

Abstract
Intestinal epithelial cell (IEC) death is typical of inflammatory bowel disease (IBD). We investigated: i) whether IEC–released necrotic cell products (proinflammatory mediators) amplify mucosal inflammation, ii) the capacity of necrotic cell lysates from HT29 cells or human IECs to induce human intestinal fibroblasts' (HIF) production of IL-6 and IL-8, and iii) whether IL-1α, released by injured colonocytes, exacerbated experimental IBD. Necrotic cell lysates potently induced HIF IL-6 and IL-8 production independent of Toll-like receptors 2 and 4, receptor for advanced glycation end-products, high-mobility group box 1, uric acid, IL-33, or inflammasome activation. IL-1α was the key IEC-derived necrotic cell product involved in HIF cytokine production. IL-1α–positive cells were identified in the epithelium in human IBD and dextran sulfate sodium (DSS)-induced colitis. IL-1α was detected in the stool of colitic mice before IL-1β. IL-1α enemas reactivated inflammation after DSS colitis recovery, induced IL-1 receptor expression in subepithelial fibroblasts, and activated de novo inflammation even in mice without overt colitis, after the administration of low-dose DSS. IL-1α amplifies gut inflammation by inducing cytokine production by mesenchymal cells. IL-1α–mediated IEC–fibroblast interaction may be involved in amplifying and perpetuating inflammation, even without obvious intestinal damage. IL-1α may be a target for treating early IBD or preventing the reactivation of IBD.
Intestinal epithelial cell (IEC) injury and death are extremely common events but only recently have been recognized as drivers of inflammation.1–3 Excessive cell death results in barrier defects and uncontrolled bacterial translocation that together induce and sustain gut inflammation.1,4 In addition, epithelial cells contain a myriad of intracellular substances normally not recognized by the immune system but, during cell necrosis, they are passively released in the surrounding microenvironment and trigger inflammation. This response may represent a novel pathogenic component of inflammatory bowel disease (IBD), since epithelial damage is a typical feature of both ulcerative colitis and Crohn disease.5,6 Cell products released by cells undergoing necrosis (passive, programed, or after apoptosis) are called damage-associated molecular patterns (DAMPs) or alarmins as they alert the immune system and trigger a sterile inflammatory response.7 These events give rise to the danger model of immunity, which suggests that inflammation is preferentially geared to defend against host damage rather than foreign signals.8,9
DAMPs represent the endogenous counterpart of exogenous pathogen-associated molecular patterns.10 DAMP signals are likely to converge with microbe-derived pathogen-associated molecular patterns to amplify inflammation, as they share various receptors and elicit similar responses.11,12 This integration is especially important to the microbiota-rich gut microenvironment, where plentiful and diversified signals that mediate key cell interactions in IBD are elicited. Evidence of the involvement of DAMPs in IBD pathogenesis is limited. IBD tissue releases abundant S100A12, S100A8/S100A9 complexes (calprotectin)13 and high-mobility group box 1 (HMGB-1), which serve as fecal biomarkers of intestinal inflammation.14 Hmgb-1 levels are elevated in Il10−/− and in dextran sulfate sodium (DSS)-induced murine colitis; blockade of Hmgb-1 ameliorates inflammation.15,16
IL-1α has been recognized as a major product of epidermal keratinocytes and enterocytes for some time17,18 but has only recently emerged as a major DAMP and inducer of sterile inflammation.19–22 This prototypical member of the IL-1 family is involved in the pathogenesis of several acute and chronic inflammatory diseases and is expressed in most cells, including IECs. However, unlike its other family member IL-1β, little information is available on the mechanisms by which IL-1α may function as a DAMP in intestinal inflammation.23,24
We report here that epithelial cell–derived DAMPs elicit a potent proinflammatory cytokine response from human intestinal fibroblasts (HIFs). These cells are active participants in intestinal inflammation,25 acting as first responders to products of epithelial cell necrosis due to their anatomical proximity. Among a number of potential DAMPs, our data indicate that IL-1α appears to be the major alarmin involved in the induction of proinflammatory cytokine production by fibroblasts. We also present evidence that IL-1α is an early mediator and reactivator of intestinal injury in vivo in experimental colitis, suggesting a key role in the pathogenesis of IBD.
Materials and Methods
Human Colonic Tissue Specimens
Human colonic tissue samples were obtained according to the protocol approved by the Cleveland Clinic Institutional Review Board (protocol 12–383; Cleveland, OH).
Cells and Reagents
HT29 cells (ATCC HTB-38) and THP1 cells (ATCC TIB-202) were purchased from the ATCC (Manassas, VA). Ultrapure lipopolysaccharide (LPS) was purchased from InvivoGen (San Diego, CA). All cytokines [tumor necrosis factor (TNF)-α, IL-1α, and IL-1β] were purchased from PeproTech Inc. (Rocky Hill, NJ). Monosodium urate crystals (5 mg/mL stock solution in phosphate-buffered saline (PBS; InvivoGen) were vortexed for 5 minutes before use to obtain a homogeneous suspension.
Cell Isolation and Culture
Primary HIFs were isolated and cultured as previously described.26 Briefly, HIFs were derived from outgrowths of minced colonic mucosa explants placed on etched polystyrene flasks containing HIF growth medium comprising Dulbecco's modified Eagle's medium, 10% fetal bovine serum (FBS), 2 mmol/L l-glutamine, 25 mmol/L HEPES, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (all purchased from Lonza, Basel, Switzerland) and used between passages 4 and 10 at 80% confluence.
IECs were isolated by a method that has been previously reported in detail.27 Peripheral blood monocytes and neutrophils and lamina propria mononuclear cells were isolated, and THP1 monocytic cells were cultured as previously described.27–29
Necrotic cell lysates (NCLs; diluted 1:10 to 1:1000 in PBS), recombinant human HMGB-1 (10 pg/mL to 5 μg/mL; R&D Systems, Minneapolis, MN), IL-33 (0.01 pg/mL to 10 ng/mL; Peprotech), or recombinant IL-1α (0.1 to 1000 pg/mL) as controls were incubated with HIFs for 24 hours at 37°C, and supernatants were harvested for cytokine enzyme-linked immunosorbent assays (ELISAs).
Preparation of NCLs
NCLs were prepared by standard methods.30,31 The IEC line HT29, fresh IECs, lamina propria mononuclear cells, primary human polymorphonuclear neutrophils, and human monocytes were suspended in RPMI, 10% FBS medium (Lonza) containing 2.5% v/v penicillin, streptomycin, and amphotericin B, 1.5% v/v 1 mol/L HEPES buffer. THP1 cells were suspended in RPMI 10% v/v FBS medium containing 0.5 mmol/L 2-mercaptoethanol. Cells were washed, resuspended at 108 cells/mL in PBS, and subjected to five freeze–thaw cycles by placement sequentially in a liquid nitrogen/37°C water bath, followed by centrifugation for 30 minutes at 15,000 × g. The supernatants were collected and are hereafter referred to as NCLs.
Cytokine Measurement
HIFs were incubated with NCL (at 1:20 dilution in the culture medium) for 24 hours, then supernatants were collected. Levels of human IL-6 and IL-8 (BD Biosciences, San Jose, CA) released into the HIF culture medium and of IL-1α (BioLegend, San Diego, CA) and IL-1β (BD Biosciences) in the NCLs were determined using ELISA kits following the manufacturer's protocols.
Reverse Transcription-PCR
For HIF Toll-like receptor (TLR) gene expression analysis, total RNA was extracted from nonstimulated HIFs and reverse-transcribed, and cDNA was amplified by RT-PCR. Five microliters of cDNA was amplified in the presence of 0.125 μmol/L each of the 5′ and 3′ primers (Biosynthesis, Lewisville, TX) and 1 U of Taq DNA polymerase (Roche Diagnostics Deutschland, Mannheim, Germany). PCR was performed in a DNA thermal cycler using preoptimized temperatures and times, with the following primers: TLR1: forward 5′-CTATACACCAAGTTGTCAGC-3′, reverse 5′-GTCTCCAACTCAGTAAGGTG-3′; TLR2: forward 5′-GCCAAAGTCTTGATTGATTGG-3′, reverse 5′-TTGAAGTTCTCCAGCTCCTG-3′; TLR3: forward 5′-AAATTGGGCAAGAACTCACAGG-3′, reverse 5′-GTGTTTCCAGCGCCGTGCTAA-3′; TLR4: forward 5′-TGGATACGTTTCCTTATAAG-3′, reverse 5′-GAAATGGAGGCACCCCTTC-3′; TLR5: forward 5′-CTAGCTCTTAATCCTGATG-3′, reverse 5′-CCATGTGAAGTCTTTGCTGC-3′; TLR6: forward 5′-GAGGAAGCCCACTAAAGGAC-3′, reverse 5′-GGGAGACAAAACAAAGATGGAC-3′; TLR7: forward 5′-TCACCCTCACCATTAACCAC-3′, reverse 5′-TTTCAGGGAGAGCACTTTTAAC-3′; TLR8: forward 5′-TGTGGAATGATGATGACAACAG-3′, reverse 5′-AGAAAGAAAGCCTTGTGCC-3′; and TLR9: forward 5′-GGAAAGAGGAAGGGGTGAAG-3′, reverse 5′-TGAGGGACAGGGATATGAGG-3′.
As a positive control, RNA was obtained from peripheral blood mononuclear cells. Fifteen microliters of the PCR product were subjected to electrophoresis on 1.5% Agarose gel and stained with 0.5 μg/mL ethidium bromide, using 100-BP DNA ladder as a marker.
Preparation of DNA, Histones, and Chromatin
DNA
Genomic DNA was extracted from HT29 NCLs by incubation in 700 μL of lysis buffer (20 mmol/L Tris, pH8, 20 mmol/L EDTA, 2% SDS, 0.5 mg/mL proteinase K) at 55°C overnight with gentle shaking. Samples were mixed with an equal volume of phenol: chloroform in phase-lock gel tubes. DNA was then precipitated with cold ethanol (100%, 700 μL) and incubated for 1 hour at room temperature or overnight at −20°C. DNA was pelleted at maximum speed in a microcentrifuge at 4°C for 5 minutes and decanted, and 500 μL of 70% ethanol was added to wash the pellet. Samples were spun in a microcentrifuge at maximum speed at room temperature for 5 minutes, and DNA pellets were air-dried and resuspended in distilled water.
Histones
Histones were extracted from HT29 nuclei (0.5 to 1 × 108) by resuspension of the nuclear pellet in 0.4 normality sulfuric acid and gentle shaking for 2 to 16 hours at 4°C as previously described.32
Chromatin
Native chromatin was extracted from HT29 with the ChIP-It Express enzymatic kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions, with modifications as detailed previously.33
DNA, histones, and chromatin (5 × 106 cell equivalents/mL, as this was equal to the cell equivalents in 1:20 dilution of NCLs), were incubated with HIFs for 24 hours at 37°C.
Preparation of Nuclear and Cytosolic Extracts
Nuclear and cytosolic cell extracts were prepared as previously described,34 using reagents from Sigma (St. Louis, MO). Briefly, cells were washed in ice-cold phosphate-buffered saline and scraped, on ice, in a hypotonic buffer containing additional proteinase and phosphatase inhibitors [buffer A: 10 mmol/L HEPES, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol (DTT), 100 μmol/L phenanthroline, 1 μg/mL pepstatin, 100 μmol/L E64, 100 μmol/L DCI, 10 mmol/L NaF, 100 μmol/L Na3VO4, and 25 mmol/L β-glycerophosphate]. Cells were then lysed by incubating for 10 minutes on ice in 60 to 80 μL hypotonic buffer containing 0.2% NP40. Lysates were then centrifuged (10,000 × g at 4°C) for 10 minutes, and the supernatants were discarded. The pelleted nuclei were resuspended in 60 to 80 μL of a lysis buffer [buffer C: 20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L ethylenediaminetetraacetic acid (EDTA), 25% glycerol, and 100 μmol/L DCI] and were incubated at 4°C for 15 minutes. Lysed nuclei were vortexed then centrifuged (10,000 × g at 4°C) for 10 minutes and supernatants were snap frozen and stored at −80°C.
Treatment of NCL with RNase and Proteinase K
RNA and/or proteins were depleted from NCLs by digestion with RNase (7 U/106 cells; 5 PRIME, Gaithersburg, MD) or proteinase K (20 μg/106 cells, 5 PRIME) for 1 hour at 37°C. RNA and protein content was evaluated by Agarose gel electrophoresis and SDS-PAGE, respectively. Complete digestion was confirmed in each case (data not shown). Spectrophotometry (NanoDrop; Thermo Scientific, Rochester, NY), was used to quantify RNA levels.
Inhibition of DAMPs and Related Pathways
