

Abstract
Buprenorphine is a successful analgesic and treatment for opioid abuse, with both activities relying on its partial agonist activity at mu opioid receptors. However, there is substantial interest in its activities at the kappa opioid and nociceptin/orphanin FQ peptide receptors. This has led to an interest in developing compounds with a buprenorphine-like pharmacological profile but with lower efficacy at mu opioid receptors. The present article describes aryl ring analogues of buprenorphine in which the standard C20-methyl group has been moved to the C7β position, resulting in ligands with the desired profile. In particular, moving the methyl group has resulted in far more robust kappa opioid antagonist activity than seen in the standard orvinol series. Of the compounds synthesized, a number, including 15a, have a profile of interest for the development of drug abuse relapse prevention therapies or antidepressants and others (e.g., 8c), as analgesics with a reduced side-effect profile.
Introduction
Buprenorphine (1a, Chart 1) is widely used for the treatment of opioid abuse and as an analgesic. Its mu opioid receptor (MOPr) partial agonist character is of primary importance for both indications, but it is apparent that activity at other receptors, in particular, antagonism at the kappa opioid receptor (KOPr), may play an important role in its clinical utility.1
Chart 1.

The very high rate of relapse to drug use after a period of abstinence is a major problem in substance abuse treatment, with ∼70% of treated addicts relapsing within the first year following treatment.2 The fact that many drug users use more than one drug further complicates the situation. A variety of factors play a role in precipitating relapse, but stress and/or a priming dose of the drug are particular risks. In preclinical studies, inhibition or genetic ablation of KOPrs has been shown to inhibit stress-induced relapse, but this approach has not been effective in blocking drug-prime-induced reinstatement to drug-seeking behavior.3
In contrast to the studies using selective KOPr antagonists, our own preclinical findings4,5 suggest that a combination of buprenorphine and naltrexone can be effective in blocking drug-prime-induced reinstatement to both cocaine- and opioid-seeking behavior. The combination acts as a KOP and delta opioid (DOP) receptor antagonist,6 a lower potency nociceptin/orphanin FQ peptide (NOP) receptor partial agonist,7 and a MOP antagonist.
Encouraging results have also been observed in two small clinical trials using a buprenorphine and naltrexone combination therapy in a ratio that should block most, or all, of buprenorphine’s MOPr agonist activity.8,9 The combination proved to be effective in reducing relapse in recovering opiate addicts and also caused a significant reduction in cocaine use. The beneficial effect on cocaine use is being more thoroughly evaluated within The National Drug Abuse Treatment Clinical Trials Network in the Cocaine Use Reduction with Buprenorphine (CURB: CTN-0048) study.
Thus, preclinical and clinical data suggest that the buprenorphine–naltrexone combination has significant therapeutic potential for the treatment of relapse and polydrug addiction, but it has substantial issues that need to be resolved. First, delivery of the combination is problematic; buprenorphine has poor oral bioavailability and is given sublingually, whereas naltrexone is active after oral administration but much less so after sublingual administration. The two compounds are therefore not amenable to formulation as a single tablet, and it is not desirable to use the two drugs separately due to potential compliance issues and the possibility of buprenorphine being diverted for illicit, nonmedical use. Second, the exact pharmacological profile provided by the combination is not clear, particularly whether there is a “pure” antagonist or a low-efficacy MOPr component. Provision of single chemical entities that provide buprenorphine-like activity at KOP, DOP, and NOP receptors but with substantially reduced efficacy at MOPr will allow these issues to be resolved and will provide compounds with potential therapeutic utility for relapse prevention and poly drug abuse.
Buprenorphine has also found widespread use as an analgesic and appears to be effective in neuropathic pain conditions that are not sensitive to other opioids, although the mechanism for this difference is not clear.10 There has been speculation that buprenorphine’s unique clinical profile might be linked to its ability to act as a low-efficacy partial agonist at NOPr in addition to its MOPr partial agonism,11 although Cremeans et al. found no evidence for activation of NOPr being important for buprenorphine’s analgesic activity in primates.12 On the other hand, they did find that coactivation of MOPr and NOPr produced synergistic antinociception. This suggests that bifunctional ligands with low efficacy at MOPr and moderate efficacy at NOPr might have utility as analgesics with a reduced side-effect profile. Efforts to develop such bifunctional ligands are being made.13
Orvinols, the series of compounds to which buprenorphine belongs, typically display high efficacy at one, or both, of the MOPr and KOPr.14 However, we recently reported on a series of aryl analogues of buprenorphine, some of which displayed substantially lower efficacy at the opioid receptors while retaining high affinity for opioid receptors and moderate (equivalent to buprenorphine) affinity for NOPr.15 We now report on a series of orvinol analogues in which the C20-methyl group has been moved to the C7-β position, resulting in more robust KOPr antagonism, enhanced NOPr activity, and low-efficacy partial agonism, or antagonism, at MOPr. A number of the ligands can be considered as single compound alternatives to the buprenorphine/naltrexone combination, while others offer potential as analgesics with a reduced side-effect profile.
