

Abstract
Hereditary multiple exostoses is a pediatric skeletal disorder characterized by benign cartilaginous tumors called exostoses that form next to growing skeletal elements. Hereditary multiple exostoses patients carry heterozygous mutations in the heparan sulfate (HS)-synthesizing enzymes EXT1 or EXT2, but studies suggest that EXT haploinsufficiency and ensuing partial HS deficiency are insufficient for exostosis formation. Searching for additional pathways, we analyzed presence and distribution of heparanase in human exostoses. Heparanase was readily detectable in most chondrocytes, particularly in cell clusters. In control growth plates from unaffected persons, however, heparanase was detectable only in hypertrophic zone. Treatment of mouse embryo limb mesenchymal micromass cultures with exogenous heparanase greatly stimulated chondrogenesis and bone morphogenetic protein signaling as revealed by Smad1/5/8 phosphorylation. It also stimulated cell migration and proliferation. Interfering with HS function both with the chemical antagonist Surfen or treatment with bacterial heparitinase up-regulated endogenous heparanase gene expression, suggesting a counterintuitive feedback mechanism that would result in further HS reduction and increased signaling. Thus, we tested a potent heparanase inhibitor (SST0001), which strongly inhibited chondrogenesis. Our data clearly indicate that heparanase is able to stimulate chondrogenesis, bone morphogenetic protein signaling, cell migration, and cell proliferation in chondrogenic cells. These properties may allow heparanase to play a role in exostosis genesis and pathogenesis, thus making it a conceivable therapeutic target in hereditary multiple exostoses.
Heparanase is a multifunctional protein that is involved in a variety of physiologic and pathologic processes and represents the only entity of its kind encoded in the mammalian genome.1,2 The enzyme can cleave heparan sulfate (HS) chains present in syndecans, glypicans, and other HS-rich proteoglycans and, in so doing, affects proteoglycan homeostasis, function, mobility, and internalization and can influence various processes, including cell spreading, migration, and proliferation.3,4 Importantly, cleavage of HS chains can also release HS-bound cytokines, growth factors, and signaling proteins, thus enhancing their bioavailability, range of action, and effects on target cells.5,6 Latent heparanase on the cell surface can interact with syndecans, and this interaction leads to rapid internalization of the complex and delivery to lysosomes where the enzyme is activated by cathepsin L.7 Heparanase has additional important functions. It can lead to syndecan clustering and activation of downstream effector pathways that involve protein kinase C, Src, and Rac14 and can activate β1-integrin,8 mechanisms all facilitating cell spreading and migration.9 Because some of these actions do not appear to require enzymatic activity and can be elicited by inactive heparanase as well, they are likely to involve and rely on the nonenzymatic domains of the protein.10 As a reflection of its multiple and potent biological activities, heparanase is often up-regulated in human cancers and is closely associated with, and may lead to, neoplastic cell behavior and metastasis.2,8,11 The protein is believed to facilitate the invasive behavior of cancer cells and tumor growth by release of extracellular matrix-bound angiogenic factors, including vascular endothelial growth factor and by up-regulating expression of important genes such as HGF, MMP9, and VEGFA itself.1,6,12 Indeed, inhibitors of heparanase administered systemically were found to reduce progression of tumor xenografts in mice.13 These and other studies have led to the current notion that heparanase inhibition by pharmacologic strategies may represent a promising and effective cancer therapy.14
Benign ectopic cartilaginous/bony tumors called exostoses characterize the pediatric skeletal disorder hereditary multiple exostoses (HME).15,16 The exostoses are growth plate-like structures that form next to, but never within, the growth plates of long bones, ribs, pelvis, and other skeletal elements. Because of size and location, the exostoses can cause a variety of health problems, including skeletal growth retardation and deformities, chronic pain, compression of nerves and blood vessels, and psychological concerns.15,17,18 In the majority of HME patients the exostoses remain benign through life, but in approximately 2% to 5% of them the exostoses progress to malignancy, turn into osteosarcomas or chondrosarcomas, and thus become life threatening.19 Most HME patients carry heterozygous loss-of-function mutations in EXT1 or EXT2 that encode glycosyltransferases responsible for HS synthesis.20,21 EXT1 and EXT2 form protein complexes in the Golgi and are both required for HS synthesis.20 HME patients thus have reduced levels, but not lack, of HS in their tissues. Surprisingly however, the cartilaginous portions of human exostoses display barely detectable levels of HS,22 indicating that exostosis formation may require a severe loss of HS beyond what would be caused by mere EXT haploinsufficiency. This requirement was confirmed in transgenic mouse studies that involved conditional Ext1 and/or Ext2 ablation.23–25 Studies have suggested mechanisms that could account for a severe drop of HS in human exostoses, including EXT loss-of-heterozygosity, large and encompassing genomic deletions, a second hit in another gene, and background genetic traits such as modifiers.21,26,27 However, there are still no clear answers nor obvious genotype-phenotype correlations in HME, despite that the syndrome can vary significantly in severity within affected family members and among persons from different families.28
We, therefore, sought to investigate possible additional pathogenic pathways in exostosis formation and progression in HME and focused on heparanase. We do find that the protein is widespread in the growth plate-like cartilaginous portions of human exostoses but displays a restricted distribution in normal growth plates from unaffected control patients. We show also that the enzyme greatly stimulates chondrogenesis, a mechanism that could promote and facilitate inception and growth of exostoses.
Materials and Methods
Human Exostosis Samples
Exostosis tissue samples from four consenting HME patients were taken after surgical removal and were processed by the centralized Children's Hospital of Philadelphia Pathology Laboratory. Control growth plate tissue was harvested from surgically removed toes of children who were undergoing treatment for polydactyly. Fixed tissue was embedded in paraffin and sectioned (6 μm). After specimens were used for clinical diagnosis, extra slides were de-identified and provided to us for the present study. This practice was reviewed and approved by Children's Hospital of Philadelphia's Institutional Review Board.