One hour before stimulation with the NCLs, HIFs were incubated with the following antibodies: anti-TLR2 (5 μg/mL; InvivoGen), anti-TLR4 (5 μg/mL; maba2-htlr4; InvivoGen), anti–receptor for advanced glycation end-products (RAGE; 5 μg/mL; AF1145; R&D Systems), anti–IL-33 (0.01 pg/mL to 10 ng/mL; Peprotech), anti–IL-1 receptor (IL-1RI; 2.5 μg/mL; AF269; R&D Systems), anti–HMGB-1 (5 or 10 μg/mL), soluble RAGE (5 or 10 μg/mL), or an IgG control antibody (5 or 10 μg/mL). NCLs were also digested with uricase (10 U/mL) for 30 minutes at 25°C for removal of monosodium urate crystals before being diluted 1:20 and incubated with HIFs. Supernatants were harvested 24 hours later, and IL-6 and IL-8 ELISAs were performed.
Function and Blockade of TLR2 and TLR4
The functionality of TLR2 and TLR4 expressed by HIFs was tested by exposing the cells to their respective specific ligands, synthetic diacylated glycoprotein 1, a TLR2/TLR6 agonist (1 μg/mL), and LPS (1 μg/mL), in the presence or absence of neutralizing antibodies against TLR2 and TLR4. HIFs were preincubated for 1 hour with 5 μg/mL neutralizing anti-human TLR2 antibody (InvivoGen) followed by 1 μg/mL ligand fibroblast-stimulating ligand 1, or with 5 μg/mL neutralizing anti-human TLR4 antibody (InvivoGen) followed by 1 μg/mL ligand LPS for 24 hours. IL-6 and IL-8 levels in the supernatants were measured by ELISA.
Neutralization of DAMPs in HT29 NCLs
To neutralize IL-1α and IL-1β, NCLs were diluted 1:20 in cell medium and incubated for 1 hour with each specific anti–IL-1 antibody [1 μg/mL; anti–IL-1α (AF-200-NA) or anti–IL-1β (AF-201-NA); R&D Systems]. The IL-1–neutralized NCLs were then incubated with HIFs for 24 hours. The optimal antibody concentration was determined for each antibody by using concentrations ranging from 0.1 to 5 μg/mL. For neutralization of uric acid, NCLs were incubated with 10 U/mL uricase for 30 minutes at 25°C. At the end of each treatment, lysates were diluted 1:20 and incubated with HIFs for 24 hours.
RNA Interference
Transfection of HIFs and HT29 cells was performed by Nucleofection technology (Amaxa, Koln, Germany) according to the manufacturer's instructions, using a control siRNA (2 μmol/L; ON-TARGETplus nontargeting pool, D-001810-10-05; GE Dharmacon, Lafayette, CO) or an siRNA targeting human IL-1α (ON-TARGETplus SMARTpool siRNA, L-007952-00-0005; GE Dharmacon). Transfected HIFs were incubated with NCL (at 1:20 dilution in growth medium) 24 hours after transfection, and then supernatants were collected 24 hours later for cytokine measurements. Transfected HT29 cells were rendered necrotic 48 hours after transfection, as described in the Preparation of NCLs section. The efficiency of silencing was evaluated 48 hours after transfection by measurement of the levels of IL-1α and β-actin mRNA by real-time RT-PCR. The following primers were used: IL1A: forward 5′-AGTGCTGCTGAAGGAGATGCCTG-3′, reverse 5′-CCCTGCCAAGCACACCCAGTAG-3′; ACTB: forward 5′-CTGGACTTCGAGCAAGAGATG-3′, reverse 5′-AGTTGAAGGTAGTTTCGTGGATG-3′.
HT29 Cell Infection with Shigella flexneri
Confluent HT29 cells were infected with log-phase cultures of either Shigella flexneri strain M90T or the noninvasive strain BS176 (gifts from Philippe Sansonetti, Institute Pasteur, Paris, France) at a multiplicity of infection of 50 for 8 hours. Cells were washed with PBS, and infection was continued in Dulbecco's modified Eagle's medium, 10% FBS supplemented with 100 μg/mL gentamicin, for an additional 16 hours. Medium was collected and centrifuged for 5 minutes at 250 × g, and supernatants were snap-frozen at −80°C before vacuum concentration and assessment of IL-1α release by ELISA. Infected cells were analyzed by fluorescence flow cytometry on an LSR II FACScan (BD Biosciences) to determine the number of necrotic cells using the Live/Dead Fixable Cell Stain Kit (L23105; Life Sciences, Grand Island, NY) according to manufacturer's instructions. Noninfected and dead cells were used as positive and negative controls, respectively, and 10,000 events were captured. The percentage of cells undergoing necrosis was calculated relative to infected control cells.
Activation of the Inflammasome
HT29 cells (1 × 106) were seeded into 6-well plates for 24 hours. Cells were then washed and grown in medium containing 2% FCS for 24 hours before either being incubated in 2% growth medium or primed with 10 ng/mL to 1 μg/mL ultrapure LPS (InvivoGen) for 3 hours at 37°C. HT29 cultures were then incubated with either 100 ng/mL TNF-α, 5 mmol/L ATP, or 0.1 to 0.6 μg/mL monosodium urate crystals for 24 hours at 37°C. Supernatants were harvested, cytokine (IL-1α, IL-1β, and IL-8) levels were quantified by ELISA, and the remaining cells were lysed. Protein in cell lysates was quantified (BCA protein assay; Pierce, Rockford, IL), and all ELISA assays were normalized to lysate protein content.
DSS Murine Model of Colitis
Mice (male; C57Bl/6; Jackson ImmunoResearch Laboratories, West Grove, PA) were conventionally housed, and all husbandry and treatments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic. For the induction of DSS colitis, mice were allowed to drink sterile, acidified water in the presence or absence of added 1.5% or 3.0% w/v DSS (MP Biomedicals, Solon, OH) ad libitum. Mice were euthanized on days 0, 3, 5, and 7 (three mice per group). In some experiments, mice were given water for 21 days after 7 days of DSS. On day 28, mice were administered a 45% ethanol enema 30 minutes before receiving an IL-1α enema (10 ng in 150 μL PBS). Mice were then euthanized 24 hours after the IL-1α enema. In each mouse, the colon from the cecum to the rectum was dissected and removed. The tissue was then fixed whole in Histochoice (Sigma, St. Louis, MO), paraffin-embedded, and sectioned for hematoxylin and eosin staining by the Lerner Research Institute, Histological Examination Core, Cleveland Clinic. The histopathological degree of inflammation in the colonic sections was scored as previously reported,35 and images were obtained using a microscope (Leica Microsystems, GmbH, Wetzlar, Germany), with Image-Pro Plus version 7 (Media Cybernetics, Rockville, MD) and Photoshop version 9 (Adobe Systems, San Jose, CA) software.
Fluorescence IHC and Immunocytochemical Staining for IL-1α
IL-1α Staining
Paraffin sections of mouse colon were rehydrated and incubated in a solution of Hanks' balanced salt solution (HBSS) containing 2% FBS for 30 minutes to block nonspecific antibody binding. Subsequently, the slides were incubated overnight at 4°C with 15 μg/mL specific goat polyclonal antibody against murine IL-1α (R&D Systems) diluted in HBSS + 2% FBS. The slides were washed three times in HBSS and then incubated with a solution containing Alexa 488–tagged donkey anti-goat IgG (1:1000; Molecular Probes, Eugene, OR) in HBSS + 2% FBS, for 1 hour at 25°C. The slides were washed three times in HBSS and coverslips mounted in Vectashield containing DAPI (Vector Laboratories, Inc., Burlingame, CA), sealed, and stored at −20°C until visualization. Images were obtained using a TCS-SP laser scanning confocal microscope (Leica).
Deidentified human colonic tissue was obtained from individuals undergoing colonic resection, fixed in Histochoice (AMRESCO, Solon, OH), and embedded in paraffin. Antibody staining was performed on serial 4-μm sections on a Benchmark XT automated immunostainer (Ventana Medical Systems, Inc., Tucson, AZ) using an iVIEW DAB Detection Kit (Ventana) with no cell conditioning and minus the provided secondary. Primary antibody, polyclonal rabbit anti–IL-1α (catalog #PA5-25921; Thermo Scientific–Pierce) was diluted 1:50 and incubated with sections for 60 minutes followed by goat anti-rabbit biotinylated secondary antibody (Vector Laboratories) used at 1:200 for 8 minutes. Paraffin-embedded sections of HeLa cells were used as a positive control (not shown), and negative controls comprised HeLa and colon tissue incubated with secondary antibody only. Images were taken on a DMR upright microscope (Leica) with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter (QImaging, Surrey, BC, Canada) using Image-Pro Plus software version 7 (Media Cybernetics).
IL-1 Receptor Staining
Histochoice-fixed paraffin-embedded mouse colon tissue or thymus (positive control; not shown) was stained with a goat polyclonal antibody to the extracellular domain of the murine IL-1RI (GT15109; Neuromics, Edina, MN). Tissue sections were deparaffinized and rehydrated, and slides were incubated in 3.0% hydrogen peroxide for 20 minutes. Slides were washed and incubated with 3.0% normal rabbit serum/PBS (catalog #S-5000, Vector Laboratories) for 20 minutes at room temperature before Avidin D blocking solution (catalog #SP-2001; Vector Laboratories) was added for a further 15 minutes at room temperature. After rinsing three times in PBS, biotin-blocking solution (catalog #SP-2001; Vector Laboratories) was added for a further 15 minutes at room temperature, followed by rinsing as before in PBS. The polyclonal goat anti–IL-1RI was added at 2 μg/mL diluted in 3.0% normal rabbit serum/PBS overnight at 4°C. Slides were rinsed in PBS/0.05% Tween 20 (10 minutes, three times each). The secondary biotinylated goat anti-rabbit IgG (catalog #BA-1000; Vector Laboratories) diluted 1:200 in PBS with 3% rabbit serum was added for 30 minutes at room temperature, and tissue sections were rinsed with PBS/0.05% Tween 20 (10 minutes, three times each). Freshly prepared avidin–biotin complex peroxidase reagent was incubated with the slides for 30 minutes at room temperature before rinsing with PBS/0.05% Tween 20 as before. ImmPACT DAB peroxidase substrate (catalog #SK-4105; Vector Laboratories) was added to the tissue sections for 2 minutes for appropriate color development and then rinsed thoroughly in water. Slides were counterstained in hematoxylin for 3 minutes and rinsed in warm water for 3 minutes. Sections were dehydrated and tissue sections cleared in ethanol and xylene before being mounted in Cytoseal XYL (Thermo Scientific–Richard-Allan Scientific). IL-1R staining was visualized and imaging performed as described for IL-1α staining.
Stool Collection and Sample Preparation
Stools were collected from mice treated with or without DSS. They were suspended in an equal volume of PBS and protease inhibitor cocktail 1× (Thermo Scientific) and centrifuged at 15,000 × g for 30 minutes. Supernatants were collected and total protein content measured by a BCA protein assay kit (Thermo Scientific). Samples were stored at −80°C until assayed for murine IL-1α and IL-1β.
Statistical Analysis
The means ± SEM of the data were calculated. The statistical significance of the differences between the two groups exposed to different experimental conditions was determined by the paired Student's t-test. When mentioned, n indicates the sample size and number of biological or independent replicates. P < 0.05 was considered statistically significant.
Results
Intestinal Epithelial Necrotic Cell Lysates Induce a Potent Proinflammatory Response in Human Intestinal Fibroblasts
Injury of the gut epithelium releases endogenous danger signals that may be recognized by nearby cells, such as subepithelial mesenchymal cells, and results in amplification of inflammation. To investigate the potential proinflammatory effect of epithelial cell–derived DAMPs, HIFs were incubated with NCLs from HT29 cells, which resulted in dose-dependent increases in IL-6 and IL-8 production by HIFs (Figure 1A). At a dilution of 1:10, >10-fold inductions in both IL-6 and IL-8 proteins were observed. Notably, the increases in IL-6 and IL-8 in response to NCL were greater than or equivalent to those induced by potent inflammatory stimuli such as TNF-α or LPS at optimal doses. Only background levels of IL-6 and IL-8 (23.8 ± 0.9 pg/mL and 185.3 ± 4.7 pg/mL, respectively; n = 4) were detected in nondiluted NCLs. NCLs prepared from a variety of other human cell types, such as freshly isolated IECs, lamina propria mononuclear cells, polymorphonuclear neutrophils, peripheral blood monocytes, and the monocytic THP1 line, all enhanced HIF production of IL-6 or IL-8 or both, to varying degrees (Figure 1B).
Figure 1.

