

Abstract
Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP) is a major resistance factor for the tumor necrosis factor-related apoptosis-inducing ligand TRAIL and in drug resistance in human malignancies. c-FLIP is an antagonist of caspases-8 and -10, which inhibits apoptosis and is expressed as long (c-FLIPL) and short (c-FLIPS) splice forms. c-FLIP is often overexpressed in various human cancers, including breast cancer. Several studies have shown that silencing c-FLIP by specific siRNAs sensitizes cancer cells to TRAIL and anticancer agents. However, systemic use of siRNA as a therapeutic agent is not practical at present. In order to reduce or inhibit c-FLIP expression, small molecules are needed to allow targeting c-FLIP without inhibiting caspases-8 and -10. We used a small molecule inhibitor of c-FLIP, 4-(4-chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH), and show that CMH, but not its inactive analog, downregulated c-FLIPL and c-FLIPS mRNA and protein levels, caused poly(ADP-ribose) polymerase (PARP) degradation, reduced cell survival, and induced apoptosis in MCF-7 breast cancer cells. These results revealed that c-FLIP is a critical apoptosis regulator that can serve as a target for small molecule inhibitors that downregulate its expression and serve as effective targeted therapeutics against breast cancer cells.
Keywords: c-FLIP, Breast cancer, Apoptosis, 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH), Caspases, Death receptors
Introduction
A key inhibitor of death receptor signaling is c-FLIP, which is expressed in various cancers. c-FLIP is expressed as 55-kDa long (c-FLIPL) and 26-kDa short (c-FLIPS) splice forms and is an antagonist of caspases-8 and -10 that inhibits apoptosis downstream of the death receptors TNF-receptor 1, Fas, DR4, DR5, and FADD [1–3]. Moreover, c-FLIP expression is associated with enhanced tumorgenicity and poor clinical outcome in various cancers because it plays a major role in chemotherapeutic drug and TRAIL resistance [3–10]. Previous studies have revealed that high c-FLIP expression is associated with unresponsive tumors, and downregulating its expression should be considered for death receptor targeted and chemotherapy [3, 11–13]. c-FLIP isoforms contain two N-terminal DED domains, but only c-FLIPL contains a C-terminal caspase domain with structural similarity to caspase-8. In the caspase-like domain of c-FLIPL, the catalytically active cysteine is replaced by tyrosine, rendering the molecule proteolytically inactive [14]. c-FLIP isoforms are recruited to the DISC by their DED domains and compete with the initiator caspases for FADD binding sites [3, 10, 15]. c-FLIPS inhibits death signaling at the DISC by inhibiting caspase-8 and -10 activation [3, 16]. At high concentration in the cells, the function of c-FLIPL at the DISC can be anti-apoptotic like c-FLIPS, but at low levels it acts pro-apoptotically by directly heterodimerizing and activating caspase-8 [17–21]. The pro-apoptotic function of c-FLIP is supported by studies showing that c-FLIP-deficient mice die early in embryonic development due to heart failure, which is a defect also seen in caspase-8- and FADD-deficient mice [22,–23]. However, many studies in various cancer types have found that the role of c-FLIPL is generally anti-apoptotic in cancer cells. Importantly, we [24] and others [12, 25, 26] have shown that silencing of c-FLIP expression using small interfering RNAs (siRNAs) promotes spontaneous apoptosis in breast cancer, colorectal, lung, and lymphoma cancer cell lines, and augments TRAIL-, anti-DR5 monoclonal antibody-, and drug-induced apoptosis in various cancer cell types [12, 25–29]. We recently discovered that c-FLIPL interacts with DR5, FADD, and caspase-8 forming an apoptotic inhibitory complex (AIC) in MCF-7 breast cancer cells [24]. Moreover, silencing the c-FLIP gene by a specific siRNA leads to death ligand-independent but DR5-, FADD-, and caspases-8- and -9-dependent apoptosis in these cells. Furthermore, we showed that the knockdown of c-FLIP expression triggers spontaneous apoptosis by activating both the death receptor and mitochondrial pathways and inhibiting breast cancer cell proliferation [24]. Moreover, a recent report identified a checkpoint of the autophagy pathway where cellular and viral FLIPs could limit the Atg3-mediated step of LC3 ubiquitin-like protein conjugation to regulate autophagosome biogenesis. Furthermore, the FLIP-derived short peptides induced growth inhibition and cell death with autophagy by binding to and effectively suppressing Atg3-FLIP interaction, representing new agents as potential anti-cancer therapies [30].