Synthesis
In theory, a C7β-methyl group could be introduced into orvinol type compounds by methylation of the keto-intermediate (2, Chart 1). In practice, this leads to a number of rearrangement products resulting from the instability of the C7-anion.16,17 The alternative approach is to introduce the methyl group during the Diels–Alder reaction. Methacrylonitrile is known to undergo the Diels–Alder reaction with thebaine and N-cyclopropylnorthebaine, but it gives the C7α-methyl adduct (3, Chart 1), the opposite to that desired and a result of the methyl group being bulkier than the nitrile.18 Methacrolein should give a more favorable distribution of product due to the increased bulk of the aldehyde, but it has been reported that methacrolein does not undergo Diels–Alder reaction with thebaine.19 We have previously shown that lithium tetrafluoroborate catalyzes Diels–Alder reactions between N-cyclopropylcarbonylnorthebaine and cycloalkenones20 and have now applied this method to the reaction with methacrolein (Scheme 1). The reaction was successful, giving a 1.4:1 mixture of the C7α-methyl (4a) and C7β-methyl (4b) adducts that could be separated by silica gel chromatography. Addition of phenyl Grignard, in the presence of tetrabutylammonium bromide, to 4b gave the secondary alcohol 5a, with opposite stereochemistry at C20 to that desired. This bias toward the S-isomer (5a) parallels our findings on Grignard addition to the aldehyde of the related C7β-H series14 and is opposite to that observed on addition of Grignard reagent to the methyl ketone in the C7β-H series.14,15 Treatment of 5a with LiAlH4 to reduce the amide before 3-O-demethylation gave 10a. To obtain the desired diastereoisomer, 5a was oxidized to the phenyl ketone (6a) before LiAlH4 reduction of both the cyclopropylcarbonyl group and the ketone, the latter occurring stereoselectively, to give 7a. 3-O-Demethylation yielded 8a. The analogues (8b–8e; 10b–10e) were prepared in an identical manner. The 4-fluoro analogues (7e, 9e) were not fully stable to propanethiolate-mediated 3-O-demethylation, which, in the case of 7e, resulted in the formation of isolable amounts of 8f where the fluoro substituent was substituted by propanethiol.
Scheme 1.

The etheno-bridged aldehyde 4b was also hydrogenated to 11, and the same sequence of steps was carried out to provide the ethano-bridged analogues 15 and 17 (Scheme 2).
Scheme 2.

Results
In order to streamline the development process and avoid unnecessary full evaluation of ligands of limited interest to this project, the compounds were screened in a [35S]GTPγS assay for MOPr, KOPr, and NOPr efficacy at a very high concentration (10 μM) to determine peak efficacy at each receptor (Table 1). Potencies as agonists (EC50) or antagonists (Ke) were then determined for the most interesting compounds (chosen based on efficacies at the receptors and their fit with the target profiles) and reported in Table 1. A range of compounds were also evaluated for affinity at MOP, DOP, and KOP receptors by measuring displacement of [3H]diprenorphine binding from membranes of C6-rat glioma cells expressing recombinant rat MOPr and DOPr and CHO cells expressing recombinant human KOPr (Table 2). NOPr binding affinity was measured by displacement of [3H]nociceptin from membranes of HEK cells expressing recombinant NOPr. Details of these assays have been described previously.6,21−24
Table 1. Maximal Stimulation of [35S]GTPγS Binding of the New C7β-Methyl Orvinol Analogues to Opioid and NOP Receptors.
| [35S]GTPγS, % stimulation,a and,
in brackets, agonist EC50/nM or antagonist Ke/nMb |
||||
|---|---|---|---|---|
| MOPr | KOPr | NOPr | DOPr | |
| 1a | 20 ± 6% (EC50: 0.7 ± 0.3nM) | 0 ± 6% (Ke = 0.14 ± 0.06 nM) | 39 ± 12% (EC50: 1480 ± 980 nM) | 7 ± 3% |
| 1b | 6.0 ± 1 | 19 ± 4 | 14 ± 4 | |
| 8a | 4 ± 6% | 3 ± 7% | 56 ± 2.5% (EC50: 416 ± 74 nM) | 7 ± 3% |
| 8b | 12 ± 5% | –4 ± 1% | 17 ± 1% | |
| 8c | 22 ± 5% (EC50: 1.4 ± 0.4 nM) | 6 ± 2% | 54 ± 11% (EC50: 14 ± 5 nM) | 15 ± 3% |
| 8d | 35 ± 11% (EC50: 0.24 ± 0.04 nM) | –6 ± 1% | 14 ± 2% | |
| 8e | 26 ± 8% (EC50: 0.22 ± 0.06 nM) | 9 ± 1% | 51 ± 20% (EC50: 7.3 ± 3 nM) | |
| 8f | 49 ± 2% | 4 ± 10% | 20 ± 4% | |
| 10a | 33 ± 6% | 62 ± 4% | 5 ± 6% | |
| 10b | 48 ± 1% | 72 ± 4% | 12 ± 2% | |
| 10c | 40 ± 13% | 92 ± 4% | 11 ± 3% | |
| 10d | 49 ± 3% | 8 ± 5% | 4 ± 5% | |
| 10e | 55 ± 7% | 45 ± 6% | 7 ± 3% | |
| 15a | 2 ± 4% (Ke = 0.28 ± 0.04 nM) | –2 ± 1% (Ke = 0.09 ± 0.04 nM) | 56 ± 1% (EC50: 147 ± 33 nM) | 0 ± 4% |
| 15b | 15 ± 7% | –4 ± 4% (Ke = 0.13 ± 0.05 nM) | 15 ± 4% | |
| 15c | 7 ± 1% | –1 ± 1% (Ke = 0.12 ± 0.08 nM) | 61 ± 15% (EC50: 331 ± 223 nM) | |
| 15d | 14 ± 4% | –5 ± 4% (Ke = 0.10 ± 0.05nM) | 4 ± 3% | |
| 15e | 17 ± 2% | –5 ± 3% (Ke = 0.10 ± 0.05 nM) | 13 ± 5% | 6 ± 3% |
| 15f | 15 ± 1% | 2 ± 1% | 3 ± 1% | |
| 15g | 7 ± 1% | 7 ± 4% | 42 ± 3% (EC50: 230 ± 37 nM) | |
| 15h | 2 ± 1% | 15 ± 4% | 31 ± 2% (EC50: 160 ± 70 nM) | |
| 15i | 10 + 1 | 28 + 6 | 2 + 1 | |
| 15j | 31 + 4 | 26 + 2 | 15 + 1 | |
| 15k | 61 ± 2% | 10 ± 2% | 16 ± 3% | |
| 15l | 10 ± 4% | 20 ± 2% | 6 ± 3% | |
| 15m | 1 ± 1% | 2 ± 4% | 29 ± 3% (EC50: 230 ± 70 nM) | |
| 17 | 33 ± 7% | 97 ± 5% | 10 ± 4% | |
Percent maximal stimulation (% stim) at a single high concentration (10 μM) with respect to the standard agonists DAMGO (MOPr) and U69,593 (KOPr) and nociceptin (NOPr); values are an average ± SEM from three separate experiments.