Intestinal epithelial necrotic cell lysates (NCLs) elicit IL-6 and IL-8 production by human intestinal fibroblasts (HIFs). A: HIFs were incubated in medium alone or 10-fold serial dilutions (1:0 to 1:1000) of HT29 NCLs, 1 μg/mL lipopolysaccharide (LPS), or 100 U/mL tumor necrosis factor (TNF)-α for 24 hours. B: HIFs were incubated in medium alone or in 1:20 dilution of NCLs from HT29, primary human intestinal epithelial cells (IECs), lamina propria mononuclear cells (LPMCs), human blood polymorphonuclear neutrophils (PMNs), human blood monocytes, or THP1 cells for 24 hours. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 3 (A); n = 7 (B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
Identification of the Epithelial Cell–Derived Bioactive Factor Inducing Fibroblast Activation
The findings presented in Figure 1 are compatible with the assumption that epithelial DAMPs were accountable for the observed effect. Therefore, a sequential series of experiments was performed to identify the nature of the DAMP(s) involved in HIF activation. As an initial step, NCLs were pretreated with nuclease and/or proteinase to deplete RNA, DNA, and protein content (Supplemental Figure S1A). NCL, with or without prior RNase treatment, resulted in the induction of IL-6 and IL-8, suggesting that the removal of RNA did not alter the ability of NCLs to activate the inflammatory response in HIFs (Supplemental Figure S1A). However, the removal of proteins from NCLs by proteinase K essentially abolished HIF cytokine production (Supplemental Figure S1A). Notably, HIFs failed to respond to residual DNA in RNase combined with proteinase K–treated NCLs. Further fractionation of NCLs into cytosolic and nuclear fractions revealed the cytosolic localization of the protein(s) contributing to the induction of IL-6 and IL-8 by HIFs (Supplemental Figure S1B). Likewise, stimulation with chromatin or human genomic DNA alone failed to induce IL-6 and IL-8 release (Supplemental Figure S2).
HIF Responses to Necrotic Epithelial Products Are Independent of TLR2, TLR4, RAGE, or IL-33 Ligation
To identify the cytosolic DAMP(s) that triggered the inflammatory response in HIFs, we focused on TLR2 and TLR4, as these are two of the best-characterized receptors shared by various DAMPs and pathogen-associated molecular patterns.36 We first confirmed that: i) the HIF cell lines tested for the production of IL-6 and IL-8 in response to the NCLs and all other stimuli expressed TLR1 to TLR9 by RT-PCR (Supplemental Figure S3), ii) HIF TLR2 and TLR4 are functional, as specifically demonstrated by the induction of IL-6 and IL-8 production on ligation by the respective ligands (Supplemental Figure S4), and iii) the function of TLR2 and TLR4 induced by the ligands could be specifically blocked by neutralizing antibodies, but not IL-6 or IL-8 secretion induced by the cell lysates (Supplemental Figures S4–S6). Surprisingly, these experiments also demonstrated that the NCLs were remarkably powerful in activating HIFs, as shown by the far greater amounts of IL-6 and IL-8 that they induced compared with those induced by TLR2 and TLR4 stimulation (Supplemental Figure S5). The lack of a significant inhibitory effect observed when HIFs were exposed to NCLs in the presence of antibodies to TLR2 and TLR4 (Supplemental Figure S6, A and B) indicated that TLR2 and TLR4 are dispensable for IL-6 and IL-8 induction.
Similar results were obtained after the treatment of NCLs with an anti-RAGE antibody (Supplemental Figure S6C), or soluble RAGE, an antibody to HMGB-1, or an IgG control (Supplemental Figure S7A), and by the failure of recombinant HMGB-1 to induce IL-6 and IL-8 from HIFs (Supplemental Figure S7B). Removal of urate crystals, another classic DAMP,37 by the treatment of NCLs with uricase, also did not alter the ability of the NCLs to induce cytokine production by HIFs (Supplemental Figure S8). Finally, HIF secretion of IL-6 and IL-8 induced by NCL stimulation occurred independently of the DAMP IL-33, as shown by the inability of an IL-33 antibody to block, and failure of recombinant IL-33 to induce, HIF cytokine production (Supplemental Figure S9).
Necrotic Epithelial Cell Products Activate HIFs in an IL-1α−Dependent but Not in an IL-1β−Dependent Manner
We next investigated whether the IL-1 pathway was involved in the NCL activation of HIFs because cytosolic IL-1α is a prototypical DAMP, is only intracellular under physiological conditions, and is universally expressed along with its receptor, IL-1RI.38 Incubation of NCLs with a specific antibody to IL-1RI completely blocked cytokine production (Figure 2A). This occurred in a dose-dependent manner (Supplemental Figure S10A), suggesting that the IL1-RI pathway is required for the sterile inflammatory response triggered by NCLs. To confirm this possibility and to identify the involved IL-1 species, we measured IL-6 and IL-8 secretion after treatment with neutralizing antibody to IL-1α or IL-1β. Interestingly, neutralization of IL-1α inhibited HIF production of IL-6 by 92.0% and of IL-8 by 92.5% (Figure 2B and Supplemental Figure S10B), whereas the antibody to IL-1β failed to significantly decrease the capacity of NCLs to induce HIF cytokine secretion (Figure 2C and Supplemental Figure S10C). Recombinant human IL-1α incubated with HIFs at different doses (0.1 to 1000 pg/mL) for 24 hours was found to induce IL-6 and IL-8 production in a dose-dependent manner (Supplemental Figure S11). Comparison of the quantity of cytokines induced by NCLs (1:10) to that produced in response to recombinant IL-1α (10 pg/mL) indicated that approximately 0.4 ng/mL IL-1α was present in the NCLs in vitro.
Figure 2.