In addition to c-FLIP’s key role in preventing apoptosis, c-FLIP variants have also been shown to be involved in proliferation, cell cycle progression, and carcinogenesis. The overexpression of c-FLIPL inhibited the proteosomal degradation of β-catenin, and the elevated β-catenin levels were shown to either promote Wnt signaling or induce cyclin D expression, leading to the proliferation and cell cycle progression of cancer cells [31, 32]. In both studies, the c-FLIP/β-catenin signals contributing to cell growth were reversed by the selective silencing of c-FLIP expression. Recent results also suggest a role for nuclear c-FLIPL in modulating Wnt signaling [33]. Caspase-8-dependent cleavage of c-FLIP has been shown to produce an N-terminal p22 fragment that directly induced NFκB activation and promoted the proliferation of lymphocytic cells [34]. Furthermore, several studies have shown that c-FLIP overexpression can promote carcinogenesis and aggressiveness of endometrial and cervical cancers [3, 35–38]. These studies highlight the functional importance of c-FLIP in the proliferation of cancer cells.
Inhibition of c-FLIP either by compounds that degrade it or c-FLIP-specific small interfering RNA (siRNA) sensitizes a wide range of cancer cell types to TNF-related apoptosis-inducing ligand (TRAIL) and chemotherapy-induced apoptosis [3, 11–13, 27, 39]. We recently reported that an apoptotic inhibitory complex (AIC) comprised of DR5, FADD, caspase-8, and c-FLIPL exists in MCF-7 cells, and the absence of c-FLIPL from this complex induces ligand-independent caspase-8 activation in the death-inducing signaling complex (DISC), leading to apoptosis [24]. These results clearly show that c-FLIP prevents death signaling in MCF-7 cells. Similarly, ectopic expression of c-FLIP variants decreased apoptosis caused by anticancer agents [3, 12, 13, 16], indicating that over-expression of these proteins may cause resistance to multiple anticancer drugs. Therefore, c-FLIP is a critical apoptosis regulator that can serve as a target for small molecule inhibitors that downregulate its expression and serve as effective targeted therapeutics for cancer treatment. In oder to support this hypothesis, our in vivo results showed that injecting liposomal complexes of c-FLIP-specific siRNA into MCF-7 xenografts eliminated the neoplastic cells without affecting the normal stromal and fibroblastic cells [28].
There does not appear to be a “handle” to inhibit c-FLIP function with small molecule ligands since as mentioned above, c-FLIP has significant structural similarity to caspase-8 [3]. This resemblance with caspase-8 makes c-FLIP protein a very difficult target for drugs to inhibit its function, since small molecules capable of blocking c-FLIP’s recruitment to the DISC could also inhibit the recruitment of caspase-8 and as a result inhibit apoptosis. Therefore, to reduce or inhibit c-FLIP expression, small molecules which target c-FLIP without inhibiting caspases-8 and -10 are needed. Employing a high-throughput chemical screening strategy, a small molecule inhibitor of c-FLIP, 4-(4-chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) (5809354), that downregulates c-FLIP mRNA expression has been identified [40, 41]. Our results show that CMH, but not its inactive analog 4-(4-chloro-2-methylphenoxy)-N-(3-ethoxypropyl) butanamide (CMB) (6094911), downregulates c-FLIPL and c-FLIPS mRNA and protein levels in MCF-7 cells, caused PARP degradation, reduced cell survival, and induced apoptosis.
Materials and methods
Cell culture, materials, reagents and antibodies
The MCF-7 human breast cancer cell lines were obtained from American Type Culture Collection (ATTC, Manassas, VA). MCF-7 human breast cancer cells were maintained in RPMI 1640 medium containing 10% fetal calf serum (FCS) and 100 ng/ml each of penicillin and streptomycin (Invitrogen, Carlsbad, CA) at 37°C in 5% CO2. The MCF-7 multidrug resistant variant MCF-7/ADR20 cells were selected for resistance to increasing concentrations of doxorubicin (DOX) in the growth medium. These cells were maintained in 10 nM DOX and kept in the growth medium without DOX 1–2 weeks before performing experiments.
4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) (5809354) and its inactive analog 4-(4-chloro-2-methylphenoxy)-N-(3-ethoxypropyl) butanamide (CMB) (6094911) were purchased from ChemBridge Corporation (San Diego, CA). Stock solutions (50 mM) of these agents were prepared in dimethyl sulfoxide (DMSO) and stored at 4°C in polystyrene tubes. The final concentration of DMSO in growth medium for survival and apoptosis assays was less than 0.05%.
The following primary antibodies were used: anti-c-FLIP clone G-11 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-c-FLIP clone Dave-2 (ProSci, Poway, CA), anti-DR5 clone B-D37 (IgG2b), anti-Bcl-2 antibody, anti-PARP antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti-DR5 (Cell Signaling Technology, Danvers, MA), anticaspase-8 clone 3-1-9 (BD Biosciences, San Jose, CA), and anti-β-actin clone AC-74 (Sigma-Aldrich, St. Louis, MO).