In brackets for selected compounds, agonist EC50/nM or antagonist Ke (the antagonist dissociation constant determined against the standard agonists listed above) are given.
Table 2. Binding Affinities (Ki/nM) of of the New C7β-Methyl Orvinol Analogues to Opioid and NOP Receptors.
| binding/nMa |
||||
|---|---|---|---|---|
| MOPr | KOPr | NOPr | DOPr | |
| 1a | 0.13 ± 0.02 | 0.089 ± 0.02 | 212 ± 7 | 0.48 ± 0.26 |
| 1b | 0.17 ± 0.05 | 0.044 ± 0.015 | 43.2 ± 13.4 | NT |
| 8a | 0.08 ± 0.02 | 0.08 ± 0.03 | 97 ± 12 | 0.48 ± 0.05 |
| 8b | NT | NT | 1270 ± 170 | NT |
| 8c | 0.17 ± 0.11 | 0.04 ± 0.01 | 79 ± 8 | 0.40 ± 0.16 |
| 8e | 0.16 ± 0.12 | 0.05 ± 0.01 | 34 ± 6 | NT |
| 15a | 0.10 ± 0.02 | 0.04 ± 0.01 | 80 ± 10 | 0.25 ± 0.18 |
| 15b | 0.20 ± 0.07 | 0.12 ± 0.03 | 820 ± 60 | NT |
| 15c | 0.098 ± 0.022 | 0.11 ± 0.04 | 240 ± 50 | NT |
| 15d | 0.14 ± 0.06 | 0.16 ± 0.05 | 56 ± 5 | NT |
| 15e | 0.071 ± 0.014 | 0.10 ± 0.03 | 56 ± 3 | 0.47 ± 0.38 |
| 15f | NT | NT | 26 ± 6 | NT |
| 15g | 0.052 ± 0.007 | 0.094 ± 0.04 | 47.8 ± 17 | NT |
| 15h | 0.09 ± 0.03 | 0.13 ± 0.05 | 62.2 ± 31 | NT |
| 15k | NT | NT | 44 ± 11 | NT |
| 15l | NT | NT | 571 ± 77 | NT |
| 15m | 0.11 ± 0.011 | 0.14 ± 0.10 | 1455 ± 469 | NT |
Ki (nM) versus [3H]diprenorphine (for MOPr, KOPr, and DOPr) and [3H]nociceptin (for NOPr); values are an average ± SEM from three separate experiments. NT: not tested
All ligands from series 8 are KOPr antagonists with maximal percent stimulation <10%. Efficacy at MOPr ranges from zero (8a) to moderate efficacy partial agonism (8d, 8f: 35 and 49% of DAMGO, respectively) with clear SAR relating to the location of the phenyl ring substituent (4′ > 3′ > 2′). At NOPr, the compounds were partial agonists somewhat similar in efficacy to buprenorphine, with the highest efficacy demonstrated by the unsubstituted (8a; 56%), 3′-methyl substituted (8c; 43%), and 4′fluoro (8e, 51%) analogues. Potency at NOPr was higher than seen with buprenorphine. Series 10, the diastereomers of 8, were consistently of higher efficacy at the KOPr with only the 4′-Me (10d) substituted analogue profiling as a KOPr antagonist. The others in the series were moderate to high efficacy KOPr agonists (45–92% of U69,593). At MOPr, they were all of moderate efficacy (33–55%) and very low efficacy at NOPr.
The phenyl and substituted phenyl, ethano-bridge analogues (15a–h) had very similar SAR to series 8 in that they also had very low, or no, efficacy at KOPr. A number (15a–e) were evaluated as antagonists at this receptor and were found to be as potent as buprenorphine (all having Ke < 1 nM). As with series 8, they were also antagonists (e.g., 15a) or low-efficacy partial agonists at MOPr (≤17% of DAMGO). While SAR at this receptor was not absolutely clear cut, 4′-substituents again gave rise to higher efficacy than 3′-substituents (e.g., 15e vs 15h, 15d vs 15c). This series was extended to include heterocyclic analogues 15i–15m. Those having a thiophene ring (15i–15l) had partial agonist activity at KOPr (10–28%) and at MOPr (10–61%), whereas 15m, having a 3-furanyl group, was devoid of efficacy at KOPr and MOPr. Only 15m had appreciable efficacy at NOPr within the series of heterocyclic analogues. 17, the C20-diastereoisomer of 15a, was a full agonist at KOPr (97% stim) and a partial agonist at MOPr, with little efficacy at NOPr.