Monolayer and Micromass Cell Culture
ATDC5 cells, derived from mouse embryonal carcinoma, were cultured in 10-cm dishes with a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 (Life Technologies, Carlsbad, CA) supplemented with 5% fetal bovine serum (ATCC, Manassas, VA), 10 μg/mL human transferrin (Mediatech, Herndon, VA), 3 × 10−8 mg/L sodium selenite (Mediatech), and 20 μg/mL insulin at 37°C under 5% CO2. Experiments were conducted once cells reached confluence.
Micromass cultures were prepared from the mesenchymal cells of embryonic day 11.5 mouse embryo limb buds.29 Dissociated cells were suspended at a concentration of 5 × 106 cells/mL in Dulbecco's modified Eagle's medium that contained 3% fetal bovine serum and antibiotics. Micromass cultures were initiated by spotting 20 μL of the cell suspensions (1.5 × 105 cells) onto the surface of 24-well tissue culture dishes. After a 90-minute incubation at 37°C in a humidified carbon dioxide incubator to allow for cell attachment, the cultures were supplied with 0.25 mL of medium. After 24 hours, medium supplemented with the indicated concentrations of bis-2-methyl-4-aminoquinolyl-6-carbamide (Surfen; obtained from the Open Chemical Repository in the Developmental Therapeutic Program at the National Cancer Institute, NSC12155), bacterial heparitinase III (Sigma-Aldrich, St. Louis, MO), recombinant human bone morphogenetic protein (BMP)2 (R&D Systems, Minneapolis, MN), recombinant human heparanase (kindly provided by Drs. Israel Vlodavsky, Technion, Haifa, Israel, and Ralph Sanderson, University of Alabama at Birmingham, Birmingham, AL), or SST0001 (Sigma-Tau Pharmaceuticals Inc., Gaithersburg, MD). Fresh reagents (drug and/or protein) were given with medium change every other day. Equivalent amounts of vehicle were added to control cultures. Cultures were stained with Alcian Blue (pH 1.0) after 4 and 6 days to monitor chondrogenic cell differentiation by standard methods. Micromass analysis was performed with ImageJ version 1.47 (NIH, Bethesda, MD). Images were made binary under an RGB threshold, and particle analysis was used to measure nodule size, number, and Alcian Blue–positive area.
Cell Proliferation and Migration Analysis
ATDC5 cells were treated at day 0 with human heparanase (400 ng/mL) or a vehicle control. At this time, one culture dish was harvested for DNA. DNA was also isolated from control and treated cells at day 3, 7, and 14. DNA isolation was performed with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocols and measured with a NanoDrop (ThermoScientific, Marietta, OH). ATDC5 migration was analyzed via a scratch test. Cells were replated on a six-well dish and allowed to reach confluence before treatment. After 24 hours of treatment, a straight line scratch was made with a P10 pipette tip. Reference points were made with a fine-tipped marker on the bottom of the dish. Images at time 0 were taken with a phase-contrast microscope (Leica, Wetzlar, Germany). Cells were cultured at 37°C in a humidified carbon dioxide incubator for 4 hours, with images in the same field of view taken at 2 and 4 hours. The open scratch area was quantified with ImageJ (n = 6 per treatment group; NIH). In micromass cultures, migration was determined by expansion of the micromass itself from day 0 to day 4 or 6. The diameters of untreated micromasses (n = 10) were measured and averaged for a baseline size. This baseline was subtracted from the average diameter of treated cultures (n = 6) to estimate cell migration.
Semiquantitative PCR Analyses
Total RNA was extracted from cells by the guanidine phenol method with the use of TRIzol reagent (Invitrogen) according to the manufacturer's protocols and was measured with a NanoDrop. One microgram of total RNA was reverse transcribed with the SuperScript III First-Strand Synthesis System (Life Technologies). Band intensities for semiquantitative PCR were normalized to glyceraldehyde-3-phosphate dehydrogenase and compared, whereas band intensities and cycle numbers were linear. The following primer sets were used: Gapdh forward primer (5′-CGTCCCGTAGACAAAATGGT-3′) and reverse primer (5′-TTGATGGCAACAATCTCCAC-3′); Runx2 forward primer (5′-CGCACGACAACCGCACCAT-3′) and reverse primer (5′-AACTTCCTGTGCTCCGTGCTG-3′); Agg forward primer (5′-GGAGCGAGTCCAACTCTTCA-3′) and reverse primer (5′-CGCTCAGTGAGTTGTCATGG-3′); Col2a1 forward primer (5′-CTACGGTGTCAGGGCCAG-3′) and reverse primer (5′-GTGTCACACACACAGATGCG-3′); BMP2 forward primer (5′-TCTTCCGGGAACAGATACAGG-3′) and reverse primer (5′-TCTCCTCTAAATGGGCCACTT-3′); BMPRI forward primer (5′-GCTTGCGGCAAtCGTGTCTAA-3′) and reverse primer (5′-GCAGCCTGTGAAGATGTAGAGG-3′); and BMPRII forward primer (5′-CACACCAGCCTTATACTCTAGATA-3′) and reverse primer (5′-CACATATCTGTTATGAAACTTGAG-3′). Band intensities were quantified by computer-assisted image analysis (ImageQuant TL; GE Healthcare, Chalfont St. Giles, UK).
Protein Analysis
ATDC5 cells were grown to 100% confluence in six-well plates and treated with indicated concentrations of Surfen. Micromass cultures were treated with control vehicle, Surfen, human heparanase, SST0001, or recombinant human BMP2. At specified time points, total cellular proteins were harvested in SDS-PAGE sample buffer, electrophoresed on 4% to 15% SDS-Bis-Tris gels (40 μg per lane), and transferred to polyvinylidene difluoride membranes (Invitrogen). Membranes were incubated overnight at 4°C with dilutions of antibodies against phospho-Smad1/5/8 (dilution 1:1000; 9511; Cell Signaling Technology Inc., Danvers, MA), Smad1 (dilution 1:500; 63439; Abcam Inc., Cambridge, MA), or human heparanase (dilution 1:750; 59787; Abcam Inc.). Enhanced chemiluminescent immunoblotting detection system (ThermoScientific) was used to detect the antigen–antibody complexes. The membranes were re-blotted with antibodies to α-tubulin (dilution 1:2000; T-5168; Sigma-Aldrich) for normalization, and band intensities were quantified by computer-assisted image analysis.