Western blotting
Cells were harvested, rinsed in cold PBS, and lysed in an M-PER (Pierce Biotechnology, Rockford, IL) containing 1% protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined using the BCA/Cu2SO4 protein assay (Sigma-Aldrich) as described by the manufacturer. Protein lysates (70 μg) were separated on a 10% Bis–Tris gel (Invitrogen) and transferred to an Immobilon-P membrane (Fisher Scientific, Pittsburgh, PA). Membranes were incubated in blocking buffer (PBS, 0.05% Tween 20, 5% skim milk) with specific antibodies, followed by the addition of horseradish peroxidase-conjugated sheep anti-mouse or anti-rabbit secondary antibodies (Amersham Biosciences, Piscataway, NJ). For detecting the anti-c-FLIP clone Dave-2 antibody, a horseradish peroxidase-conjugated goat anti-rat secondary antibody was used (Santa Cruz Biotechnology).
Detection of apoptosis by DAPI staining
DAPI staining was performed on untreated and CMH-treated MCF-7 cells as described previously (42). Briefly, prior to staining, the cells were fixed with 4% paraformaldehyde for 30 min at RT. After washing with PBS by centrifugation at 650×g, DAPI was added to the fixed cells for 5 min, and then the cells were examined by fluorescence microscopy. Apoptotic cells were identified by condensation and fragmentation of nuclei. A minimum of 300 cells were counted for each treatment, and the percentage of apoptotic cells was calculated as the ratio of apoptotic cells to total cells counted × 100. The DAPI staining experiments were performed in triplicate.
Determination of apoptosis and cell death using annexin V and propidium iodide staining
Cells were plated at a density of 2 × 105 cells/ml and treated with 30–100 μM CMH for 48 h, harvested, and stained with fluorescein isothiocyanate-labeled annexin V (BD Biosciences) and propidium iodide according to the manufacturer’s protocol. Cells were analyzed using FAC-Scan (BD Biosciences) flow cytometer and CellQuest software (BD Biosciences). Apoptotic and necrotic cells were analyzed by quadrant statistics on the propidium iodide-positive, annexin V-positive, and the propidium iodide-negative and annexin V-positive cells, respectively.
Effects of the general caspase inhibitor z-VAD-fmk on CMH-induced apoptosis
MCF-7 cells (2 × 104/well in 8-well glass slide) were pretreated with or without 100 μM z-VAD-fmk for 2 h. After that cells were grown in the presence of 10–100 μM PMH at 37°C for 48 h. Numbers of apoptotic cells in the z-VAD-fmk treated and control untreated cells were determined by DAPI staining as described.
RNA isolation and RT-PCR analysis
Total RNA from treated and untreated MCF-7 cells was isolated by Tri Reagent TR-118 (Molecular Research Center, Cincinnati, OH) as described by the manufacturer. One microgram of total RNA was used in reverse transcription reactions with M-MLV reverse transcriptase and oligo (dT) 15 primer (Promega, Madison, WI) as described by the manufacturer. Two microliters of the resulting total cDNA were then used as the template in PCR to measure the mRNA level of interest by using following primers: for c-FLIPL forward, 5′-GCTGAAGTTATCCATCAGGT-3′; reverse, 5′-CATACTGAGATGCAAGAATT-3′; a 840-bp band was produced. For c-FLIPS, the forward primer was the same as c-FLIPL; reverse, 5′-GATCAGGACAATGGGCATAG-3′; a 662-bp band was produced. For β-actin, the forward primer was 5′-CAGAGCAAGAGAGGCATCCT-3′; reverse 5′-TTGAAGGTCTCAAACATGAT-3′; these produce a 200-bp band. The reactions were performed at 94°C for denaturation, 58°C for annealing, and 72°C for extension for 30 cycles. β-Actin mRNA levels were used as internal controls. The amplified fragments were separated on 1.5% agarose gels and visualized by ethidium bromide staining.
Statistical analysis
Data are expressed as the mean ± standard deviation (SE). The statistical significance of intergroup differences was determined using Student’s t test. P values <0.05 were considered statistically significant.
Results
CMH induces robust apoptosis
We used the small molecule inhibitor of c-FLIP, CMH, and showed that CMH, but not its inactive analog CMB (Fig. 1a), robustly reduces cell survival, but the inactive analog had little effect on cell survival (Fig. 1b). The concentration of the drug that reduced cell survival by 50% (IC50 value) in MCF-7 cells was obtained from the CMH dose–response curves using methylene blue cytotoxicity assay after 72-h treatment with increasing concentrations of the drug; the IC50 was approximately 30 μM. We also determined whether CMH inhibits cell survival in MCF-7/ADR20, a drug resistant variant of MCF-7 which expresses 20-, 2.5-, and 1.9-fold resistance to DOX, Taxol, and vincristine, respectively. The data shown in Fig. 1c demonstrate that CMH had even a greater inhibitory effect on cell survival in these drug resistant cells. The IC50 value for 72 h treatment with CMH for MCF-7/ADR20 was 10 μM.