Binding affinities were determined for a range of compounds, including the more interesting analogues from series 8 and 15. As would be expected, all had high affinity for MOPr and KOPr (and DOPr when tested) with subnanomolar Ki values. Of particular note was the range of affinities at NOPr, where a number of the compounds had substantially higher affinity than that of buprenorphine (e.g 8e, 15d, 15e, 15f, 15k), whereas others had roughly equivalent affinity to that of buprenorphine (e.g., 8a, 8c, 15a, 15b, 15c).
In order to help explain the unusually low efficacy at KOPr of this series, the docking of 15a to the crystal structure of the KOPr was examined. The crystal structure of the KOPr was determined in the presence of the selective KOPr antagonist JDTic and presumably represents the inactive conformation of the receptor and, importantly, the conformation favored when binding 15a.2515a sits in the hydrophobic binding pocket with the expected interaction between the protonated nitrogen and the side chain of Asp138, while the C7β-methyl projects toward Tyr139 in TM3 (Figure 1).
Figure 1.
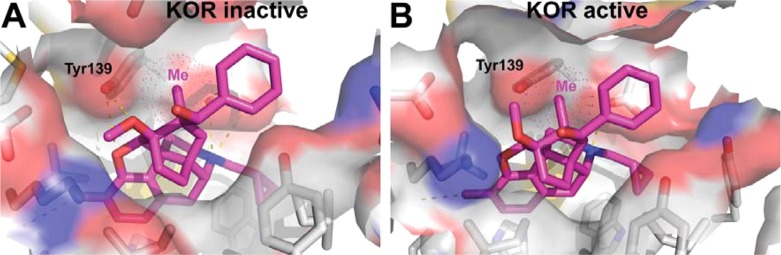
Proposed binding modes for 15a (purple) in the models of inactive (A) and active (B) conformations of human KOPr. The C7β-methyl-group of 15a negatively interacts with Tyr139 in the active receptor conformation but not in the inactive one.
Discussion
Members of the orvinol series of compounds are mostly known for their nonselective binding to MOPr, KOPr, and DOPr and potent agonist activity at one or more of these receptors.26 Even those with a cyclopropylmethyl (CPM) group attached to the basic nitrogen, which has the effect of reducing efficacy particularly at MOPr,27 tend to be efficacious and potent agonists, primarily at KOPr.14 Buprenorphine, being a KOPr antagonist, is a very distinct member of the series, with the lack of efficacy at KOPr being attributed to the significant steric bulk adjacent to C20.14 Initially, it was believed that buprenorphine was also unusual in having measurable affinity for NOPr,28 an activity that has been postulated to explain some of buprenorphine’s in vivo pharmacology.29,30 However, in recent years, it has become apparent that some other members of the orvinol family also interact with NOPr, particularly those with a bulky group attached to C20 (the t-butyl group of buprenorphine fulfils this role).31−33 While it has been possible to introduce good affinity for NOPr, it has proven to be difficult to couple this with moderate efficacy for this receptor without also introducing efficacy for classical opioid receptors, in particular KOPr. The orvinol TH-030418 (18; Chart 1) is reported as having affinity for NOPr virtually equivalent to its affinity for MOPr, KOPr, and DOPr; however, it also displays very potent and efficacious antinociceptive activity in vivo. This activity is blocked by the opioid antagonist naloxone, indicating high efficacy at MOPr and/or KOPr.33 Even more closely related to the current series are analogues of 1a having alternative bulky groups at C20.31 In that series, moderate affinity and efficacy for NOPr was always associated with partial agonism at MOPr and/or KOPr. The robust KOPr antagonist activity of series 8 and 15, especially when coupled with improved affinity for NOPr, is therefore unprecedented within the orvinols and close analogues. That they are 2° alcohols, which typically have very much higher efficacy than their 3° analogues,14 confirms the very substantial effect of moving the methyl group from C20 to the C7β-position. A comparison with the recently reported 1b and aryl substituted analogues confirms the importance of this methyl group. 1b and analogues differ from 15 only in the location of this single methyl, yet in the 1b series, only the parent compound (R = Ph) and the 3′-chloro analogue (1c: R = 3-ClPh) had low efficacy at KOPr (19 and 26%, respectively), with other substituents (2′-, 3′-, and 4′-methyl, 3′- and 4′-F) all resulting in efficacy in the range 77–102% at this receptor.15
Docking of 15a to the published crystal structure of the KOPr25 allows a hypothesis to be formulated to explain the very low efficacy of this series to the KOPr. In the model of the KOPr active and inactive states,23 the methyl group of 15a projects toward Tyr139 in TM3. In the inactive conformation, Tyr139 and the whole of TM3 move away from this position, whereas in the active conformation, they are much closer, possibly too close to allow the active conformation to be taken up; i.e., through an interaction between the C7β-methyl group and Tyr139, 15a and analogues may disfavor the active state of the receptor.
Distinct SAR for affinity and efficacy at NOPr is found in series 8 and 15. In both cases, the unsubstituted parent or a 3′-substituent gives highest efficacy at NOPr, whereas highest affinity is associated with a 4′-substituent, although this latter effect is not large.
Conclusions
Moving the methyl group from C20 to the C7β position has resulted in one of the most striking pieces of SAR in the orvinol series. In particular, the robust reduction in efficacy at KOPr coupled with a retention, or increase, in NOPr affinity and efficacy has resulted in a series of compounds worthy of consideration as (i) relapse prevention agents and (ii) low abuse liability analgesics. 15a is currently undergoing in vivo evaluation as part of its preclinical development.