Immunohistochemistry
Immunostaining for heparanase was performed with paraffin sections that were deparaffinized and de-masked with 5 mg/mL hyaluronidase for 1 hour at 37°C to de-mask. Sections were incubated with anti-heparanase polyclonal antibodies (59787; Abcam Inc.) at 1:200 dilution in 3% normal goat serum in phosphate-buffered saline overnight at 4°C. After rinsing, sections were then incubated with biotinylated goat anti-rabbit secondary antibody, and the signal was visualized with a horseradish peroxidase/diaminobenzidine detection immunohistochemistry kit according to the manufacturer's instructions (Abcam Inc.). For collagen X staining, companion serial sections were treated with 0.1% pepsin in 0.02N HCl for 10 minutes at 37°C, incubated with 1:1000 dilution of rabbit collagen X antibodies (Cosmo Bio, Carlsbad, CA) in 10% normal goat serum for 1 hour, followed by treatment with ABC reagent (Vector Laboratories, Burlingame, CA) for 1 hour for antibody detection. Bright-field images were taken with a SPOT insight camera (Diagnostic Instruments, Inc., Sterling Heights, MI) operated with SPOT software version 4.0 (Diagnostic Instruments, Inc.).
Statistical Analysis
Data were validated by two-tailed Student's t-tests. The threshold for significance for all tests was set as P < 0.05.
Results
Broad Heparanase Distribution in Human Exostoses
Mature and symptomatic exostoses removed at surgery are typically composed of a cartilaginous cap and an underlying bony stem that connects with the affected skeletal element, be it a long bone or a rib. The cartilaginous cap often displays a growth plate-like structure and organization, with small chondrocytes located at its distal end and surrounded by a connective/perichondrium-like tissue layer and with large hypertrophying chondrocytes located more proximally near the bony portion.15,16 To determine whether heparanase presence and distribution were altered in exostoses, we obtained exostoses from consenting HME patients undergoing surgical treatment and processed them for section immunostaining with rabbit antibodies against human heparanase. For comparison, we used longitudinal sections of growth plates from control nonaffected persons who had undergone surgical treatment for polydactyly. In these control specimens, heparanase staining was clearly and strongly detected in the hypertrophic chondrocytes at the chondro-osseous junction (Figure 1, A and B) and in the perichondrium (Figure 1C), but little to no staining was observed in resting and proliferative zones (Figure 1, A–C). Blood vessels that invaded the secondary ossification center displayed high heparanase staining as expected,30 thus acting as an internal positive control of staining specificity and sensitivity (Figure 1D). In contrast, heparanase staining was found in all chondrocytes within the exostoses, regardless of location within the tissue and apparent maturation stage (Figure 1, E–G). Tissue portions that contained enlarged and hypertrophying cells in clusters were even more strongly stained, particularly in their pericellular compartment (Figure 1H). However, staining for collagen X in companion serial sections showed that it was restricted to mature and hypertrophic chondrocytes (Figure 1I).
Figure 1.
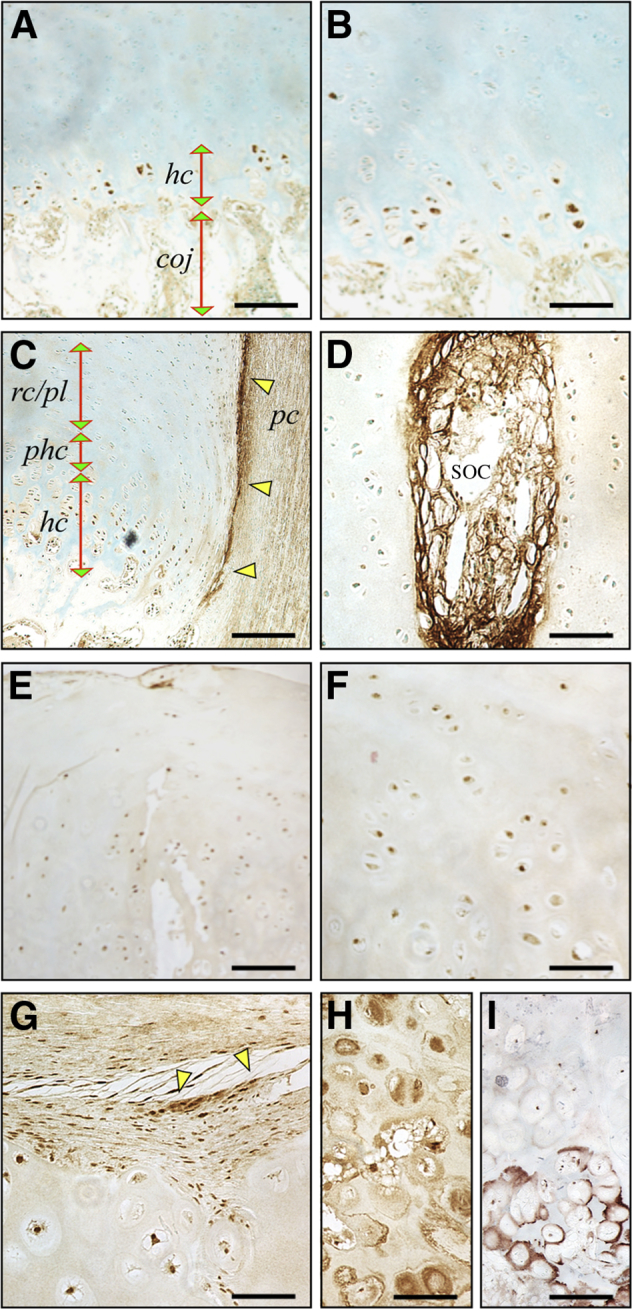
Heparanase is broadly distributed in human exostoses but restricted in control growth plate. A–D: Immunohistochemical staining of longitudinal sections of control human growth plate shows that heparanase is detectable in hcs at the coj and is also prominent in perichondrium (C, arrowheads). Strong staining is appreciable in a blood vessel present in the SOC as expected (D). E–H: Sections from human exostoses were stained in parallel. Heparanase staining is clear in nearly every chondrocyte regardless of location within tissue and apparent phenotypic maturation status (E and F) and also in neighboring perichondrium-like tissue (G, arrowheads). Clusters of large hypertrophying chondrocytes display strong staining, particularly in their pericellular compartment (H). I: Continuous sections from the above exostosis specimen were stained with collagen X antibodies. Note that only mature and hcs stain positively. B and F are higher magnification images of areas in A and E, respectively. Scale bars: 250 μm (A and E); 75 μm (B, D, F, G, H, and I); 300 μm (C). Coj, chondro-osseous junction; hc, hypertrophic chondrocyte; pc, perichondrium; phc, pre-hypertrophic cartilage; rc/pl, resting-proliferating cartilage; SOC, secondary ossification center.