Intestinal epithelial necrotic cell lysate (NCL)–induced IL-6 and IL-8 production by human intestinal fibroblasts (HIFs) is mediated by IL-1 receptor (IL-1R) and IL-1α but not IL-1β. A: HIFs were incubated with 2.5 μg/mL neutralizing antibody to type I IL-1R (IL-1RI) for 1 hour, followed by HT29 NCLs (diluted 1:20) for 24 hours. B and C: HIFs were incubated for 24 hours with HT29 NCLs (diluted 1:20) preincubated for 1 hour with 0.1 μg/mL neutralizing anti-human IL-1α antibody (B) or with 2.5 μg/mL neutralizing anti-human IL-1β antibody (C). IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 3 (A–C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
HIF Cytokine Production Depends on Paracrine Activation by Necrotic Epithelial Cell–Derived IL-1α
To distinguish between intracellular IL-1α released on epithelial cell necrosis and the endogenous IL-1α present in HIFs that could exert autocrine effects on cytokine gene expression,21,39,40 we used siRNA to silence IL-1α in either HT29 cells or HIFs. IL-1α siRNA or a scrambled siRNA control was transfected into HIFs or HT29 cells. The degree of silencing compared with a scrambled siRNA control was validated by real-time quantitative PCR in both HIFs and HT29 cells (Figure 3A). When HIFs were transfected with IL-1α siRNA and then incubated with NCL, no change in the levels of IL-6 and IL-8 production by HIFs was observed (Figure 3B). The cytokine levels were similar to those after transfection of the scrambled siRNA control. However, when NCLs were prepared from IL-1α–silenced HT29 cells and then incubated with HIFs, decreased production of both IL-6 and IL-8 was observed (Figure 3C).
Figure 3.

Intestinal epithelial, but not mesenchymal, cell–derived IL-1α induces cytokine production by human intestinal fibroblasts (HIFs). A: HIFs and HT29 cells were transfected with siRNA specific for IL-1α or scrambled siRNA as control. siRNA-mediated silencing was validated 48 hours after transfection by real-time quantitative PCR for IL-1α mRNA. B: HIFs transfected with IL-1α siRNA or scrambled siRNA were incubated with HT29 necrotic cell lysate (NCL) (dilution 1:20) for 24 hours. C: HT29 cells transfected with IL-1α siRNA or scrambled siRNA were rendered necrotic 24 hours after transfection; the resulting NCLs were diluted 1:20 and incubated with HIFs for 24 hours. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 5 (A); n = 4 (B and C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
The Gut Pathogen S. flexneri Induces Intestinal Epithelial Cell Necrosis and IL-1α Release
The importance of cell death in intestinal pathology has been demonstrated by the ability of gut pathogens to induce death of host cells on infection.36,41 To investigate whether IL-1α released by necrotic IECs after bacterial infection could activate HIFs, we incubated HT29 cells with invasive (M90T) and noninvasive (BS176) strains of S. flexneri.41 Only the invasive form of the S. flexneri M90T strain significantly increased IEC necrosis and concomitantly induced IL-1α but not IL-1β release (Figure 4A).
Figure 4.

Intestinal epithelial cell IL-1α is released during bacterial infection–induced necrosis but not exposure to lipopolysaccharide (LPS), inflammasome agonists, or tumor necrosis factor (TNF)-α. A: HT29 cells were incubated with the invasive Shigella flexneri strain M90T or the noninvasive strain BS176 for 8 hours and cultured in 100 μg/mL gentamicin containing medium for an additional 16 hours. Supernatants were analyzed for IL-1α and IL-1β. The percentages of necrotic cells relative to the number of cells incubated with the noninvasive strain BS176 strain as a control are shown. B and C: HT29 cells were incubated in growth medium or primed with 100 ng/mL to 1 μg/mL ultrapure LPS for 3 hours and then incubated with 100 ng/mL TNF-α or 5 mmol/L ATP or 0.1 to 0.6 μg/mL monosodium urate (MSU) crystals for 24 hours; levels of cytokines in the necrotic cell lysate (B) and supernatants (C) are shown. IL-1α and IL-8 levels were quantified by enzyme-linked immunosorbent assay. All cytokine levels were normalized to lysate protein content. n = 3 (A); n = 3 (B and C). ∗P < 0.05, ∗∗∗P < 0.001.
Lack of IL-1α Secretion in Response to TNF-α or Inflammasome Activators
Findings from recent studies have suggested that IL-1α can be secreted in a nonconventional cell- and stimulus-specific manner after priming with LPS and incubation with activators of the NOD (nucleotide-binding oligomerization domain-containing protein)-like receptor protein 3 (NLRP3) inflammasome.39,42,43 To determine whether epithelial cells can be activated to secrete IL-1α, HT29 cells were primed by incubation with 10 ng/mL to 1 μg/mL ultrapure LPS for 3 hours. At the end of this period, cell cultures were incubated with TNF-α or soluble and particulate activators of the NLRP3 inflammasome: 5 mmol/L ATP or 0.1 to 0.6 μg/mL monosodium urate crystals, respectively, for 24 hours.43 Intracellular and secreted levels of IL-1α were quantified in whole NCLs and supernatants collected from the HT29 cultures. TNF-α caused an increase in intracellular levels of IL-1α in the NCLs (Figure 4B) but failed to induce secretion of IL-1α irrespective of whether the cells were primed with LPS (Figure 4C). In contrast, under these same conditions, TNF-α alone and TNF-α after LPS priming induced a large increase in IL-8 secretion in the HT29 supernatants (Figure 4C). In addition, IL-1α was also not secreted in response to ATP or monosodium urate crystals with or without LPS priming (Figure 4C). Similar experiments with human peripheral blood monocytes confirmed that activation of the NLRP3 inflammasome could induce IL-1β release by these cells (not shown).44
Presence of IL-1α–Positive Cells in the Epithelium in Murine Colitis and Human IBD
Immunofluorescence staining of colon sections of mice with DSS colitis revealed intense expression of IL-1α in the epithelium (Figure 5A) compared with controls (Figure 5C), and shedding of IL-1α–positive IECs in the lumen was evident at days 3 through 7 (Figure 5, B and D). Immunostaining of colonic tissue sections from patients with Crohn disease and ulcerative colitis also showed intense IL-1α staining in the epithelium (Figure 5E). IL-1α was also detected in the control colonic mucosa sections from non-IBD control patients but at substantially lower intensity.
Figure 5.