Experimental Section
General Procedures
Reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar and used as received. Buprenorphine (1a) was supplied by the National Institute on Drug Abuse, Bethesda, Maryland. 1H and 13C NMR spectra were obtained with a Bruker 400 MHz instrument (1H at 400 MHz, 13C at 100 MHz); δ is given in ppm, J, in Hz, with TMS as an internal standard. ESIMS: microTOF (BRUKER), EIMS: Fisons Autosampler. Microanalysis: PerkinElmer 240C analyzer. Column chromatography was performed using RediSep prepacked columns with a Teledyne Isco CombiFlash instrument. Most ligands were tested as their hydrochloride salts, prepared by adding 5 equiv of HCl (1 N solution in diethyl ether) to a solution of compound in anhydrous methanol. Alternatively, the oxalate salt was formed by adding 1 equiv of oxalic acid in EtOH to the ligand in EtOH. All reactions were carried out under an inert atmosphere of nitrogen unless otherwise indicated. All compounds were >95% pure, as determined by microanalysis. A representative synthesis is reported here.
Arylmagnesium Halide Addition (General Procedure A)
To a solution of aldehyde 4b or 11 in dry THF (10 mL/mmol of aldehyde) were added 3 equiv of Bu4NBr followed by 2 equiv of arylmagnesium halide as solution in THF. The solution was then heated at reflux for 48 h, cooled to RT, and quenched with 0.05 mL of water. The mixture was allowed to stir for 5 min and then filtered over Celite. The solids were washed with hot THF, and the solution was removed of its solvent by rotary evaporation. The remaining residue was partitioned between EtOAc (20 mL) and water (10 mL). The aqueous layer was extracted twice with 5 mL of EtOAc. The pooled organic solvent was washed twice with water (5 mL) and once with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The residue was dissolved in a minimum amount of Et2O to induce crystallization. The crystals were collected by filtration and dried under vacuum.
Swern Oxidation/LiAlH4 Reduction of Secondary Alcohols (General Procedure B)
A solution of oxalyl chloride (1.25 equiv) in CH2Cl2 (3 mL/mmol) was cooled to −78 °C in a one-necked flask. Into this flask was added dropwise a solution of dry DMSO (2.6 equiv) in CH2Cl2 (3 mL/mmol). The solution stirred for 5 min, and then a solution of thevinol 5 or 12 in CH2Cl2 (2 mL/mmol) was added. The mixture stirred for 20 min, and then Et3N (5 equiv) was added. The reaction was removed from the cold bath and stirred for 1 h, and water was added. The mixture was shaken, and the organic layer was separated and washed with a saturated solution of NH4Cl and then with a concentrated solution of NaHCO3. The solution was washed once more with brine, dried over magnesium sulfate, and filtered, and the solvents were removed under reduced pressure to yield crude 6 or 13 as a clear solid.
This residue was dissolved in dry THF (10 mL/mmol) and added to a stirring suspension of LiAlH4 (4 equiv) in dry THF (5 mL/mmol) at 0 °C. The suspension was allowed to warm to RT and was stirred for 24 h. The reaction was cooled to 0 °C and quenched with water in THF. The mixture was filtered, rinsing the solids with hot THF. The solution was subjected to rotary evaporation to yield an oil that was subjected to silica gel column chromatography, eluting with 15% EtOAc in petroleum ether to yield two constituents, 7 or 14 (as the major product), as a high Rf component, and 9 or 16 (as the minor product), with lower Rf.
O-Demethylation Using NaSPr/HMPA (General Procedure C)
A solution of thevinol 7, 9, 14, or 16 in dry HMPA (6 mL/mmol) was added sodium propanethiolate (6 equiv). The reaction was stirred for 3 h at 115 °C and then cooled to RT and quenched with 7 mL/mmol of a concentrated solution of NH4Cl. The mixture was extracted three times with Et2O. The organic layer was then extracted five times with water and once with brine, dried over MgSO4, and filtered, and the solvents were removed under reduced pressure. The residue was then subjected to silica gel flash column chromatography, eluting with a gradient of EtOAc in petroleum ether. The fractions containing the compound of interest were then evaporated to dryness, dissolved in a 2 M solution of HCl in EtOH, and then induced to crystallize upon addition of EtOAc. The crystals were collected by filtration and dried under vacuum.