Fig. 1.

CMH but not its inactive analog (CMB) robustly inhibited cell survival in MCF-7 breast cancer cell line. a Chemical structures of 4-(4-chloro-2-methylphenoxy) N-hydroxybutanamide (CMH) and its inactive analog 4-(4-chloro-2-methylphenoxy) N-(3-ethoxypropyl) butanamide (CMB). b MCF-7 or c MCF-7/ADR20 cells were treated with 10–100 μM of CMH or its inactive analog CMB for 72 h. After harvesting the cells, survival was determined by cell survival assay. For each treatment, 1 × 104 cells were seeded in a 96-well plate and treated with or without CMH. Cells were stained with 1% methylene blue in 50% methanol. After the plates were washed by immersing in dH2O, and excess of water was removed the dye was dissolved in 100 μl of 0.5 M HCl. Absorbance was determined by an automated scanning photometer at a wavelength of 630 nm. The results were obtained from triplicate experiments. Percent inhibitions of cell survival by various CMH concentrations compared to the control untreated cells (0) ± SD are shown (P < 0.001)
Next, we determined the levels of CMH-induced apoptosis in MCF-7 cells by DAPI staining. Apoptotic cells were identified by condensation and fragmentation of nuclei. The data shown in Fig. 2a clearly show CMH induced apoptosis visualized by DAPI staining of MCF-7 cells treated with 10–100 μM CMH. Quantitation of apoptosis assessed by DAPI staining revealed that treating the cells with 10–100 μM for 48 h induced 5–28% apoptosis, compared to untreated MCF-7 cells (Fig. 2a). Similar results were obtained when apoptosis was quantified by annexin V binding assay (Fig. 2b). Moreover, treating the cells with 100 μM of the general caspase inhibitor z-VAD-fmk for 48 h significantly inhibited CMH-induced apoptosis as shown in Fig. 3A and B.
Fig. 2.
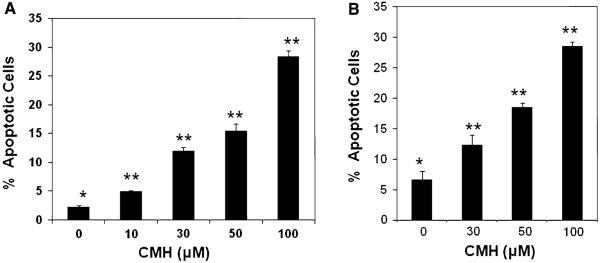
CMH triggered dose-dependent apoptosis in MCF-7 breast cancer cells. a MCF-7 cells were treated with 10–100 μM of CMH for 48 h and apoptotic cell death was determined by DAPI staining as previously described [42]. Apoptotic cells were identified by condensation and fragmentation of nuclei. A minimum of 300 cells were counted for each treatment, and the percentage of apoptotic cells was calculated. b Apoptosis was determined by FACScan analysis as described under “Materials and methods” section. Compensation was executed for each experiment using untreated cells stained with Annexin V and propidium iodide. Experiments were performed in triplicate. Error bars show SD from triplicate measurements (P < 0.005)
Fig. 3.

CMH triggered caspase-dependent apoptosis in MCF-7 breast cancer cells. DAPI staining was carried out as described in “Materials and methods” section. A fluorescence microscopy of untreated (a), 50 and 100 μM CMH treated (b, c), MCF-7 cells compared with z-VAD-fmk pre-treated (d), plus 50 and 100 μM CMH treated (e, f) cells for 48 h. B Apoptosis was quantified by counting fragmented nuclei and condensed stained among 300 cells from the control, z-VAD-fmk, and cells treated with 10–100 μM CMH. Data are average of triplicate experiments (** P < 0.01)
CMH inhibits mRNA expression of c-FLIPL and c-FLIPS variants
In order to determine if the CMH-mediated decrease in c-FLIP variant expression occurred at the mRNA level, semiquantitative RT-PCR was performed as we previously described (16). After treating MCF-7 cells with 10–100 μM CMH for 72 h, a dose-dependent decrease in c-FLIPS transcript level was observed (Fig. 4a–c). Moreover, treatment with 50 and 100 μM CMH for 72 h very effectively decreased the c-FLIPL transcript level (Fig. 3A–C). Our data clearly showed that CMH mediated the reduction of both c-FLIP variants at the mRNA level.
Fig. 4.