Heparanase Stimulates Cell Migration and Proliferation and Chondrogenesis
What could be the significance and possible pathologic implications of the findings in the previous section? Exostoses are ectopic cartilaginous outgrowths and as such, must depend on local cell proliferation and migration and on chondrogenic differentiation to initiate, sustain, and propel their development and growth.31 Thus, we asked whether heparanase would influence such processes. To analyze cell proliferation, we grew ATDC5 chondrogenic cells in monolayer culture in control medium or medium that contained 400 ng/mL human recombinant heparanase, a concentration used in previous studies and elicited maximal responses.32 DNA quantification at three time points over a 2-week culture period showed that heparanase had stimulated cell growth by nearly threefold by day 14 over control values (Figure 2A). To examine cell migration, we used the scratch and wound healing system.32 Accordingly, confluent monolayers were scratched in the middle with a pipette tip, and the repopulation of the scratch area by migrating cells was determined over a 3-hour period in the absence or presence of exogenous heparanase. Clearly, cultures treated with heparanase repopulated the wound area more rapidly and more extensively than untreated control cells by 3 hours (Figure 2B).
Figure 2.

Treatment with human recombinant heparanase stimulates cell proliferation, migration, and chondrogenesis. A: DNA quantification shows that heparanase stimulates proliferation of ATDC5 cells in monolayer as early as 3 days after treatment compared with vehicle-treated controls. B: Scratch wound-healing assays show that cell migration is increased by heparanase treatment. Wound closure was monitored by sequential phase contrast imaging up to 3 hours. C: Representative images of stained limb bud cell micromass cultures on day 4 and 6 treated with recombinant heparanase or vehicle. Note the substantial increase in the number and staining intensity of cartilage nodules in treated versus control cultures. D: Image-based quantification of Alcian Blue–positive areas shows more than twofold increase in treated cultures over control set at 1. E: Measurement of overall micromass diameter on day 4 and 6 indicates that proliferation/migration of peripheral cells was stimulated by heparanase treatment. Increase in diameter for each micromass was set relative to that at 24 hours after plating and is described in arbitrary units. F: Immunoblot images show that heparanase treatment had increased Smad1/5/8 phosphorylation protein levels in day 4 and 6 micromass cultures relative to respective controls. Membranes were re-blotted with α-tub antibodies for sample loading normalization. G: Normalized band intensity quantifications of blots in F with controls set at 1. n = 3 (A); n = 6 (B, D, and E). ∗P < 0.05, ∗∗P < 0.01. Scale bar = 1.5 mm. Cntl, control; Hep'ase, heparanase protein; α-tub, α-tubulin.
To examine chondrogenic cell differentiation, we used micromass cultures.29,33 In this popular experimental system, progenitor chondrogenic cells are isolated from embryonic limb buds and are grown at high cell density in spot cultures in which they resume their differentiation program and give rise to cartilaginous nodules over time. For these experiments, cells isolated from mouse embryo limb buds were plated in micromass and grown in medium lacking or containing exogenous heparanase as in the previous paragraph. Staining with Alcian Blue on day 4 and 6 showed that control cultures displayed several positive cartilage nodules scattered in their central portion as expected (Figure 2C), but more numerous and more strongly staining nodules were present in companion heparanase-treated cultures (Figure 2C). Computer-assisted imaging showed that the Alcian Blue–staining area was more than twofold higher in treated than in control cultures (P < 0.05) (Figure 2D). We obtained similar results previously by using exogenous bacterial heparitinase III that cleaves HS in far smaller fragments than mammalian enzyme,34 indicating that chondrogenesis is stimulated regardless of HS fragment size. The peripheral portion of micromass cultures normally contain variously shaped fibroblastic cells that actively proliferate and migrate away from the micromass over time.29 Thus, we measured the overall maximal diameter of the cultures and found that the heparanase-treated cultures had achieved a significantly larger diameter at both day 4 and day 6 than controls (Figure 2E), reaffirming that heparanase promotes cell proliferation and/or migration.
BMP signaling is a main regulator and stimulator of chondrogenesis,35 and we previously showed that it is ectopically activated during the early stages of exostosis-like tissue formation in HME mouse models that involved conditional Ext1 ablation.33 Thus, we tested whether human heparanase treatment would stimulate BMP signaling along with its stimulation of chondrogenesis as seen in the previous paragraph. Immunoblot analysis with antibodies to phosphorylated Smad1/5/8 by using whole micromass cell extracts showed that pSmad levels were increased after heparanase treatment at both day 4 and day 6 of culture compared with controls (Figure 2, F and G), likely signifying that heparanase had stimulated chondrogenesis by increasing bioavailability and/or signaling activity of endogenous cell/matrix-associated BMPs.