N-Cyclopropylcarbonyl-7α-formyl-7β-methyl-6,14-endoethenotetrahydronorthebaine (4b)
To a suspension of 13.61 g (37.29 mmol) N-CPCnorthebaine in 20 mL of methacrolein was added 3.49 g of LiBF4. The resulting solution was stirred for 16 h at RT. Into this solution was added 30 mL of CH2Cl2, and the mixture was extracted with water (10 mL × 3) and brine (5 mL). The solution was dried, filtered, and removed of solvent on a rotary evaporator to afford a dark red syrup. This material was subjected to silica gel flash column chromatography, eluting with 50% EtOAc in petroleum ether to afford 5.91 g of the faster running component 4a (N-cyclopropylcarbonyl-7β-formyl-7α-methyl-6,14-endo-ethenotetrahydronorthebaine) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 6.67 (d, 1H, J = 8.0 Hz), 6.57 (d, 1H, J = 8.0 Hz), 6.14–6.07 (2d, 1H), 5.57 (d, 1H, J = 12.0 Hz), 5.33 (d, 1H, J = 8.0 Hz), 4.76 (d, 1H, 0.4H), 4.57–4.53 (m, 1.6H), 4.15–4.10 (m, 1H), 3.85 (s, 3H), 3.72 (s, 3H), 3.42 (dt, 1H, Ja = 12.0 Hz, Jb = 4.0 Hz), 3.11–3.05 (dd, 1H, Ja = 8.0 Hz, Jb = 4.0 Hz), 2.88–2.83 (m, 1H), 2.43–2.35 (dt, 1H, Ja = 12.0 Hz, Jb = 8.0 Hz), 1.85–1.76 (m, 1H), 1.69–1.65 (m, 1H), 1.09–1.03 (m, 5H), 0.91–0.76 (m, 2H). ESIMS: m/z 436 (M + H+, 100). In addition, 4.11 g of a slower running component, N-cyclopropylcarbonyl-7α-formyl-7β-methyl-6,14-endoethenotetrahydronorthebaine (4b), was afforded as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 0.5H), 9.45 (s, 0.5H), 6.69 (d, 1H, J = 8.0 Hz), 6.59 (d, 1H, J = 8.0 Hz), 6.14 (t, 1H), 5.57 (dd, 1H, Ja = 12.0 Hz, Jb = 8.0 Hz), 5.35 (d, 0.5H, J = 4.0 Hz), 4.93 (s, 1H), 4.80 (d, 0.5H, J = 8.0 Hz), 4.64 (dd, 1H, Ja = 8.0 Hz, Jb = 4.0 Hz), 4.15 (dd, 1H, Ja = 8.0 Hz, Jb = 4.0 Hz), 3.85 (s, 3H), 3.72 (2s, 3H), 3.49 (dt, 1H), 3.31–3.21 (m, 2H), 2.37–2.26 (dt, 0.5H), 2.27–2.15 (dt, 0.5H), 2.06–1.72 (m, 3H), 1.35 (s, 3H), 1.08 (m, 2H), 0.82 (m, 2H). At RT, the 1H NMR spectra of this compound in DMSO-d6 has two signals at δ 9.408 (s, 0.5H) and δ 9.375 (s, 0.5H) that coalesce when running the 1H NMR experiment at 360 K. ESIMS: m/z 436 (M + H+, 100).
N-Cyclopropylcarbonyl-7α-formyl-7β-methyl-6,14-endoethanotetrahydronorthebaine (11)
Aldehyde 4b (500 mg) was dissolved in 15 mL of EtOH. Into this solution was added 30 mg of 10% Pd on carbon. The mixture was shaken in a Parr hydrogenator under 100 psi of H2 for 12 h. The mixture was filtered, and the solvents were removed under reduced pressure to yield 510 mg of 11 as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 0.6H), 9.69 (s, 0.4H), 6.76 (d, 1H, J = 8.1 Hz), 6.61 (d, 1H, J = 8.1 Hz), 4.9 (d, 0.4H, J = 6.9 Hz), 4.82 (s, 0.6H), 4.81 (s, 0.4H), 4.52 (dd, 0.6H, Ja = 14 Hz, Jb = 5.7 Hz), 4.34 (d, 0.6H, J = 6.8 Hz), 4.04 (dd, 0.4H, Ja = 15, Jb = 6 Hz), 3.89 (s, 3H), 3.48 (s, 1.8H), 3.46 (s, 1.2H), 3.34 (td, 0.4H, Ja = 13.4, Jb = 3.2 Hz), 3.05 (dd, 0.6H, Ja = 18.5 Hz, Jb = 6.9 Hz), 2.97 (dd, 0.4H, Ja = 18.7 Hz, Jb = 7.2 Hz), 2.87 (d, 0.6H, J = 18.7 Hz), 2.81 (td, 0.6H, Ja = 13.7, Jb = 4.1 Hz), 2.71 (d, 0.4H, J = 18.8 Hz), 2.42–2.37 (m, 1H), 2.27 (td, 0.4H, Ja = 13.1 Hz, Jb = 5.7 Hz), 2.16 (td, 0.6H, Ja = 13.2 Hz, Jb = 5.8 Hz), 1.80–1.60 (m, 3.6H), 1.5 (dd, 0.4H, Ja = 13.7 Hz, Jb = 3.7 Hz), 1.33–1.18 (m, 5H), 1.15–1.06 (m, 1H), 1.03–0.94 (m, 2H), 0.84–0.71 (m, 2H). ESIMS: m/z 438 (M + H+, 100).
(1′S,5α,6R,7R,14α)-1′-Phenyl-1′-(4,5-epoxy-7,8-dihydro-3,6-dimethoxy-7β-methyl 17-cyclopropylcarbonyl-6,14-ethano-morphinan-7-yl)-methan-1′-ol (12a)
General procedure A was followed using 515 mg of 11 to yield 336 mg of 12a as white crystals. 1H NMR (400 MHz, CDCl3) δ 7.36–7.23 (m, 5H), 6.78–6.75 (2d, 1H), 6.61–6.58 (2d, 1H), 5.06 (d, 0.55H, J = 2.6 Hz), 4.99 (d, 0.45H, J = 2.6 Hz), 4.91–4.87 (m, 1H), 4.51–4.46 (dd, 0.55H, Ja = 5.4 Hz, Jb = 13.7 Hz), 4.35–4.34 (d, 0.55H, J = 6.8 Hz), 4.03–3.98 (d, 0.45H, Ja = 4.9 Hz, Jb = 13.5 Hz), 3.92–3.914 (2s, 3H), 3.53 (s, 1.65H), 3.48 (s, 1.35H), 3.38–3.30 (dt, 0.55H, Ja = 3.8 Hz, Jb = 8.6 Hz), 3.11–3.04 (dd, 0.55H, Ja = 6.9 Hz, Jb = 18.3 Hz), 3.02–2.96 (dd, 0.45H, Ja = 6.9 Hz, Jb = 18.5 Hz), 2.92–2.88 (d, 0.55H, J = 18.3 Hz), 2.49–2.44 (m, 1H), 2.36–2.29 (dt, 0.45 Hz, Ja = 5.6 Hz, Jb = 12.9 Hz), 2.25–2.17 (dt, 0.55 Hz, Ja = 6.0 Hz, Jb = 13.2 Hz), 2.03–1.90 (m, 2H), 1.86–1.80 (m, 0.45 Hz), 1.76–1.51 (m, 6H), 1.10–0.69 (m, 7H). ESIMS: m/z 516 (M + H+, 100).
