOI] [PubMed] [Google Scholar]
- 61.Yan JX, Devenish AT, Wait R, Stone T, Lewis S, Fowler S. Fluorescence two-dimensional difference gel electrophoresis and mass spectrometry based proteomic analysis of Escherichia coli. Proteomics. 2002;2:1682–1698. doi: 10.1002/1615-9861(200212)2:12<1682::AID-PROT1682>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 62.Tannu NS, Hemby SE. Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat Protoc. 2006;1:1732–1742. doi: 10.1038/nprot.2006.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 64.Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 65.Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 66.Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I, Hochstrasser DF, Williams KL. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev. 1996;13:19–50. doi: 10.1080/02648725.1996.10647923. [DOI] [PubMed] [Google Scholar]
- 67.Hernández F, Sancho JV, Ibáñez M, Abad E, Portolés T, Mattioli L. Current use of high-resolution mass spectrometry in the environmental sciences. Anal Bioanal Chem. 2012;403:1251–1264. doi: 10.1007/s00216-012-5844-7. [DOI] [PubMed] [Google Scholar]
- 68.Provenzano JC, Siqueira JF, Rôças IN, Domingues RR, Paes Leme AF, Silva MR. Metaproteome analysis of endodontic infections in association with different clinical conditions. PLoS One. 2013;8:e76108. doi: 10.1371/journal.pone.0076108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Julka S, Regnier FE. Recent advancements in differential proteomics based on stable isotope coding. Brief Funct Genomic Proteomic. 2005;4:158–177. doi: 10.1093/bfgp/4.2.158. [DOI] [PubMed] [Google Scholar]
- 70.Sadygov RG, Zhao Y, Haidacher SJ, Starkey JM, Tilton RG, Denner L. Using power spectrum analysis to evaluate (18)O-water labeling data acquired from low resolution mass spectrometers. J Proteome Res. 2010;9:4306–4312. doi: 10.1021/pr100642q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye X, Luke B, Andresson T, Blonder J. 18O stable isotope labeling in MS-based proteomics. Brief Funct Genomic Proteomic. 2009;8:136–144. doi: 10.1093/bfgp/eln055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 73.Phillips KA, Van Bebber S, Issa AM. Diagnostics and biomarker development: priming the pipeline. Nat Rev Drug Discov. 2006;5:463–469. doi: 10.1038/nrd2033. [DOI] [PubMed] [Google Scholar]
- 74.Cho WC. Contribution of oncoproteomics to cancer biomarker discovery. Mol Cancer. 2007;6:25. doi: 10.1186/1476-4598-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paulovich AG, Whiteaker JR, Hoofnagle AN, Wang P. The interface between biomarker discovery and clinical validation: The tar pit of the protein biomarker pipeline. Proteomics Clin Appl. 2008;2:1386–1402. doi: 10.1002/prca.200780174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carr SA, Anderson L. Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin Chem. 2008;54:1749–1752. doi: 10.1373/clinchem.2008.114686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 78.Pan S, Aebersold R, Chen R, Rush J, Goodlett DR, McIntosh MW, Zhang J, Brentnall TA. Mass spectrometry based targeted protein quantification: methods and applications. J Proteome Res. 2009;8:787–797. doi: 10.1021/pr800538n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–1186. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 80.Makawita S, Diamandis EP. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin Chem. 2010;56:212–222. doi: 10.1373/clinchem.2009.127019. [DOI] [PubMed] [Google Scholar]
- 81.Sung HJ, Jeon SA, Ahn JM, Seul KJ, Kim JY, Lee JY, Yoo JS, Lee SY, Kim H, Cho JY. Large-scale isotype-specific quantification of Serum amyloid A 1/2 by multiple reaction monitoring in crude sera. J Proteomics. 2012;75:2170–2180. doi: 10.1016/j.jprot.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 82.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6:2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 84.Remily-Wood ER, Liu RZ, Xiang Y, Chen Y, Thomas CE, Rajyaguru N, Kaufman LM, Ochoa JE, Hazlehurst L, Pinilla-Ibarz J, et al. A database of reaction monitoring mass spectrometry assays for elucidating therapeutic response in cancer. Proteomics Clin Appl. 2011;5:383–396. doi: 10.1002/prca.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hüttenhain R, Soste M, Selevsek N, Röst H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Niméus-Malmström E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4:142ra94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keshishian H, Addona T, Burgess M, Mani DR, Shi X, Kuhn E, Sabatine MS, Gerszten RE, Carr SA. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2009;8:2339–2349. doi: 10.1074/mcp.M900140-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jenkins RE, Kitteringham NR, Hunter CL, Webb S, Hunt TJ, Elsby R, Watson RB, Williams D, Pennington SR, Park BK. Relative and absolute quantitative expression profiling of cytochromes P450 using isotope-coded affinity tags. Proteomics. 2006;6:1934–1947. doi: 10.1002/pmic.200500432. [DOI] [PubMed] [Google Scholar]