IL-1α is expressed in the intestinal epithelium and is released before IL-1β during dextran sulfate sodium (DSS)-induced colitis. A: Immunofluorescence staining shows prominent IL-1α expression (red) of murine colonic epithelium before DSS administration. B: IL-1α staining of inflamed epithelium and necrotic colonocytes (arrows) shed in the lumen during DSS colitis [days 3–7; nuclei are stained in blue (DAPI)]. C: Section adjacent to A without primary antibody. D: Hematoxylin and eosin staining of inflamed epithelium and cellular debris (arrow) shed in the lumen during DSS colitis (days 3 to 7). E: immunohistochemical staining showing prominent IL-1α expression in the epithelium and lamina propria in colons with Crohn disease (CD) and ulcerative colitis (UC); IL-1α staining of a normal control colon (NL) is shown but is substantially weaker; antibody control indicates staining in the absence of primary antibody. F: Differential and time-dependent increases in IL-1α (early) and IL-1β (late) in the stool of mice during the progression of DSS-induced colitis. Images were captured with a Leica DMR upright microscope with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter using Image-Pro Plus software. Images are representative of four experiments (A–D) and 3 mice per group (E); n = 5 to 9 mice per group (F). Images were obtained with 20× objective (A–D) or a 10× objective (E).
IL-1α Production Precedes IL-1β and Parallels Intestinal Tissue Damage in Colitic Mice
The release of IL-1α is an early event in various forms of necrosis-induced sterile inflammation,45,46 but the precise timing of the appearance of IL-1α during intestinal inflammation compared with IL-1β is not known. Stool samples were collected from mice during the progression of DSS colitis, and the extent of disease activity was evaluated. Low levels of IL-1α were detectable in the stools at baseline and day 3 and progressively increased until day 7 (Figure 5F) in parallel with progressive tissue damage (data not shown). IL-1β was not detected until day 5 and was increased on day 7 (Figure 5F), suggesting that the release of the epithelial cell DAMP IL-1α is a very early event in experimental IBD.
IL-1α Exacerbates Intestinal Inflammation after Epithelial Injury
IBD is characterized by recurrent episodes of inflammation interspersed with periods of remission, but what exacerbates or reactivates inflammation is unclear. We asked whether IL-1α could: i) trigger inflammation, ii) exacerbate ongoing inflammation, or iii) reactivate inflammation after clinical or histological remission. Three groups of mice were studied: i) control mice given no DSS, ii) mice given a standard colitis-inducing dose (3.0%) of DSS, and iii) mice given a low dose (1.5%) of DSS. The latter two groups received DSS for 7 days and then water for 21 additional days, received an enema of ethanol alone or ethanol plus IL-1α on day 28, and were sacrificed on day 29 (Figure 6A).
Figure 6.

IL-1α exacerbates intestinal injury in dextran sulfate sodium (DSS)-induced colitis. A: Percent changes in body weight of control (no DSS) mice and mice given DSS 1.5% or 3.0% w/v for 7 days and then water for 28 days. On day 28, an IL-1α enema was administered, and mice were sacrificed on day 29. B: Hematoxylin and eosin staining of colons from control (no DSS; day 0), 3.0% DSS, and 1.5% DSS mice given enemas of ethanol alone or ethanol with IL-1α (day 29). Images were captured with a Leica DMR upright microscope with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter using Image-Pro Plus software. n = 5 mice per group (A); image is representative of 3 mice per group (B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Images were obtained with a 2.5× objective.
Mice given 3.0% DSS displayed progressive weight loss and concomitant acute intestinal injury (not shown), with recovery starting at day 10 and continuing thereafter (Figure 6A). On day 28, when active intestinal inflammation had subsided, the administration of IL-1α rapidly (<24 hours) induced severe inflammation, whereas ethanol alone failed to reactivate inflammation (Figure 6B). In contrast, mice given 1.5% DSS maintained a stable weight throughout the duration of the experiment, but the weight was less than that of mice not given DSS, with no histological evidence of intestinal inflammation (Figure 6A). However, after the administration of IL-1α on day 28, in <24 hours they also developed severe intestinal inflammation (Figure 6B) that was, to a degree, comparable to that in mice given IL-1α enemas after recovery from 3.0% DSS-induced colitis or mice with acute DSS colitis (day 7; not shown). Ethanol alone failed to induce inflammation in mice given 1.5% DSS (Figure 6B). These observations were confirmed by the matching histopathological scores of inflammation (Supplemental Table S1). Administration of enemas containing ethanol alone or ethanol plus IL-1α induced no histological evidence of colitis in mice that did not receive DSS (Figure 6B).
In mice given DSS for 7 days, allowed to recover for 21 days, and administered IL-1α for 24 hours, stool levels of IL-1α and IL-1β were assessed by ELISA (Supplemental Figure S12). The levels of IL-1α increased early, before IL-1β, but remained relatively constant over the period of the experiment. In contrast, IL-1β levels increased more, particularly in the 24 hours after the IL-1α enema administration.
Immunostaining with an antibody to the IL-1RI was performed (Figure 7 and Supplemental Figures S13 and S14). The specificity of staining by the antibody to the IL-1RI was validated on mouse thymus tissue (not shown) before immunostaining of colon tissue sections. The anti–IL-1RI antibody staining showed constitutive levels of IL-1RI expression in the intestinal epithelium and progressively enhanced levels of staining in the epithelial, mononuclear, and mesenchymal cell populations with increasing number of days of treatment with both 1.5% and 3.0% DSS (Supplemental Figure S13, B and C). The subepithelial staining was evident on day 5 of DSS treatment and was particularly pronounced 24 hours after the introduction of IL-1α. Increased levels of IL-1RI detected in the lamina propria also indicated an expansion in the mesenchymal cell population (Supplemental Figures S13, A–C, and S14).
Figure 7.