(1′R,5α,6R,7R,14α)-1′-Phenyl-1′-(4,5-epoxy-7,8-dihydro-3,6-dimethoxy-7β-methyl 17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-methan-1′-ol (14a)
General procedure B was followed using 473 mg of 12a to yield a clear residue after extraction. Crystallization from CH2Cl2/Et2O gave 133 mg of 14a as white crystals. 1H NMR (400 MHz, CDCl3) δ 7.45 (m, 2H), 7.34–7.30 (m, 2H), 7.27–7.24 (m, 1H), 6.73 (d, 1H, J = 8.4 Hz), 6.57 (d, 1H, J = 8.0 Hz), 5.76 (s, 1H), 5.03 (s, 1H), 4.96 (s, 1H), 3.91 (s, 3H), 3.64 (s, 3H), 3.01–2.94 (m, 2H), 2.57 (d, 1H, J = 6.8 Hz), 2.29 (dd, 1H, Ja = 6.8 Hz, Jb = 18.3 Hz), 2.24–2.20 (m, 3H), 1.95 (dd, 1H, Ja = 4 Hz, Jb = 14.3 Hz), 1.90–1.77 (m, 2H), 1.60–1.52 (m, 2H), 1.40–1.28 (m, 4H), 0.95 (m, 1H), 0.68 (m, 1H), 0.51–0.39 (m, 2H), 0.08–0.01 (m, 2H). ESIMS: m/z 502 (M + H+, 100).
(1′R,5α,6R,7R,14α)-1′-Phenyl-1′-(4,5-epoxy-7,8-dihydro-3-hydroxy-6-methoxy-7β-methyl 17-cyclopropylmethyl-6,14-ethanomorphinan-7-yl)-methan-1′-ol (15a)
General procedure C was followed using 133 mg of 14 to yield 75 mg of 15a as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.49–7.42 (m, 2H), 7.35–7.28 (m, 2H), 7.28–7.20 (m, 1H), 6.61 (d, 1H, J = 8.0 Hz), 6.47 (d, 1H, J = 8.0 Hz), 5.98 (s, 1H), 5.05 (s, 1H), 4.95 (d, 1H, J = 1.6 Hz), 3.59 (s, 3H), 2.98 (d, 1H, J = 6.4 Hz), 2.93 (d, 1H, J = 18.3 Hz), 2.63–2.50 (m, 1H), 2.32–2.13 (m, 5H), 1.95 (dd, 1H, Ja = 14.2 Hz, Jb = 3.9 Hz), 1.89–1.71 (m, 2H), 1.59–1.44 (m, 2H), 1.35–1.21 (m, 4H), 0.97–0.81 (m, 1H,), 0.75–0.60 (m, 1H), 0.52–0.36 (m, 2H), 0.10–0.03 (m, 2H). ESIMS: m/z 488 (M + H+, 100).
Molecular Modeling
We used the crystal structure of the human KOR (PDB ID: 4djh) together with our previous active state models.23 Compound 15a was built, and the low-energy conformation was then subjected to automated rigid docking implemented in QUANTA (Accelrys Inc.).
Acknowledgments
This work was funded by the National Institutes of Health, National Institute on Drug Abuse grant DA07315 (to S.M.H.). T.M.H. was supported by T32 DA07268. Irina Pogozheva (University of Michigan) is thanked for docking studies between 15a and the KOPr.
Glossary
Abbreviations Used
- MOP receptor
mu opioid receptor
- DOP receptor
delta opioid receptor
- KOP receptor
kappa opioid receptor
- NOP receptor
nociceptin opioid peptide receptor
- EKC
ethylketocyclazocine
- N/OFQ
nociceptin/orphanin FQ
Supporting Information Available
Full characterization of final compounds; microanalysis and HPLC; pharmacological methods. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00130.
Author Present Address
§ (J.P.C.) Neurocrine Biosciences, Inc., San Diego, California 92130, United States.
Author Present Address
∥ (V.K.) Centre for Chemical and Pharmaceutical Sciences, Central University of Punjab, Bathinda, 151001, India.