Heparanase Gene Expression Is Responsive to HS Levels
The widespread presence of heparanase in exostoses and its pro-chondrogenic and signaling effects seen in Figure 2 raised the question of whether heparanase is an early response gene, able to be up-regulated soon after or concomitantly with declining HS levels and thus perpetuating or amplifying cellular responses. To test this hypothesis, we treated primary limb mesenchymal cell micromass cultures with the HS antagonist Surfen. As we and others showed previously, this compound interferes with HS function and elicits functional HS deficiency,36 triggering cellular responses similar to Ext gene ablation, including enhanced chondrogenesis.33 Interestingly, we found that treatment with an optimal concentration of Surfen (7.5 μmol/L)33 significantly increased the protein levels of endogenous heparanase as indicated by immunoblotting (Figure 3A), approximately eightfold at this dose (Figure 3B). A similar response was observed in ADTC5 cell cultures (Figure 3, C and D). Control of this response appeared to be at the transcriptional and/or RNA stabilization levels because the heparanase mRNA amounts were increased as well in Surfen-treated cultures as indicated by semiquantitative RT-PCR (Figure 3, E and F).
Figure 3.

Heparanase expression is responsive to modulation in heparan sulfate levels or function. A–D: Immunoblot images and densitometric histograms indicate that endogenous heparanase protein levels increase by Surfen treatment (+) in limb bud micromasses (A and B) and ATDC5 cell cultures (C and D) compared with respective untreated controls (−). E and F: Semiquantitative RT-PCR and densitometric analyses show that Surfen treatment increases heparanase gene expression in micromass cultures. G and H: Immunoblot and densitometric quantification analyses show up-regulation of endogenous heparanase protein levels on treatment with recombinant BMP2. Data are expressed as means ± SD. ∗P < 0.05. BMP, bone morphogenetic protein; Cntl, control; Hep'ase, heparanase protein; Surfen, bis-2-methyl-4-aminoquinolyl-6-carbamide; α-tub, α-tubulin.
A related and congruent possibility is that endogenous heparanase levels may be stimulated not only by declining HS levels/function but also by concurrent BMP bioavailability. Thus, we treated day 2 micromass cultures with exogenous 100 ng/mL recombinant human BMP2, a concentration that strongly stimulates BMP signaling and chondrogenesis,33 for 24 or 48 hours. Immunoblot analysis showed that endogenous heparanase levels were clearly increased in BMP2-treated cultures compared with companion untreated cultures (Figure 3G), approximately twofold as revealed by densitometry and normalization (Figure 3H).
Chondrogenesis Is Inhibited by a Heparanase Antagonist
Given our results that link heparanase to the stimulation of chondrogenesis and BMP signaling (and likely other growth factor signaling pathways), we asked whether inhibiting endogenous heparanase would have negative repercussions on chondrogenesis. Thus, we used the heparanase inhibitor SST0001, a modified heparin molecule without anticoagulant properties and with high affinity for heparanase and strong inhibitory activity.37 This compound was already thoroughly tested for safety and pharmacokinetics in a number of animal models and was shown to have antitumorigenic properties.38 On the basis of this groundwork, we chose three concentrations with which to treat limb bud micromass cultures, at or above the concentration that inhibits 50% for heparanase inhibition. We found that SST0001 treatment strongly reduced the formation of Alcian Blue–positive chondrogenic nodules (Figure 4A) in a dose-dependent manner as indicated by image quantification (Figure 4B). Counterstaining with hematoxylin showed that overall cell number and density had not changed significantly after treatment (Figure 4A). Whole cellular RNA and DNA content also remained similar in treated and untreated cultures at day 4 and day 6, although minor decreases were observed by these methods at the maximal 20 μg/mL dose (not shown). Whole cellular RNA samples were then processed for semiquantitative RT-PCR, and the data showed that expression of such chondrogenic marker genes as collagen II (ColII), aggrecan (Agg), and Runx2 was dose dependently inhibited by SST0001 treatment (Figure 4, C–E). Interestingly, SST0001 treatment also reduced the levels of endogenous heparanase (Figure 4F) compared with untreated control cultures (Figure 4F), whereas treatment with bacterial heparitinase increased them (Figure 4F). In addition, SST0001 treatment reduced the overall diameter of the micromasses (Figure 4G), indicating that it had inhibited migration of peripheral fibroblastic cells.
Figure 4.

Chondrogenesis is inhibited by a heparanase antagonist. A: Images of Alcian Blue–stained micromass cultures on day 4 and 6 show a dose-dependent reduction in cartilage nodule formation on treatment with the heparanase antagonist SST0001. Panels on the right show images of control and treated micromass cultures counterstained with hematoxylin. B: Image-assisted quantification of Alcian Blue–positive area in control versus SST0001-treated cultures. Statistical significance was reached at every concentration tested. C–E: Histograms depict gene expression levels of chondrogenic genes ColII, Agg, and Runx2 in controls versus SST0001-treaed micromass cultures on day 4 and 6 measured by semiquantitative RT-PCR and quantified by imaging. F: Immunoblot images show decrease in the endogenous Hep'ase levels in micromass cultures on SST0001 treatment and increase on bacterial Hep treatment compared with control. G: Histograms depict the overall absolute micromass diameter at day 4 and 6 in control versus SST0001-treated cultures. Data are expressed as means ± SD. n = 2 (C–E); n = 6 (B and G). ∗P < 0.05, ∗∗P < 0.01. Scale bar = 3 mm. Hep, heparitinase; Hep'ase, heparanase protein; SST, SST0001.