IL-1 receptor (IL-1R) immunostaining of colons after dextran sulfate sodium (DSS) induction of colitis and IL-1α administration. A and B: Microscopic images of IL-1R immunostaining of colons from control (no DSS; day 0), 3.0% DSS, and 1.5% DSS mice given enemas of ethanol alone or ethanol with IL-1α (day 29) shown in A. Images were captured with a Leica DMR upright microscope with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter using Image-Pro Plus software. Images are representative of 3 mice per group. Images were obtained with 2.5× objective (A) or 10× objective (B).
Discussion
Over the past decade, cell injury and death have emerged as potent inducers of inflammatory responses, yet knowledge of the underlying mechanisms and their importance in intestinal inflammation remains limited.36,37 Because epithelial damage is a common event in IBD, we set out to define the key DAMPs released by injured epithelial cells and to determine whether they promote proinflammatory activity by other mucosal cells. Myofibroblasts lie immediately below the epithelium, and a DAMP-initiated interaction between them could magnify intestinal inflammation.10,25
Our initial experiments demonstrated that HT29-derived DAMPs elicited IL-6 and IL-8 production by HIFs in a dose-dependent manner, and this induction was even greater than that induced by potent proinflammatory mediators such as LPS and TNF-α. This response is likely to occur in vivo since NCL from primary human IECs also induced HIFs to produce IL-6 and IL-8. The enhanced production of these cytokines would result in recruitment and activation of additional inflammatory cells. Our findings are consistent with those from studies that employed mice with conditional epithelial deletions of either caspase 8 or Fas-associated death domain (adaptor proteins required for death receptor–induced apoptosis) that identified epithelial cell necrosis as a crucial factor in the initiation and perpetuation of terminal ileitis and colitis.1,4
A variety of DAMPs from multiple sources have been described that include nuclear and cytosolic proteins as well as nucleic acids and urate crystals.37 We found that the epithelial DAMPs that mediate the response of HIFs are cytosolic proteins and not DNA or RNA. In subsequent experiments, blockade of the IL-1RI essentially abrogated the IL-6 and IL8 production by HIFs, pointing to the IL-1 pathway as vitally involved in the observed effects. This finding was enlightening because one of the two IL-1 species able to bind the IL-1RI is IL-1α, a protein abundantly expressed in epithelial cells and a known major mediator of sterile inflammation.21,24 In fact, neutralization of IL-1α, but not IL-1β, inhibited HIF production of IL-6 and IL-8 by >90%. These results, combined with the fact that all other components present in the NCLs contribute to <10% of the activation process, provide compelling evidence that IL-1α is the DAMP involved in the HIF activation process. To consolidate this conclusion, we excluded the involvement of proteins that signal through TLR2 or TLR4 and other danger signals, namely HMGB-1, IL-33, DNA, histones, chromatin, and uric acid crystals, as the mediators of HIF cytokine production. Finally, the presence of IL-1α in NCLs of HT29 cells and IEC isolates from normal and IBD mucosa was confirmed on findings of IL-1α–positive epithelial cells in the corresponding tissues on immunohistochemical analysis.
Abundant evidence indicates that IL-1α acts as an important DAMP in sterile inflammation in a variety of conditions, such as rheumatoid arthritis, gout, autoinflammatory syndromes, chronic lung disease, and atherosclerosis.22,47 Interestingly, findings from studies of infectious lung disease, such as Legionnaires pneumonia infection, suggest that IL-1α but not IL-1β is the dominant cytokine produced early in the disease.46 Nonetheless, given the importance of the IL-1 family of cytokines to innate immunity, the actions of IL-1α have been far less studied and understood than those of IL-1β.38 Emerging evidence indicates differential timing and clear functional differences between IL-1α and IL-1β,45,48 including nonredundant roles for each cytokine in Mycobacterium tuberculosis infection.49
Investigation into the mechanisms by which IL-1α regulates cytokine gene expression has shown that intracellular IL-1α can exert transcriptional effects by interacting with chromatin and activating cytokine gene expression.39,40 Thus, it was crucial to distinguish between the intracellular HIF pool of IL-1α and the one released by necrotic epithelial cells. The IL-1α siRNA experiments showed that paracrine activation by epithelial cell–derived IL-1α was involved in eliciting cytokine production by HIFs. These experiments indicate that HIF sensing of IEC injury, with subsequent activation of cytokine synthesis, is mediated by IL-1α acting as an alarmin and does not involve the intracellular IL-1α pool in HIFs. No evidence of HIF cell death was observed during these experiments, ruling out the involvement of autocrine effects due to the release of endogenous IL-1α by HIFs (not shown). The location of fibroblasts immediately below the intestinal epithelium and overlaying the immune cell compartment indicates that these mesenchymal cells are strategically located to sense superficial intestinal injury through IL-1α–dependent signaling and then produce cytokines to directly amplify the immune response in the deeper mucosa.
Some reports claim that IL-1α is released only from necrotic but not live or apoptotic cells.38,39,43,50,51 However, in specific contexts, live monocytes/macrophages, dendritic cells, and keratinocytes can atypically secrete IL-1α. Secretion of the cytokine occurs in response to NLRP3 inflammasome agonists, LPS priming, and calpain cleavage.43 Conflicting reports also suggest that inflammasome-activated caspase 1 and caspase 11 can mediate the secretion of IL-1α in response to noninfectious stimuli, whereas other studies have concluded that neither of these pathways is required.46,52,53 In our experiments with live IECs (HT29), TNF-α and both soluble and particulate activators of NLRP3 failed to induce IL-1α secretion by HT29. Our controlled experiments verified that the same stimuli could induce the release of IL-1α and IL-1β by human peripheral blood monocytes.51 Therefore, it appears that intracellular IL-1α is released by injured but not intact intestinal epithelium in response to proinflammatory or NLRP3 inflammasome stimuli.
This conclusion is supported by the identification of IL-1α in murine and human colonocytes by immunostaining and the presence of necrotic IL-1α–positive epithelial cells in the lumen of mice with DSS colitis. In addition, our experiments with the gut pathogen S. flexneri showed that the invasive, but not the noninvasive, strain caused release of IL-1α but not IL-1β, and that this release occurred only concomitantly with epithelial cell necrosis. Recent studies using a Matrigel plug containing supernatants of hypoxic cells injected into mice have shown that, during inflammation, IL-1α and IL-1β recruit distinct types of myeloid cells (neutrophils and macrophages, respectively) in a time-dependent fashion.45 To dissect the in vivo function of IL-1α, we examined the timing of the appearance of the cytokine in the stool of mice with DSS colitis. We found that IL-1α and IL-1β are separately expressed at an early and late phase, respectively, of the gut inflammatory response. These experiments clearly consolidate the role of IL-1α as an important DAMP acting as a very early mediator of intestinal inflammation, a conclusion supported by findings in other tissues such as in the lung inflammatory response to bacterial infection.46,49
To further define the role of IL-1α, we conducted in vivo experiments to determine whether it acts as a proinflammatory DAMP only in the early gut inflammation, during recovery, or even in the absence of overt inflammation. Mice were given a 3.0% dose of DSS, which induces overt clinical and histopathological evidence of colitis, or a 1.5% dose, which does not induce histopathological evidence of inflammation. After a 3-week recovery period, they were challenged with intrarectal IL-1α. Mice that had acute colitis in the first week and had essentially recovered both clinically and histologically experienced reactivation of severe colitis in a remarkably short period of time (<24 hours). Even more remarkable was the observation that mice given 1.5% DSS and without histological evidence of colitis also developed acute severe colitis on colonic exposure to IL-1α. These novel observations suggest that, whereas IL-1α is unable to induce inflammation in the healthy intestine, it acts as an inflammatory DAMP not only in active gut inflammation but also during the apparent recovery of a previous inflammatory episode. Thus, even a subtle epithelial injury that releases IL-1α may be enough to trigger overt inflammation in an intestine that is normal at the histological level but not at the molecular level. A limited number of previous studies have investigated the role of the IL-1RI specifically, in amplification of intestinal inflammation in experimental colitis or in human IBD.54–57 Inhibition of the IL-1RI with the IL-1RI antagonist in experimental colitis has been reported to both suppress inflammation and enhance susceptibility to intestinal damage. In our studies, immunostaining of the colons from mice in recovery after 3.0% or 1.5% DSS administration showed enhanced levels of IL-1RI in the lamina propria, as well as expanded numbers of fibroblasts in this region of the colon, in response to a 24-hour enema of IL-1α. Along with the increased levels of the IL-1 type I signaling receptor in 1.5% and 3.0% DSS–treated mice, high levels of IL-1α and IL-1β were released as detected in the stool, suggesting that the IL-1 pathway is pivotal to the reactivation of inflammation in DSS colitis and possibly in flare episodes of human IBD.
Interestingly, multiple inflammatory genes remain dysregulated in the colon after recovery from murine colitis58 and human ulcerative colitis.59 This is consistent with the concept that changes at the molecular level underlie the recurrence of inflammation associated with the chronic, relapsing nature of IBD. Taken together, our observations also suggest that the local, early release of IL-1α may be sufficient to reactivate inflammation, possibly by inducing the expression of IL-1β and/or IL-1RI.60 This may be the mechanism underlying the accelerated induction of severe colitis in IL-10–deficient mice treated with nonsteroidal anti-inflammatory drugs, agents known to induce epithelial cell injury.61 Combined, these observations are potentially of clinical significance in view of the well-known emergence or reactivation of human IBD after intestinal epithelial damage caused by subclinical infections or agents such as nonsteroidal anti-inflammatory drugs.62,63
In summary, our studies provide the basis of a new paradigm that epithelial DAMPs induce sterile proinflammatory responses in IBD and contribute to innate immune responses through nonimmune cell interactions in the inflamed intestine. It is likely that DAMP-induced production of large quantities of IL-8 and IL-6 would lead to neutrophil recruitment as well as T-cell and monocyte differentiation, respectively. These well-documented consequences of cytokine production are pivotal events by which the intestinal inflammatory response can be amplified and perpetuated. Hence, IL-1α is a mediator of epithelial–fibroblast interaction and plays a vital role in amplification and reactivation of intestinal inflammation. Specifically targeting IL-1α in these nonimmune cell interactions may provide alternative and effective strategies for treating and even preventing intestinal tissue damage.
Acknowledgments
We thank Dr. Judy Drazba and Denise Hatala (Lerner Research Institute) for immunohistochemical analysis; Dr. Franco Scaldaferri (Catholic University, Rome, Italy) for RT-PCR of TLRs; Eboni Ubinas, Tiffany Hollo, and all staff of the Tissue Procurement Service (Cleveland Clinic) for help with the collection of intestinal tissue specimens; and Dr. Philippe Sansonetti (Institute Pasteur, Paris, France) for M90T and BS176 S. flexneri strains.
Footnotes
Supported by NIH grants DK093630 (C.F. and E.S.), DK050984 (C.F.), DK082437 and UL1TR000439 (C.M.), and 1P01-HL107147 (C.d.M.); the Department of Pathobiology, Lerner Research Institute; and the Digestive Diseases Institute of the Cleveland Clinic.
Disclosures: None declared.
Current address of M.S., Istituto Oncologico Veneto, I.R.C.C.S., Padua, Italy.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.02.018.
Supplemental Data
Supplemental Figure S1.

Cytosolic proteins are the active damage-associated molecular patterns in necrotic intestinal epithelial cell lysates. A: HT29 necrotic cell lysates (NCLs) were depleted of RNA and/or proteins by RNase and/or proteinase K treatment, diluted 1:20 in culture medium, and incubated with human intestinal fibroblasts (HIFs) for 24 hours. B: Total protein, cytosolic protein, or nuclear protein fractions prepared from HT29 NCLs and incubated with HIFs for 24 hours. Equal amounts of total, cytosolic, and nuclear fractions were used, and cytoplasmic and nuclear extracts were normalized to 2.5 × 106 cell equivalents. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 4.
Supplemental Figure S2.

DNA, histones, and chromatin do not mediate intestinal epithelial necrotic cell lysate (NCL)–induced human intestinal fibroblast (HIF) cytokine production. HIFs were incubated in medium alone or in the presence of HT29-derived genomic DNA, soluble histones, or sheared chromatin for 24 hours; the concentrations of DNA, soluble histones, and chromatin were equivalent to those in a 1:20 dilution of the NCLs of the same cells. NCLs increased IL-6 production, but none of the three nuclear components induced HIFs to produce IL-6. IL-6 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 4.
Supplemental Figure S3.

Demonstration of Toll-like receptor (TLR) expression by human intestinal fibroblasts (HIFs). Expression of mRNA for TLR1 to TLR9 in HIF was evaluated by RT-PCR as described in Materials and Methods. Glyceraldehyde-3-phosphate dehydrogenase was used as loading control. The image shown is representative of nine different HIF lines.
Supplemental Figure S4.

Antibodies to Toll-like receptor (TLR)-2 and TLR4 inhibit cytokine production by human intestinal fibroblasts (HIFs) induced by their respective ligands, synthetic diacylated glycoprotein 1 (FSL-1) and lipopolysaccharide (LPS). HIFs were preincubated for 1 hour either with 5 μg/mL neutralizing anti-human TLR2 antibody followed by 1 μg/mL FSL-1 for 24 hours, or preincubated for 1 hour with 5 μg/mL neutralizing anti-human TLR4 antibody followed by 1 μg/mL LPS. After 24 hours, IL-8 and IL-6 levels in the supernatants were measured by enzyme-linked immunosorbent assay. ∗P < 0.05 for ligand-treated plus anti-TLR–treated HIF versus nonstimulated (no ligand) and anti-ligand antibody only–stimulated HIF. n = 3.
Supplemental Figure S5.