CMH-induced decrease of c-FLIPS and c-FLIPL levels occurs at the mRNA level. a MCF-7 cells were treated without (0) or with 10–100 μM CMH for 72 h, and the total RNA was isolated for semiquantitative RT-PCR as we previously described (2). Equivalent addition of RNA was confirmed by PCR analysis of β-actin. (b, c) c-FLIPS and c-FLIPL mRNA expression relative to β-actin expression shown in panel (a) was quantified using Image J 1.37c software. Data are average of triplicate measurements (** P < 0.05)
The molecular mechanism by which CMH downregulates c-FLIPL and c-FLIPS mRNA in MCF-7 cells is not known. Therefore, we investigated whether CMH-induced downregulation of c-FLIPL and c-FLIPS occurs at the transcription level or by altering the stability of their mRNA. We treated the cells with the transcription inhibitor actinomycin D, or actinomycin D plus CMH, as described in the legend of Fig. 5a and b. In order to suppress apoptosis while inhibiting mRNA expression with actinomycin D or actinomycin D plus CMH, we pre-treated the cells with 100 μM of the general caspase inhibitor z-VAD-fmk in DMSO for 2 h and collected them after 6, 12, and 22 h of treatment. RNA from these samples was then prepared, and semiquantitative RT-PCR analysis of c-FLIPL and c-FLIPS mRNA expression was carried out as previously described [16]. Our mRNA analysis showed that actinomycin D treatment inhibited c-FLIPL and c-FLIPS mRNA expression by about 50% after 22 h treatment. Further-more, mRNA levels in the cells treated with actinomycin D plus CMH were similar to the level of mRNA in the cells treated with actinomycin D alone (Fig. 5a, b). These data and the results shown in Fig. 4a indicate that CMH inhibits c-FLIPL and c-FLIPS mRNA expression at the transcription level and does not affect c-FLIPL mRNA stability.
Fig. 5.

Densitometric analysis of semiquantitative RT PCR of c-FLIPL (a) and c-FLIPS mRNA (b) expression following treatment with actinomycin D or actinomycin D plus CMH for 22 h. c-FLIPL and c-FLIPS gene expression were measured in MCF-7 cells treated with 5 μg/ml actinomycin D for the times indicated in the presence or absence of 50 μM CMH. In order to prevent apoptosis, cells were pretreated for 2 h with the 100 μM of the general caspase inhibitor z-VAD-fmk. Results show that while the stability of c-FLIPL mRNA is not affected by CMH, it decreases the stability of c-FLIPS mRNA. mRNA expression was normalized to β-actin mRNA levels. Data are average of triplicate measurements (** P < 0.005)
CMH inhibits expression of c-FLIPL and c-FLIPS variants at the protein level
The results of Western blot analysis corroborated our RT-PCR data and showed that treating MCF-7 cells with 10–100 μM CMH for 72 h reduced levels c-FLIPS and c-FLIPL in MCF-7 cells (Fig. 6a). In order to determine whether CMH only affects c-FLIP variants or it inhibits expression of another anti-apoptotic protein, we treated the cells with 10–100 μM CMH for 72 h and performed Western blotting to detect BCL-2 expression. These results were consistent with degradation of poly(ADP-ribose) polymerase (PARP) as shown in Fig. 6b. The results in Fig. 6b clearly show that CMH treatment did not affect the expression of BCL-2.
Fig. 6.
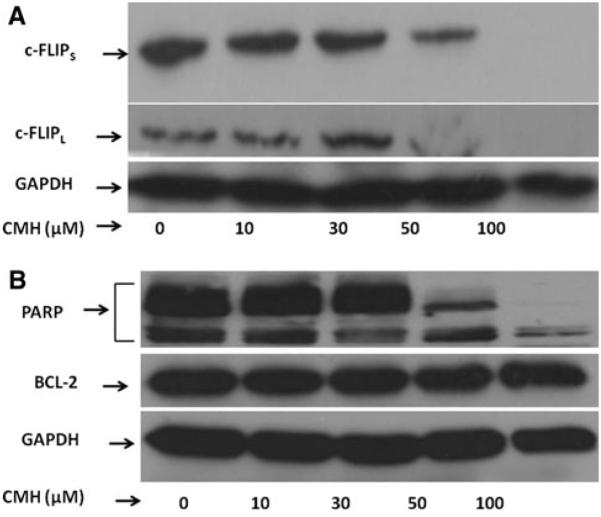
Western blot analysis of c-FLIPS, c-FLIPL, PARP, and BCL-2 in MCF-7 cells treated with CMH. Cell lysates (70 βg protein/lane) from MCF-7 cells treated without (0) or with 10, 30, 50, and 100 βM CMH were subjected to SDS polyacrylamide gel electrophoresis (PAGE) and Western blot analysis using a an anti-c-FLIP antibody (Cell Signaling Technology, Danvers, MA) and b anti-PARP antibody, anti-BCL 2 antibody, as well as anti-GAPDH antibody (Santa Cruz Biotechnology, Santa Cruz, CA)
Discussion
The overexpression of c-FLIP variants in tumor cells is a determinant of resistance to death ligands such as TRAIL and many chemotherapeutic agents in variety of tumors [3]. Moreover, silencing c-FLIP variants with specific siRNAs has sensitized various resistant tumor cells from different types of cancer to these agents [18, 19, 26, 27, 43]. However, in vivo use of siRNA as a systemic therapeutic agent is not feasible because of the lack of appropriate delivery vehicles into the cells. Therefore, agents causing degradation of c-FLIP at the protein level or therapeutics that directly target c-FLIP mRNA are potentially useful modalities for treating tumors resistant to cytokines and chemotherapeutic agents. Moreover, our previous results [28] and the current understanding of c-FLIP action in normal tissues [15, 44, 45] support the notion that c-FLIP-targeted cancer therapy will be well tolerated. However, it is not possible to inhibit c-FLIP function with small molecule ligands, since the cytoprotective DISC binding is mediated by highly conserved DEDs which function by homotypic binding. However, a small molecule CMH (5809354) that induces c-FLIP downregulation and sensitize neoplastic cells to apoptosis induction by the cytokine TRAIL have been identified in global, unbiased chemical screens [40]. CMH causes anoikis in PPC-1 prostate cancer cells cultured in suspension but not in adherent cultures [41]. In order to downregulate c-FLIP variants expression at the mRNA level and trigger apoptosis in MCF-7 and MCF-7/ADR20 cells, we used CMH, which has been shown to sensitize cells to Fas and mediated anoikis [41].