Discussion
Our data provide evidence that heparanase is detectable and broadly distributed in the chondrocytes present in benign human exostoses, a phenotype quite distinct from that seen in normal growth plate cartilage where the protein is restricted to the hypertrophic zone and perichondrium, consistent with data in a previous study.39 We show also that exogenous recombinant human heparanase is a strong stimulator of cell migration, proliferation, and chondrogenesis in mouse cell cultures, indicating that one function, and possibly a main function, of heparanase would be to promote cell recruitment for the initiation and outgrowth of the exostoses. This mechanistic sequel is reinforced by our observations that heparanase stimulates the pro-chondrogenic BMP signaling pathway and that endogenous heparanase gene expression and overall protein levels are increased as the HS levels decrease. Thus, heparanase appears to be a part of tightly controlled mechanisms that link it to HS levels and also responsive to modulations in HS levels. A significant drop in HS levels occurring in local cells in HME patients would elicit increased heparanase expression which in turn would elicit further HS degradation, stimulate growth factor release and signaling pathways, and promote chondrogenesis, all steps converging on and contributing to exostosis growth (Figure 5). Although counterintuitive and seemingly paradoxical, similar inverse relations among HS levels, Ext1 expression and heparanase expression were reported in cancer studies. Cancer cells with higher Ext1 expression exhibited lower heparanase expression, whereas cancer cells with lower Ext1 expression (and higher metastatic potential) exhibited higher heparanase expression, and Ext1 knockdown with siRNA up-regulated heparanase expression and potentiated metastatic capacity.40 Indeed, heparanase expression levels are used to help predict tumor severity and patient outcome.41–43 These and other studies suggest that one of the changes occurring during the transition from benign exostoses to osteosarcoma in HME patients could be an even steeper increase in heparanase expression.
Figure 5.

Model illustrates a series of regulatory steps that may cause inception and promotion of formation and growth of exostosis. A: The cells of most patients with hereditary multiple exostoses bear a heterozygous loss-of-function mutation in EXT1 or EXT2 (depicted here at Ext+/− cells in half white and half red). B: At one point prenatally or postnatally, some cells may undergo a second genetic change (depicted here as red cells along perichondrium and called mutant in the text), resulting in a more severe loss of overall EXT expression or function and leading to further reduction in local HS production and levels. C: This in turn may up-regulate Hep'ase expression, increase growth factor availability and cell proliferation, and induce ectopic chondrogenesis near/at perichondrium. D: The incipient exostosis cells may recruit surrounding heterozygous cells, induce them into neoplastic behavior, and promote their incorporation into the outgrowing exostosis. E: Cells within the outgrowing exostosis may assemble into a typical growth plate-like structure that protrudes away from the surface of the skeletal element (depicted here as a long bone) and cover distally by perichondrium. By inhibiting heparanase activity and possibly other processes, SST may inhibit chondrogenesis and in turn exostosis formation as indicated. Surfen (not indicated here) may elicit the opposite and stimulate chondrogenesis. Hep'ase, heparanase; HS, heparan sulfate; SST, SST0001; Surfen, bis-2-methyl-4-aminoquinolyl-6-carbamide.
Previous studies have shown that EXT haploinsufficiency per se is not sufficient for multiple exostosis formation in patients or mice and that a steeper reduction would be needed.44,45 Current possible and plausible explanations of how Ext expression and/or HS production could decrease further include EXT loss-of-heterozygosity; large genomic deletions; a second hit in another gene; background genetic variability, including gene modifiers; or compound heterozygosity in EXT1 and EXT2.21,26–28 Such changes would occur in subsets of cells within the HME patient; the cells would be prone to exostosis formation and would give rise to exostoses in conjunction with an active growth plate (Figure 5).
Our data now add another potential culprit heparanase to such pathogenic scenarios. The protein could contribute to disease in combination with any of the genetic events mentioned in the previous paragraph, together leading to a steeper decrease in EXT expression and HS levels less than those achieved by mere haploinsufficiency. Heparanase is preferentially expressed in perichondrium along the growth plates (Figure 1). Previous studies reported similar data in mouse growth plate perichondrium,46 and we provided evidence previously that progenitor cells located within perichondrium may be a primary contributor to exostosis formation.33 Thus, by possessing basal heparanase expression, perichondrial cells may be particularly sensitive to genetic changes and could require a lower threshold to tilt their homeostatic balance, boost basal heparanase expression, stimulate otherwise largely inactive growth factor signaling pathways, incite ectopic chondrogenesis, and promote exostosis formation. One additional aspect of exostosis formation stemming from mouse and human studies is that the exostoses themselves appear to be composed by a mixture of mutant cells (ie, heterozygous Ext cells that have undergone an additional genetic change) and plain heterozygous cells.33,44 The mutant cells would recruit the heterozygous cells into the incipient exostosis mass and induce them into a neoplastic behavior (Figure 5). Given that heparanase facilitates cell migration and could diffuse into the surroundings, it could certainly have a role in such recruitment and mobilization processes during exostosis growth.
The heparanase inhibitor SST0001was shown to interfere with the growth of myeloma and sarcoma cells and concomitant angiogenesis in mouse models,37,38 reaffirming that heparanase has a main role in pathogenesis and may be a particularly good target for cancer therapy. Our data with primary limb cell micromass cultures provide evidence that SST0001 also inhibits chondrogenesis and does so powerfully as revealed by major decreases in development of Alcian Blue–positive cartilage nodules and expression of such chondrogenic marker genes as collagen II, aggrecan, and Runx2. The data correlate well with a previous study using a different heparanase inhibitor, PI-88, with the ADC5 chondrogenic cell line in which, however, only modest reductions in Alcian Blue staining and cartilage gene expression were observed,46 possibly reflecting diverse drug potency.
Previous studies37,38 and our present data do not allow us to clarify exactly how SST0001 elicited such a strong effect. One possibility is that heparanase has a far more important role in chondrogenesis than currently realized, possibly being essential in mobilizing chondrogenic factors and enhancing their bioavailability and diffusion among condensed prechondrogenic cells. Its effective suppression would hamper this process and would elicit a strong antichondrogenic effect. Similarly, SST0001 could have interfered with cell mobilization and cell–cell interactions needed by the limb mesenchymal cells to initially condense and then activate the chondrogenic program.29
SST0001 is a heparin modified through glycol splitting and partial desulfation and N-acetylation47 that is no longer an anticoagulant and cannot dislodge basic fibroblast growth factor from the extracellular matrix or enhance its mitogenic activity, traits that native heparins and HS chains have.48,49 However, by still being a partially sulfated polymer, SST0001 could bind other growth factors, including BMPs, and limit their bioactivity (Figure 5). Our data also show that it decreased the endogenous levels of heparanase in the micromasses and reduced cell migration. Thus, although it will take more direct experimentation to clarify precisely how SST0001 blocked chondrogenesis so effectively, the present data do sustain the possibility that SST0001 could be a potent inhibitor of exostosis formation in vivo. Ongoing studies with HME mouse models should provide such evidence in the near future.