Comparative levels of IL-6 and IL-8 production by human intestinal fibroblasts (HIFs) in response to Toll-like receptor (TLR)-2 and TLR4 ligands and intestinal epithelial necrotic cell lysates (NCLs). HIFs were incubated in medium alone or in the presence of 1 μg/mL synthetic diacylated glycoprotein 1 (FSL)-1, 1 μg/mL lipopolysaccharide (LPS), or NCLs (1:20 dilution); the NCLs were also preincubated for 1 hour with 5 μg/mL neutralizing anti-human TLR2 or 5 μg/mL neutralizing anti-human TLR4. After 24 hours, IL-8 and IL-6 levels in the supernatants were measured by enzyme-linked immunosorbent assay. ∗∗P < 0.01 and ∗∗∗P < 0.001. n = 3.
Supplemental Figure S6.

Toll-like receptor (TLR)-2, TLR4, and receptor for advanced glycation end-products (RAGE) do not mediate intestinal epithelial necrotic cell lysate (NCL)-induced human intestinal fibroblast (HIF) cytokine production. A–C: HT29 NCLs (diluted 1:20) were preincubated for 1 hour with 5 μg/mL neutralizing anti-human antibodies for TLR2 (A), TLR4 (B), or RAGE (C), and then added to the HIFs for 24 hours. None of the antibodies block the NCL-induced cytokine production by HIF. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 3.
Supplemental Figure S7.

Soluble receptor for advanced glycation end-products (sRAGE) and high-mobility group box (HMGB)-1 do not mediate intestinal epithelial necrotic cell lysate (NCL)-induced human intestinal fibroblast (HIF) cytokine production. A: HIFs were incubated with 0, 5, and 10 μg/mL soluble RAGE, 5 and 10 μg/mL neutralizing anti-human HMGB-1 antibody, and 5 and 10 μg/mL IgG for 1 hour followed by an incubation with medium alone or NCLs (diluted 1:20) for 24 hours. IL-6 levels in the supernatants were measured by enzyme-linked immunosorbent assay (ELISA). B: HIFs were incubated with 0 to 1000 ng/mL recombinant human HMGB-1 for 24 hours, and IL-6 and IL-8 levels in the supernatants were measured by ELISA. n = 3 (A and B).
Supplemental Figure S8.

Uricase treatment of intestinal epithelial necrotic cell lysates (NCLs) does not abrogate human intestinal fibroblast (HIF) cytokine production. NCLs (1:20) were treated with 10 U/mL uricase for 30 minutes at 25°C, diluted 1:20, and incubated with HIFs for 24 hours. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. No significant changes in the level of cytokines were observed after uricase treatment. n = 5 (IL-6); n = 3 (IL-8).
Supplemental Figure S9.

Anti-human IL-33 does not abrogate, and IL-33 does not mediate, intestinal epithelial necrotic cell lysate (NCL)-induced human intestinal fibroblast (HIF) cytokine production. A: HIFs were incubated for 1 hour with 1 to 10 μg/mL anti–IL-33, and NCLs were added to the culture medium (dilution 1:20). B: Recombinant IL-33 (0.01 to 10,000 μg/mL) was added to HIFs, and cells were incubated for 24 hours. IL-6 and IL-8 levels in the supernatants were measured by enzyme-linked immunosorbent assay. n = 3.
Supplemental Figure S10.

Dose-dependent inhibition of human intestinal fibroblast (HIF) cytokine production by antibodies to type I IL-1 receptor (IL-1RI) and IL-1α but not IL-1β. A: A neutralizing anti-human IL-1RI (0.1 to 2.5 μg/mL) antibody was added to HIFs for 1 hour followed by epithelial necrotic cell lysates (NCLs) (diluted 1:20), and cells were incubated for 24 hours. B and C: Neutralizing anti-human antibodies to IL-1α (0 to 100 ng/mL) (B) or IL-1β (0 to 5000 ng/mL) (C) were incubated with NCLs for 1 hour, and then lysates were added to HIFs for 24 hours. The levels of IL-6 and IL-8 were measured in HIF supernatants by enzyme-linked immunosorbent assay. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. n = 3 (A and C); n = 4 (B).
Supplemental Figure S11.
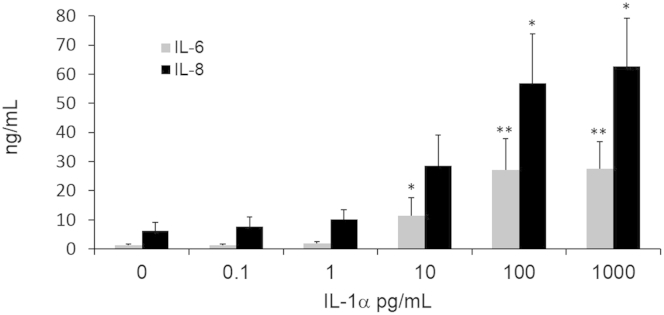
Recombinant IL-1α induces cytokine production in human intestinal fibroblasts (HIFs). Confluent HIFs were incubated with increasing doses of recombinant IL-1α (0.1 to 1000 pg/mL) for 24 hours. Supernatants were harvested and levels of IL-6 and IL-8 measured by enzyme-linked immunosorbent assay. ∗P < 0.05 and ∗∗P < 0.01. n = 6.
Supplemental Figure S12.

Cytokine levels in the stool of colitic mice after dextran sulfate sodium (DSS) induction of colitis. Stools from control mice (no DSS; day 0) and mice given 1.5% w/v or 3.0% w/v DSS for 7 days followed by water for 21 days and an enema of IL-1α for 24 hours on day 28 were assessed for IL-1α and IL-1β by enzyme-linked immunosorbent assay. The cytokine values are plotted in pg/mL on a logarithmic scale. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
Supplemental Figure S13.

IL-1 receptor (IL-1R) expression in murine colon during dextran sulfate sodium (DSS)-induced colitis. Mice were given either water alone [no DSS (control); day 0] or 3.0% w/v DSS for 3, 5, or 7 days. A: Hematoxylin and eosin (H&E) staining of colons. B and C: Immunostaining of murine IL-1R. Images were captured with a Leica DMR upright microscope with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter using Image-Pro Plus software. n = 5 mice per group (images are representative of three mice per group) (A). Images were obtained with 2.5× objective (A and B) or 10× objective (C).
Supplemental Figure S14.