We found that CMH decreased c-FLIPL and c-FLIPS mRNA and protein expression and thereby sensitized MCF-7 and MCF-7/ADR20 cells to apoptosis. A previous report showed that CMH was only able to cause anoikis in PPC-1 prostate cancer cells cultured in suspension and did not affect the adherent cells [41]. However, our previous data using c-FLIPL-specific siRNA [24] and the data shown here using CMH demonstrated that these agents induce apoptosis in adherent cells as well. In this study, we also investigated mechanism by which CMH downregulates c-FLIPL and c-FLIPS at the mRNA level. Our data suggest that CMH decreased transcription of the c-FLIPL and c-FLIPS mRNA.
We also investigated the apoptosis mechanism by which decreased the level of c-FLIPL and c-FLIPS sensitized cells. We found that CMH at lower concentrations (10–30 μM) caused little inhibition of the c-FLIP variants while it decreased cell survival and caused cell death. Moreover, CMH triggered more cell death in the resistant variant compared to sensitive MCF-7 cells at these concentrations. However, CMH-induced cell death occured via a caspase 8-dependent mechanism only at 100 μM. Moreover, PARP degradation was observed at 50 and 100 μM CMH treatment. Our data collectively suggest that at 10–30 μM, CMH induced inhibition of cell survival by a mechanism(s) independent of c-FLIP variants expression.
To our knowledge, this study is the first to show that (a) malignant breast cancer cells can undergo apoptosis when expression of c-FLIPL and c-FLIPS variants is decreased by the small molecule inhibitor CMH and (b) CMH downregulates c-FLIPL and c-FLIPS at the transcriptional level and by decreasing the stability of RNA, respectively. In summary, our data suggest that downregulation of c-FLIPL and c-FLIPS by the small molecule inhibitor CMH causes inhibition of cell survival and triggers cell death. Since c-FLIP variants are master regulators of cell survival and apoptosis, as well contributing to cell proliferation and metastasis, inhibiting the expression of c-FLIP variants by small molecule inhibitors such as CMH may provide a useful strategy to treat malignancies.
Acknowledgments
We thank Dr. Mary D. Kraeszig for her editorial assistance. This work was in part supported by research grant R01CA101743 to ARS from the National Cancer Institute.