Acknowledgments
We thank Drs. Ralph Sanderson (University of Alabama at Birmingham, Birmingham, AL) and Israel Vlodavsky (Technion-Israel Institute of Technology, Haifa, Israel) for supplying recombinant human heparanase; Drs. Marta Guttenberg and Tricia Bhatti (Children's Hospital of Philadelphia Pathology Laboratory) for patient exostosis tissue diagnostics and preparation; Dr. Alessandro Noseda (Sigma-Tau Pharmaceuticals, Inc., Gaithersburg, MD) for advice and for providing SST0001; and the Multiple Hereditary Exostoses Research Foundation, an organization dedicated to the support of families and patients with MHE and for advocating MHE public awareness and biomedical research, for their passionate efforts.
Footnotes
Supported by the NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR061758 (M.P. and E.K.). J.H. was the recipient of the National Research Service Awards predoctoral award F31DE022204 from the NIH National Institute of Dental and Craniofacial Research during the course of the study.
Disclosures: SST0001 used in this study was provided by Sigma-Tau Pharmaceuticals, Inc. (Gaithersburg, MD).
Current address for J.H., McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, University of Pennsylvania, Philadelphia, PA.
References
- 1.Fux L., Ilan N., Sanderson R.D., Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vreys V., David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman C., Parish C.R. Human platelet heparanase: purification, characterization and catalytic activity. Biochem J. 1998;330:1341–1350. doi: 10.1042/bj3301341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy-Adam F., Feld S., Suss-Toby E., Vlodavsky I., Ilan N. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS ONE. 2008;3:e2319. doi: 10.1371/journal.pone.0002319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel V.N., Knox S.M., Likar K.M., Lathrop C.A., Hossain R., Eftekhari S., Whitelock J.M., Elkin M., Vlodavsky I., Hoffman M.P. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development. 2007;134:4177–4186. doi: 10.1242/dev.011171. [DOI] [PubMed] [Google Scholar]
- 6.Ramani V.C., Yang Y., Ren Y., Nan L., Sanderson R.D. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbouud-Jarrous G., Atzmon R., Perets T., Palermo C., Gadea B.B., Joyce J.A., Vloadavsky I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008;283:18167–18176. doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zetser A., Bashenko Y., Miao H.Q., Vlodavsky I., Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 9.Woods A. Syndecans: transmembrane modulators of adhesion and matrix assembly. J Clin Invest. 2001;107:935–941. doi: 10.1172/JCI12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldshmidt O., Zcharia E., Cohen M.M., Aingorn H., Cohen I., Nadav L., Katz B.Z., Geiger B., Vlodavsky I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003;17:1015–1025. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- 11.Hulett M.D., Freeman C., Hamdorf B.J., Baker R.T., Harris M.J., Parish C.R. Cloning of mammalian heparanase: an important enzyme in tumor invasion and metastasis. Nat Med. 1999;5:803–809. doi: 10.1038/10525. [DOI] [PubMed] [Google Scholar]
- 12.Purushothaman A., Chen L., Yang Y., Sanderson R.D. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype of myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casu B., Vlodavsky I., Sanderson R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol Haemost Thromb. 2008;36:195–203. doi: 10.1159/000175157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenzie E.A. Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol. 2007;151:1–14. doi: 10.1038/sj.bjp.0707182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones K.B. Glycobiology and the growth plate: current concepts in multiple hereditary exostoses. J Pediatr Orthop. 2011;31:577–586. doi: 10.1097/BPO.0b013e31821c7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter D.E., Simpson A.H. The neoplastic pathogenesis of solitary and multiple osteochondromas. J Pathol. 1999;188:119–125. doi: 10.1002/(SICI)1096-9896(199906)188:2<119::AID-PATH321>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.Goud A.L., de Lange J., Scholtes V.A., Bulstra S.K., Ham S.J. Pain, physical and social functioning, and quality of life in individuals with multiple hereditary exostoses in The Netherlands: a national cohort study. J Bone Joint Surg Am. 2012;94:1013–1020. doi: 10.2106/JBJS.K.00406. [DOI] [PubMed] [Google Scholar]
- 18.Hosalkar H., Greenberg J., Gaugler R.L., Garg S., Dormans J.P. Abnormal scarring with keloid formation after osteochondroma excision in children with multiple hereditary exostoses. J Pediatr Orthop. 2007;27:333–337. doi: 10.1097/BPO.0b013e3180326732. [DOI] [PubMed] [Google Scholar]
- 19.Bovee J.V., Cleton-Jansen A.M., Wuyts W., Caethoven G., Taminiau A.H., Bakker E., Van Hul W., Cornelisse C.J., Hogendoorn P.C. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcoma. Am J Hum Genet. 1999;65:689–698. doi: 10.1086/302532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esko J.D., Selleck S.B. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 21.Jennes I., Pedrini E., Zuntini M., Modenti M., Balkassmi S., Asteggiano C.G., Casey B., Bakker B., Sangiogi L., Wuyts W. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb) Hum Mutat. 2009;30:1620–1627. doi: 10.1002/humu.21123. [DOI] [PubMed] [Google Scholar]
- 22.Hecht J.T., Hayes E., Haynes R., Cole W.G., Long R.J., Farach-Carson M.C., Carson D.D. Differentiation-induced loss of heparan sulfate in human exostosis derived chondrocytes. Differentiation. 2005;73:212–221. doi: 10.1111/j.1432-0436.2005.00025.x. [DOI] [PubMed] [Google Scholar]