IL-1 receptor (IL-1R) expression in murine colon during dextran sulfate sodium (DSS)-induced colitis (higher magnification of Supplemental Figure S13C). Red arrows indicate staining for IL-1R, which appears to increase in intensity from day 0 to days 5 and 7. Yellow arrows indicate staining in the lamina propria; day 0 shows an essential lack of IL-1R staining; day 3 shows minimal staining and slight enlargement of the lamina propria; days 5 and 7 show intense staining of mononuclear and mesenchymal cells and diffuse (day 5) and gross (day 7) enlargement of the lamina propria by increased cellularity. On days 5 and 7, staining for IL-1R is also observed in the muscularis mucosae. Images were captured with a Leica DMR upright microscope with a Retiga EXi Cooled CCD camera with liquid crystal tunable RGB filter using Image-Pro Plus software. Images were obtained with a 20× objective.
References
- 1.Gunther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H., Waldner M.J., Hedrick S.M., Tenzer S., Neurath M.F., Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunther C., Neumann H., Neurath M.F., Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–1071. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 3.Linkermann A., Green D.R. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welz P.S., Wullaert A., Vlantis K., Kondylis V., Fernandez-Majada V., Ermolaeva M., Kirsch P., Sterner-Kock A., van Loo G., Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 5.Dourmashkin R.R., Davies H., Wells C., Shah D., Price A., O'Morain C., Levi J. Epithelial patchy necrosis in Crohn's disease. Hum Pathol. 1983;14:643–648. doi: 10.1016/s0046-8177(83)80207-x. [DOI] [PubMed] [Google Scholar]
- 6.Barkla D.H., Gibson P.R. The fate of epithelial cells in the human large intestine. Pathology. 1999;31:230–238. doi: 10.1080/003130299105043. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y., Zheng W., Rock K.L. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci U S A. 2000;97:14590–14595. doi: 10.1073/pnas.260497597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 9.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C., Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi M.E., Manfredi A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi M.E. HMGB1 loves company. J Leukoc Biol. 2009;86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 13.Foell D., Wittkowski H., Ren Z., Turton J., Pang G., Daebritz J., Ehrchen J., Heidemann J., Borody T., Roth J., Clancy R. Phagocyte-specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J Pathol. 2008;216:183–192. doi: 10.1002/path.2394. [DOI] [PubMed] [Google Scholar]
- 14.Foell D., Wittkowski H., Roth J. Monitoring disease activity by stool analyses: from occult blood to molecular markers of intestinal inflammation and damage. Gut. 2009;58:859–868. doi: 10.1136/gut.2008.170019. [DOI] [PubMed] [Google Scholar]
- 15.Maeda S., Hikiba Y., Shibata W., Ohmae T., Yanai A., Ogura K., Yamada S., Omata M. Essential roles of high-mobility group box 1 in the development of murine colitis and colitis-associated cancer. Biochem Biophys Res Commun. 2007;360:394–400. doi: 10.1016/j.bbrc.2007.06.065. [DOI] [PubMed] [Google Scholar]
- 16.Dave S.H., Tilstra J.S., Matsuoka K., Li F., DeMarco R.A., Beer-Stolz D., Sepulveda A.R., Fink M.P., Lotze M.T., Plevy S.E. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol. 2009;86:633–643. doi: 10.1189/jlb.1008662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kupper T.S., Ballard D.W., Chua A.O., McGuire J.S., Flood P.M., Horowitz M.C., Langdon R., Lightfoot L., Gubler U. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J Exp Med. 1986;164:2095–2100. doi: 10.1084/jem.164.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stadnyk A.W., Sisson G.R., Waterhouse C.C. IL-1 alpha is constitutively expressed in the rat intestinal epithelial cell line IEC-6. Exp Cell Res. 1995;220:298–303. doi: 10.1006/excr.1995.1319. [DOI] [PubMed] [Google Scholar]
- 19.Elkon K.B. IL-1alpha responds to necrotic cell death. Nat Med. 2007;13:778–780. doi: 10.1038/nm0707-778. [DOI] [PubMed] [Google Scholar]
- 20.Eigenbrod T., Park J.H., Harder J., Iwakura Y., Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol. 2008;181:8194–8198. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C.J., Kono H., Golenbock D., Reed G., Akira S., Rock K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 22.Suwara M.I., Green N.J., Borthwick L.A., Mann J., Mayer-Barber K.D., Barron L., Corris P.A., Farrow S.N., Wynn T.A., Fisher A.J., Mann D.A. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cominelli F., Nast C.C., Llerena R., Dinarello C.A., Zipser R.D. Interleukin 1 suppresses inflammation in rabbit colitis. Mediation by endogenous prostaglandins. J Clin Invest. 1990;85:582–586. doi: 10.1172/JCI114476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bersudsky M., Luski L., Fishman D., White R.M., Ziv-Sokolovskaya N., Dotan S., Rider P., Kaplanov I., Aychek T., Dinarello C.A., Apte R.N., Voronov E. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut. 2014;63:598–609. doi: 10.1136/gutjnl-2012-303329. [DOI] [PubMed] [Google Scholar]
- 25.Danese S. Nonimmune cells in inflammatory bowel disease: from victim to villain. Trends Immunol. 2008;29:555–564. doi: 10.1016/j.it.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Strong S.A., Pizarro T.T., Klein J.S., Cominelli F., Fiocchi C. Proinflammatory cytokines differentially modulate their own expression in human intestinal mucosal mesenchymal cells. Gastroenterology. 1998;114:1244–1256. doi: 10.1016/s0016-5085(98)70431-7. [DOI] [PubMed] [Google Scholar]
- 27.Youngman K.R., Simon P.L., West G.A., Cominelli F., Rachmilewitz D., Klein J.S., Fiocchi C. Localization of intestinal interleukin 1 activity and protein and gene expression to lamina propria cells. Gastroenterology. 1993;104:749–758. doi: 10.1016/0016-5085(93)91010-f. [DOI] [PubMed] [Google Scholar]
- 28.Binion D.G., West G.A., Ina K., Ziats N.P., Emancipator S.N., Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112:1895–1907. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- 29.El Gazzar M., Yoza B.K., Hu J.Y., Cousart S.L., McCall C.E. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. J Biol Chem. 2007;282:26857–26864. doi: 10.1074/jbc.M704584200. [DOI] [PubMed] [Google Scholar]
- 30.Iyer S.S., Pulskens W.P., Sadler J.J., Butter L.M., Teske G.J., Ulland T.K., Eisenbarth S.C., Florquin S., Flavell R.A., Leemans J.C., Sutterwala F.S. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauter B., Albert M.L., Francisco L., Larsson M., Somersan S., Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shechter D., Dormann H.L., Allis C.D., Hake S.B. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 33.Sadler T., Scarpa M., Rieder F., West G., Stylianou E. Cytokine-induced chromatin modifications of the type I collagen alpha 2 gene during intestinal endothelial-to-mesenchymal transition. Inflamm Bowel Dis. 2013;19:1354–1364. doi: 10.1097/MIB.0b013e318281f37a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugiyama H., Savill J.S., Kitamura M., Zhao L., Stylianou E. Selective sensitization to tumor necrosis factor-alpha-induced apoptosis by blockade of NF-kappaB in primary glomerular mesangial cells. J Biol Chem. 1999;274:19532–19537. doi: 10.1074/jbc.274.28.19532. [DOI] [PubMed] [Google Scholar]
- 35.Kessler S., Rho H., West G., Fiocchi C., Drazba J., de la Motte C. Hyaluronan (HA) deposition precedes and promotes leukocyte recruitment in intestinal inflammation. Clin Transl Sci. 2008;1:57–61. doi: 10.1111/j.1752-8062.2008.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaczmarek A., Vandenabeele P., Krysko D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Kono H., Rock K.L. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinarello C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 39.Cohen I., Rider P., Carmi Y., Braiman A., Dotan S., White M.R., Voronov E., Martin M.U., Dinarello C.A., Apte R.N. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werman A., Werman-Venkert R., White R., Lee J.K., Werman B., Krelin Y., Voronov E., Dinarello C.A., Apte R.N. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci U S A. 2004;101:2434–2439. doi: 10.1073/pnas.0308705101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carneiro L.A., Travassos L.H., Soares F., Tattoli I., Magalhaes J.G., Bozza M.T., Plotkowski M.C., Sansonetti P.J., Molkentin J.D., Philpott D.J., Girardin S.E. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe. 2009;5:123–136. doi: 10.1016/j.chom.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Mohan S.K., Yu C. The IL1alpha-S100A13 heterotetrameric complex structure: a component in the non-classical pathway for interleukin 1alpha secretion. J Biol Chem. 2011;286:14608–14617. doi: 10.1074/jbc.M110.201954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gross O., Yazdi A.S., Thomas C.J., Masin M., Heinz L.X., Guarda G., Quadroni M., Drexler S.K., Tschopp J. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 44.Guarda G., Braun M., Staehli F., Tardivel A., Mattmann C., Forster I., Farlik M., Decker T., Du Pasquier R.A., Romero P., Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Rider P., Carmi Y., Guttman O., Braiman A., Cohen I., Voronov E., White M.R., Dinarello C.A., Apte R.N. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 46.Barry K.C., Fontana M.F., Portman J.L., Dugan A.S., Vance R.E. IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol. 2013;190:6329–6339. doi: 10.4049/jimmunol.1300100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rock K.L., Latz E., Ontiveros F., Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freigang S., Ampenberger F., Weiss A., Kanneganti T.D., Iwakura Y., Hersberger M., Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 49.Mayer-Barber K.D., Andrade B.B., Barber D.L., Hieny S., Feng C.G., Caspar P., Oland S., Gordon S., Sher A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi Y., Yamamoto K., Saido T., Kawasaki H., Oppenheim J.J., Matsushima K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc Natl Acad Sci U S A. 1990;87:5548–5552. doi: 10.1073/pnas.87.14.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fettelschoss A., Kistowska M., LeibundGut-Landmann S., Beer H.D., Johansen P., Senti G., Contassot E., Bachmann M.F., French L.E., Oxenius A., Kundig T.M. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci U S A. 2011;108:18055–18060. doi: 10.1073/pnas.1109176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller M., Ruegg A., Werner S., Beer H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 53.Zheng Y., Humphry M., Maguire J.J., Bennett M.R., Clarke M.C. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity. 2013;38:285–295. doi: 10.1016/j.immuni.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferretti M., Casini-Raggi V., Pizarro T.T., Eisenberg S.P., Nast C.C., Cominelli F. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest. 1994;94:449–453. doi: 10.1172/JCI117345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cominelli F., Nast C.C., Duchini A., Lee M. Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology. 1992;103:65–71. doi: 10.1016/0016-5085(92)91096-m. [DOI] [PubMed] [Google Scholar]
- 56.Lebeis S.L., Powell K.R., Merlin D., Sherman M.A., Kalman D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2009;77:604–614. doi: 10.1128/IAI.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez-Navajas J.M., Law J., Nguyen K.P., Bhargava M., Corr M.P., Varki N., Eckmann L., Hoffman H.M., Lee J., Raz E. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9-driven antiinflammatory cytokine production. J Exp Med. 2010;207:2799–2807. doi: 10.1084/jem.20101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breynaert C., Dresselaers T., Perrier C., Arijs I., Cremer J., Van Lommel L., Van Steen K., Ferrante M., Schuit F., Vermeire S., Rutgeerts P., Himmelreich U., Ceuppens J.L., Geboes K., Van Assche G. Unique gene expression and MR T2 relaxometry patterns define chronic murine dextran sodium sulphate colitis as a model for connective tissue changes in human Crohn's disease. PLoS One. 2013;8:e68876. doi: 10.1371/journal.pone.0068876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Planell N., Lozano J.J., Mora-Buch R., Masamunt M.C., Jimeno M., Ordas I., Esteller M., Ricart E., Pique J.M., Panes J., Salas A. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut. 2013;62:967–976. doi: 10.1136/gutjnl-2012-303333. [DOI] [PubMed] [Google Scholar]
- 60.Turner N.A., Das A., Warburton P., O'Regan D.J., Ball S.G., Porter K.E. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. 2009;297:H1117–H1127. doi: 10.1152/ajpheart.00372.2009. [DOI] [PubMed] [Google Scholar]
- 61.Berg D.J., Zhang J., Weinstock J.V., Ismail H.F., Earle K.A., Alila H., Pamukcu R., Moore S., Lynch R.G. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 62.Stallmach A., Carstens O. Role of infections in the manifestation or reactivation of inflammatory bowel diseases. Inflamm Bowel Dis. 2002;8:213–218. doi: 10.1097/00054725-200205000-00009. [DOI] [PubMed] [Google Scholar]
- 63.Felder J.B., Korelitz B.I., Rajapakse R., Schwarz S., Horatagis A.P., Gleim G. Effects of nonsteroidal antiinflammatory drugs on inflammatory bowel disease: a case-control study. Am J Gastroenterol. 2000;95:1949–1954. doi: 10.1111/j.1572-0241.2000.02262.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.