References
- 1.Cereghetti GM, Scorrano L. The many shapes of mitochondrial death. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- 2.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 3.Safa AR, Day TW, Wu CH. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr Cancer Drug Targets. 2008;8:37–46. doi: 10.2174/156800908783497087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valnet-Rabier MB, Challier B, Thiebault S, Angonin R, Margueritte G, Mougin C, Kantelip B, Deconinck E, Cahn JY, Fest T. c-Flip protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome. Br J Haematol. 2005;128:767–773. doi: 10.1111/j.1365-2141.2005.05378.x. [DOI] [PubMed] [Google Scholar]
- 5.Gao S, Wang H, Lee P, Melamed J, Li CX, Zhang F, Wu H, Zhou L, Wang Z. Androgen receptor and prostate apoptosis response factor-4 target the c-FLIP gene to determine survival and apoptosis in the prostate gland. J Mol Endocrinol. 2006;36:463–483. doi: 10.1677/jme.1.01991. [DOI] [PubMed] [Google Scholar]
- 6.Morales JC, Ruiz-Magana MJ, Ruiz-Ruiz C. Regulation of the resistance to TRAIL-induced apoptosis in human primary T lymphocytes: role of NF-kappaB inhibition. Mol Immunol. 2007;44:2587–2597. doi: 10.1016/j.molimm.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Park SJ, Kim MJ, Kim HB, Kang CD, Kim SH. Sensitization of imatinib-resistant CML cells to TRAIL-induced apoptosis is mediated through down-regulation of Bcr-Abl as well as c-FLIP. Biochem J. 2009;420:73–81. doi: 10.1042/BJ20082131. [DOI] [PubMed] [Google Scholar]
- 8.Christian PA, Thorpe JA, Schwarze SR. Velcade sensitizes prostate cancer cells to TRAIL induced apoptosis and suppresses tumor growth in vivo. Cancer Biol Ther. 2009;8:73–80. doi: 10.4161/cbt.8.1.7132. [DOI] [PubMed] [Google Scholar]
- 9.Kim JY, Kim EH, Park SS, Lim JH, Kwon TK, Choi KS. Quercetin sensitizes human hepatoma cells to TRAIL-induced apoptosis via Sp1-mediated DR5 up-regulation and proteasome-mediated c-FLIPS down-regulation. J Cell Biochem. 2008;105:1386–1398. doi: 10.1002/jcb.21958. [DOI] [PubMed] [Google Scholar]
- 10.Bagnoli M, Canevari S, Mezzanzanica D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: a key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int J Biochem Cell Biol. 2009 doi: 10.1016/j.biocel.2009.11.015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Day TW, Safa AR. RNA interference in cancer: targeting the anti-apoptotic protein c-FLIP for drug discovery. Mini Rev Med Chem. 2009;9:741–748. doi: 10.2174/138955709788452748. [DOI] [PubMed] [Google Scholar]
- 12.Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005;4:2026–2036. doi: 10.1158/1535-7163.MCT-05-0262. [DOI] [PubMed] [Google Scholar]
- 13.Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L, Johnston PG. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene. 2006;25:488–838. doi: 10.1038/sj.onc.1209122. [DOI] [PubMed] [Google Scholar]
- 14.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 2007;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Day TW, Najafi F, Wu CH. Cellular FLICE-like inhibitory protein (c-FLIP): a novel target for Taxol-induced apoptosis. Biochem Pharmacol. 2006;71:1551–1561. doi: 10.1016/j.bcp.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Du X, Bao G, He X, Zhao H, Yu F, Qiao Q, Lu J, Ma Q. Expression and biological significance of c-FLIP in human hepatocellular carcinomas. J Exp Clin Cancer Res. 2009;28:24. doi: 10.1186/1756-9966-28-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grütter MG. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 19.Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:10409–19401. doi: 10.1074/jbc.M413962200. [DOI] [PubMed] [Google Scholar]
- 20.Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valmiki MG, Ramos JW. Death effector domain-containing proteins. Cell Mol Life Sci. 2009;66:814–830. doi: 10.1007/s00018-008-8489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 23.Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 24.Day TW, Huang S, Safa AR. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem Pharmacol. 2008;76:1694–1704. doi: 10.1016/j.bcp.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers KM, Thomas M, Galligan L, Wilson TR, Allen WL, Sakai H, Johnston PG, Longley DB. Cellular FLICE-inhibitory protein regulates chemotherapy-induced apoptosis in breast cancer cells. Mol Cancer Ther. 2007;6:1544–1551. doi: 10.1158/1535-7163.MCT-06-0673. [DOI] [PubMed] [Google Scholar]
- 26.Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KM, Redmond KM, Johnston PG, Longley DB. c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res. 2007;67:5754–5762. doi: 10.1158/0008-5472.CAN-06-3585. [DOI] [PubMed] [Google Scholar]
- 27.Cheung HH, Mahoney DJ, Lacasse EC, Korneluk RG. Down-regulation of c-FLIP Enhances death of cancer cells by smac mimetic compound. Cancer Res. 2009;69:7729–7738. doi: 10.1158/0008-5472.CAN-09-1794. [DOI] [PubMed] [Google Scholar]