- 88.Han B, Higgs RE. Proteomics: from hypothesis to quantitative assay on a single platform. Guidelines for developing MRM assays using ion trap mass spectrometers. Brief Funct Genomic Proteomic. 2008;7:340–354. doi: 10.1093/bfgp/eln032. [DOI] [PubMed] [Google Scholar]
- 89.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 91.Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27:190–198. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9:429–434. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mathivanan S, Ahmed M, Ahn NG, Alexandre H, Amanchy R, Andrews PC, Bader JS, Balgley BM, Bantscheff M, Bennett KL, Björling E, Blagoev B, Bose R, Brahmachari SK, Burlingame AS, Bustelo XR, Cagney G, Cantin GT, Cardasis HL, Celis JE, Chaerkady R, Chu F, Cole PA, Costello CE, Cotter RJ, Crockett D, DeLany JP, De Marzo AM, DeSouza LV, Deutsch EW, Dransfield E, Drewes G, Droit A, Dunn MJ, Elenitoba-Johnson K, Ewing RM, Van Eyk J, Faca V, Falkner J, Fang X, Fenselau C, Figeys D, Gagné P, Gelfi C, Gevaert K, Gimble JM, Gnad F, Goel R, Gromov P, Hanash SM, Hancock WS, Harsha HC, Hart G, Hays F, He F, Hebbar P, Helsens K, Hermeking H, Hide W, Hjernø K, Hochstrasser DF, Hofmann O, Horn DM, Hruban RH, Ibarrola N, James P, Jensen ON, Jensen PH, Jung P, Kandasamy K, Kheterpal I, Kikuno RF, Korf U, Körner R, Kuster B, Kwon MS, Lee HJ, Lee YJ, Lefevre M, Lehvaslaiho M, Lescuyer P, Levander F, Lim MS, Löbke C, Loo JA, Mann M, Martens L, Martinez-Heredia J, McComb M, McRedmond J, Mehrle A, Menon R, Miller CA, Mischak H, Mohan SS, Mohmood R, Molina H, Moran MF, Morgan JD, Moritz R, Morzel M, Muddiman DC, Nalli A, Navarro JD, Neubert TA, Ohara O, Oliva R, Omenn GS, Oyama M, Paik YK, Pennington K, Pepperkok R, Periaswamy B, Petricoin EF, Poirier GG, Prasad TS, Purvine SO, Rahiman BA, Ramachandran P, Ramachandra YL, Rice RH, Rick J, Ronnholm RH, Salonen J, Sanchez JC, Sayd T, Seshi B, Shankari K, Sheng SJ, Shetty V, Shivakumar K, Simpson RJ, Sirdeshmukh R, Siu KW, Smith JC, Smith RD, States DJ, Sugano S, Sullivan M, Superti-Furga G, Takatalo M, Thongboonkerd V, Trinidad JC, Uhlen M, Vandekerckhove J, Vasilescu J, Veenstra TD, Vidal-Taboada JM, Vihinen M, Wait R, Wang X, Wiemann S, Wu B, Xu T, Yates JR, Zhong J, Zhou M, Zhu Y, Zurbig P, Pandey A. Human Proteinpedia enables sharing of human protein data. Nat Biotechnol. 2008;26:164–167. doi: 10.1038/nbt0208-164. [DOI] [PubMed] [Google Scholar]
- 94.Jones P, Côté RG, Martens L, Quinn AF, Taylor CF, Derache W, Hermjakob H, Apweiler R. PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 2006;34:D659–D663. doi: 10.1093/nar/gkj138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang M, Block TM, Marrero J, Di Bisceglie AM, Devarajan K, Mehta A. Improved biomarker performance for the detection of hepatocellular carcinoma by inclusion of clinical parameters. Proceedings (IEEE Int Conf Bioinformatics Biomed) 2012;2012 doi: 10.1109/BIBM.2012.6392612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Menon R, Bhat G, Saade GR, Spratt H. Multivariate adaptive regression splines analysis to predict biomarkers of spontaneous preterm birth. Acta Obstet Gynecol Scand. 2014;93:382–391. doi: 10.1111/aogs.12344. [DOI] [PubMed] [Google Scholar]
- 98.Spratt H, Ju H, Brasier AR. A structured approach to predictive modeling of a two-class problem using multidimensional data sets. Methods. 2013;61:73–85. doi: 10.1016/j.ymeth.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Finnerty CC, Ju H, Spratt H, Victor S, Jeschke MG, Hegde S, Bhavnani SK, Luxon BA, Brasier AR, Herndon DN. Proteomics improves the prediction of burns mortality: results from regression spline modeling. Clin Transl Sci. 2012;5:243–249. doi: 10.1111/j.1752-8062.2012.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brasier AR, Ju H, Garcia J, Spratt HM, Victor SS, Forshey BM, Halsey ES, Comach G, Sierra G, Blair PJ, et al. A three-component biomarker panel for prediction of dengue hemorrhagic fever. Am J Trop Med Hyg. 2012;86:341–348. doi: 10.4269/ajtmh.2012.11-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Skates SJ, Gillette MA, LaBaer J, Carr SA, Anderson L, Liebler DC, Ransohoff D, Rifai N, Kondratovich M, Težak Ž, et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J Proteome Res. 2013;12:5383–5394. doi: 10.1021/pr400132j. [DOI] [PMC free article] [PubMed] [Google Scholar]