- 23.Jones K.B., Piombo V., Searby C., Kurriger G., Yang B., Grabellus F., Roughley P.J., Morcuende J.A., Buckwalter J.A., Capecchi M.R., Vortkamp A., Sheffield V.C. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A. 2010;107:2054–2059. doi: 10.1073/pnas.0910875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto K., Irie F., Mackem S., Yamaguchi Y. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci USA. 2010;107:10932–10937. doi: 10.1073/pnas.0914642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zak B.M., Schuksz M., Koyama E., Mundy C., Wells D.E., Yamaguchi Y., Pacifici M., Esko J.D. Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone. 2011;48:979–987. doi: 10.1016/j.bone.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waaijer C.J., Winter M.G., Reijnders C.M., de Jong D., John Ham S., Bovee J.V., Szuhai K. Intronic deletion and duplication proximal of the EXT1 gene: a novel causative mechanism for multiple osteochondromas. Genes Chromosomes Cancer. 2013;52:431–436. doi: 10.1002/gcc.22041. [DOI] [PubMed] [Google Scholar]
- 27.Wuyts W., Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat. 2000;15:220–227. doi: 10.1002/(SICI)1098-1004(200003)15:3<220::AID-HUMU2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 28.Pedrini E., Jennes I., Tremosini M., Milanesi A., Mordenti M., Parra A., Sgariglia F., Zuntini M., Campanacci L., Fabbri N., Pignotti E., Wuyts W., Sangiorgi L. Genotype-phenotype correlation study in 529 patients with hereditary multiple exostoses: identification of “protective” and “risk” factors. J Bone Joint Surg Am. 2011;93:2294–2302. doi: 10.2106/JBJS.J.00949. [DOI] [PubMed] [Google Scholar]
- 29.Ahrens P.B., Solursh M., Reiter R.S., Singley C.T. Position-related capacity for differentiation of limb mesenchyme in cell culture. Dev Biol. 1979;69:436–450. doi: 10.1016/0012-1606(79)90303-8. [DOI] [PubMed] [Google Scholar]
- 30.Elkin M., Ilan N., Ishai-Michaeli R., Friedmann Y., Papo O., Pecker I., Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. FASEB J. 2001;15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 31.Huegel J., Sgariglia F., Enomoto-Iwamoto M., Koyama E., Dormans J.P., Pacifici M. Heparan sulfate in skeletal development, growth, and pathology: the case of hereditary multiple exostoses. Dev Dyn. 2013;242:1021–1032. doi: 10.1002/dvdy.24010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gingis-Velitski S., Zetser A., Flugelman M.Y., Vlodavsky I., Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–23541. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- 33.Huegel J., Mundy C., Sgariglia F., Nygren P., Billings P.C., Yamaguchi Y., Koyama E., Pacifici M. Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: a mechanism likely deranged in Hereditary Multiple Exostoses. Dev Biol. 2013;377:100–112. doi: 10.1016/j.ydbio.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parish C.R., Freeman C., Hulett M.D. Heparanase; a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–M108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 35.Kronenberg H.M. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 36.Schuksz M., Fuster M.M., Brown J.R., Crawford B.E., Ditto D.P., Lawrence R., Glass C.A., Wang L., Tor Y., Esko J.D. Surfen, a small molecule antagonist of heparan sulfate. Proc Natl Acad Sci USA. 2008;105:13075–13080. doi: 10.1073/pnas.0805862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritchie J.P., Ramani V.C., Ren Y., Naggi A., Torri G., Casu B., Penco S., Pisano C., Carminati P., Tortoreto M., Zunino F., Vlodavsky I., Sanderson R.D., Yang Y. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17:1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cassinelli G., Lanzi C., Tortoreto M., Cominetti D., Petrangolini G., Favini E., Zaffaroni N., Pisano C., Penco S., Vlodavsky I., Zunino F. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem Pharmacol. 2013;85:1424–1432. doi: 10.1016/j.bcp.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 39.Trebicz-Geffen M., Robinson D., Evron Z., Glaser T., Fridkin M., Kollander Y., Vlodavsky I., Ilan N., Law K.F., Cheah K.S., Chan D., Werner H., Nevo Z. The molecular and cellular basis of exostosis formation in hereditary multiple exostoses. Int J Exp Pathol. 2008;89:321–331. doi: 10.1111/j.1365-2613.2008.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y., Yang X., Yamagata S., Yamagata T., Sato T. Involvement of Ext and heparanase in migration of mouse FBJ osteosarcoma cells. Mol Cell Biochem. 2013;373:63–72. doi: 10.1007/s11010-012-1475-8. [DOI] [PubMed] [Google Scholar]
- 41.Ilan N., Elkin M., Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiongenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 42.Quiros R.M., Rao G., Plate J., Harris J.E., Brunn G.J., Platt J.L., Gattuso P., Prinz R.A., Xu X. Elevated serum heparanase-1 levels in patients with pancreatic carcinoma are associated with poor survival. Cancer. 2006;106:532–540. doi: 10.1002/cncr.21648. [DOI] [PubMed] [Google Scholar]
- 43.Vlodavsky I., Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reijnders C.M., Waaijer C.J., Hamilton A., Buddingh E.P., Dijkstra S.P., Ham J., Bakker E., Szuhai K., Karperien M., Hogendoorn P.C., Stringer S.E., Bovee J.V. No haploinsufficiency but loss of heterozygosity for EXT in multiple osteochondromas. Am J Pathol. 2010;177:1946–1957. doi: 10.2353/ajpath.2010.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stickens D., Zak B.M., Rougier N., Esko J.D., Werb Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 2005;132:5055–5068. doi: 10.1242/dev.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown A., Alicknavitch M., D'Souza S.S., Daikoku T., Kirn-Safran C.B., Marchetti D., Carson D.D., Farach-Carson M.C. Heparanase expression and activity influences chondrogenic and osteogenic processes during endochondral bone formation. Bone. 2008;43:689–699. doi: 10.1016/j.bone.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naggi A., Casu B., Perez M., Torri G., Cassinelli G., Penco S., Pisano C., Giannini G., Ishai-Michaeli R., Vlodavsky I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 48.Bernfield M., Gotte M., Park P.W., Reizes O., Fitzgerald M.L., Lincecum J., Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 49.Whitelock J.M., Iozzo R.V. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105:2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]