- 28.Day TW, Sinn AL, Huang S, Pollok KE, Sandusky GE, Safa AR. c-FLIP gene silencing eliminates tumor cells in breast cancer xenografts without affecting stromal cells. Anticancer Res. 2009;29:3883–3886. [PubMed] [Google Scholar]
- 29.Chen F, Guo J, Zhang Y, Zhao Y, Zhou N, Liu S, Liu Y, Zheng D. Knockdown of c-FLIP(L) enhanced AD5–10 anti-death receptor 5 monoclonal antibody-induced apoptosis in human lung cancer cells. Cancer Sci. 2009;100(5):940–947. doi: 10.1111/j.1349-7006.2009.01119.x. Epub 2009 Feb 19; Erratum in: Cancer Sci. 2009 Dec;100(12):2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naito M, Katayama R, Ishioka T, Suga A, Takubo K, Nanjo M, Hashimoto C, Taira M, Takada S, Takada R, Kitagawa M, Matsuzawa S, Reed JC, Tsuruo T. Cellular FLIP inhibits beta-catenin ubiquitylation and enhances Wnt signaling. Mol Cell Biol. 2004;24:8418–8427. doi: 10.1128/MCB.24.19.8418-8427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimada K, Nakamura M, Matsuyoshi S, Ishida E, Konishi N. Specific positive and negative effects of FLIP on cell survival in human prostate cancer. Carcinogenesis. 2006;27:1349–1357. doi: 10.1093/carcin/bgi380. [DOI] [PubMed] [Google Scholar]
- 33.Katayama R, Ishioka T, Takada S, Takada R, Fujita N, Tsuruo T, Naito M. Modulation of Wnt signaling by the nuclear localization of cellular FLIP-L. J Cell Sci. 2010;123:23–28. doi: 10.1242/jcs.058602. [DOI] [PubMed] [Google Scholar]
- 34.Rathore N, Matta H, Chaudhary PM. An evolutionary conserved pathway of nuclear factor-kappaB activation involving caspase-mediated cleavage and N-end rule pathway-mediated degradation of IkappaBalpha. J Biol Chem. 2004;279:39358–39365. doi: 10.1074/jbc.M406712200. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Wang S, Song X, Sima N, Xu X, Luo A, Chen G, Deng D, Xu Q, Meng L, Lu Y, Ma D. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogenesis. Gynecol Oncol. 2007;105:571–577. doi: 10.1016/j.ygyno.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 36.Gilot D, Serandour AL, Ilyin GP, Lagadic-Gossmann D, Loyer P, Corlu A, Coutant A, Baffet G, Peter ME, Fardel O, Guguen-Guillouzo C. A role for caspase-8 and c-FLIPL in proliferation and cell-cycle progression of primary hepatocytes. Carcinogenesis. 2005;26:2086–2094. doi: 10.1093/carcin/bgi187. [DOI] [PubMed] [Google Scholar]
- 37.Chen HX, Liu YJ, Zhou XD, Luo RY. Expression of cellular FLICE/caspase-8 inhibitory protein is associated with malignant potential in endometrial carcinoma. Int J Gynecol Cancer. 2005;15:663–670. doi: 10.1111/j.1525-1438.2005.00122.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhou XD, Yu JP, Chen HX, Yu HG, Luo HS. Expression of cellular FLICE-inhibitory protein and its association with p53 mutation in colon cancer. World J Gastroenterol. 2005;11:2482–2485. doi: 10.3748/wjg.v11.i16.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlisi D, Lauricella M, D’Anneo A, Emanuele S, Angileri L, Di Fazio P, Santulli A, Vento R, Tesoriere G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apop tosis by TRAIL-DISC activation. Eur J Cancer. 2009;45:2425–2438. doi: 10.1016/j.ejca.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR, Konopleva M, Gurfinkel D, Mawji IA, Brown E, Reed JC. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006;66(4):2367–2375. doi: 10.1158/0008-5472.CAN-05-1061. [DOI] [PubMed] [Google Scholar]
- 41.Mawji IA, Simpson CD, Hurren R, Gronda M, Williams MA, Filmus J, Jonkman J, Da Costa RS, Wilson BC, Thomas MP, Reed JC, Glinsky GV, Schimmer AD. Critical role for Fas-associated death domain-like interleukin-1 converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J Natl Cancer Inst. 2007;99:811–822. doi: 10.1093/jnci/djk182. [DOI] [PubMed] [Google Scholar]
- 42.Gordon J, Wu CH, Rastegar M, Safa AR. Beta2-micro globulin induces caspase-dependent apoptosis in the CCRF-HSB-2 human leukemia cell line independently of the caspase-3, -8 and -9 pathways but through increased reactive oxygen species. Int J Cancer. 2003;103:316–327. doi: 10.1002/ijc.10828. [DOI] [PubMed] [Google Scholar]
- 43.Dutton A, O’Neil JD, Milner AE, Reynolds GM, Starczynski J, Crocker J, Young LS, Murrat PG. Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin’s lymphoma cells from autonomous Fas-mediated death. Proc Natl Acad Sci USA. 2004;101:6611–6616. doi: 10.1073/pnas.0400765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kataoka T. The caspase-8 modulator c-FLIP. Crit Rev Immunol. 2005;25:31–58. doi: 10.1615/critrevimmunol.v25.i1.30. [DOI] [PubMed] [Google Scholar]
- 45.Burns TF, El-Deiry WS. Identification of inhibitors of TRAIL-induced death (ITIDs) in the TRAIL sensitive colon carcinoma cell line SW480 using a genetic approach. J Biol Chem. 2001;276:37879–37886. doi: 10.1074/jbc.M103516200. [DOI] [PubMed] [Google Scholar]