The authors declare the following competing financial interest(s): Subsequent to preparation of the manuscript the compounds disclosed have been licensed to a biopharmaceutical company.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Husbands S. M.Buprenorphine and related orvinols. Research and Development of Opioid-Related Ligands; ACS Symposium Series: New York, 2013; Vol. 1131, pp 127–144. [Google Scholar]
- McLellan A. T.; Lewis D. C.; O’Brien C. P.; Kleber H. D. Drug dependence, a chronic medical illness: implications for treatment, insurance, and outcomes evaluation. J. Am. Med. Assoc. 2000, 284, 1689–1695. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Arias M.; Aguilar M. A.; Manzanedo C.; Minarro J. Preclinical evidence of new opioid modulators for the treatment of addiction. Expert Opin. Invest. Drugs 2010, 19, 977–994. [DOI] [PubMed] [Google Scholar]
- Cordery S. F.; Taverner A.; Ridzwan I. E.; Guy R. H.; Delgado-Charro M. B.; Husbands S. M.; Bailey C. P. A non-rewarding, non-aversive buprenorphine/naltrexone combination attenuates drug-primed reinstatement to cocaine and morphine in rats in a conditioned place preference paradigm. Addict. Biol. 2014, 19, 575–586. [DOI] [PubMed] [Google Scholar]
- Taverner A.; Cordery S.; Husbands S. M.; Guy R. H.; Delgado-Charro B. M.; Bailey C. P. Transdermal delivery of a buprenorphine/naltrexone combination for the treatment of polydrug abuse. Pharmacol. Rep. 2011, 63, 257–257. [Google Scholar]
- Lee K. O.; Akil H.; Woods J. H.; Traynor J. R. Differential binding properties of oripavines at cloned mu- and delta-opioid receptors. Eur. J. Pharmacol. 1999, 378, 323–330. [DOI] [PubMed] [Google Scholar]
- Bloms-Funke P.; Gillen C.; Schuettler A. J.; Wnendt S. Agonistic effects of the opioid buprenorphine on the nociceptin/OFQ receptor. Peptides 2000, 21, 1141–1146. [DOI] [PubMed] [Google Scholar]
- Rothman R. B.; Gorelick D. A.; Heishman S. J.; Eichmiller P. R.; Hill B. H.; Norbeck J.; Liberto J. G. An open-label study of a functional opioid kappa antagonist in the treatment of opioid dependence. J. Subst. Abuse Treat. 2000, 18, 277–281. [DOI] [PubMed] [Google Scholar]
- Gerra G.; Fantoma A.; Zaimovic A. Naltrexone and buprenorphine combination in the treatment of opioid dependence. J. Psychopharmacol. 2006, 20, 806–814. [DOI] [PubMed] [Google Scholar]
- Hans G. H.Buprenorphine in the treatment of neuropathic pain. Research and Development of Opioid-Related Ligands; ACS Symposium Series: New York, 2013; Vol. 1131, pp 103–123. [Google Scholar]
- McCann D. J. Potential of buprenorphine/naltrexone in treating polydrug addiction and co-occurring psychiatric disorders. Clin. Pharmacol. Ther. 2008, 83, 627–630. [DOI] [PubMed] [Google Scholar]
- Cremeans C. M.; Gruley E.; Kyle D. J.; Ko M.-C. Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J. Pharmacol. Exp. Ther. 2012, 343, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhtankar D. D.; Zaveri N. T.; Husbands S. M.; Ko M.-C. Effects of spinally administered bifunctional NOP/MOP ligands in mouse models of neuropathic and inflammatory pain. J. Pharmacol. Exp. Ther. 2013, 346, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greedy B. M.; Bradbury F.; Thomas M. P.; Grivas K.; Cami-Kobeci G.; Archambeau A.; Bosse K.; Clark M. J.; Aceto M.; Lewis J. W.; Traynor J. R.; Husbands S. M. Orvinols with mixed kappa/mu opioid receptor agonist activity. J. Med. Chem. 2013, 56, 3207–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Ridzwan I. E.; Grivas K.; Lewis J. W.; Clark M. J.; Meurice C.; Jimenez-Gomez C.; Pogozheva I.; Mosberg H.; Traynor J. R.; Husbands S. M. Selectively promiscuous opioid ligands: discovery of high affinity/low efficacy opioid ligands with substantial nociceptin opioid peptide receptor affinity. J. Med. Chem. 2014, 57, 4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley K. W.; Hardy D. G.; Meek B.; Taylor J. B.; Brown J. J.; Morton G. O. Novel analgesics and molecular rearrangements in the morphine-thebaine group. Part X. Further acid-catalysed rearrangements of alcohols in the 6,14-endo-ethenotetrahydrothebaine series. J. Chem. Soc. C 1969, 2229–2232. [DOI] [PubMed] [Google Scholar]
- Coop A.; Grivas K.; Husbands S.; Lewis J. W. Methylation of the enolates of thevinone and some analogs. Tetrahedron 1995, 51, 9681–9698. [Google Scholar]
- Husbands S. M.; Lewis J. W. Morphinan cyclic imines and pyrrolidines containing a constrained phenyl group: high affinity opioid agonists. Bioorg. Med. Chem. Lett. 1995, 5, 2969–2974. [Google Scholar]
- Lewis J. W.; Readhead M. J.; Selby I. A.; Smith A. C. B.; Young C. A. Novel analgesics and molecular rearrangements in the morphine-thebaine group. Part XIX. Further Diels–Alder adducts of thebaine. J. Chem. Soc. C 1971, 1158–1161. [DOI] [PubMed] [Google Scholar]
- Barton J. W.; Coop A.; Lewis J. W. Diels–Alder Reactions of thebaines with cycloalkenones—lithium tetrafluoroborate as a novel Diels–Alder catalyst. Tetrahedron Lett. 1993, 34, 6777–6778. [Google Scholar]
- Spagnolo B.; Calo G.; Polgar W. E.; Jiang F.; Olsen C. M.; Berzetei-Gurske I.; Khroyan T. V.; Husbands S. M.; Lewis J. W.; Toll L.; Zaveri N. T. Activities of mixed NOP and mu-opioid receptor ligands. Br. J. Pharmacol. 2008, 153, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husbands S. M.; Neilan C. L.; Broadbear J.; Grundt P.; Breeden S.; Aceto M. D.; Woods J. H.; Lewis J. W.; Traynor J. R. BU74, a complex oripavine derivative with potent kappa opioid receptor agonism and delayed opioid antagonism. Eur. J. Pharmacol. 2005, 509, 117–125. [DOI] [PubMed] [Google Scholar]
- Anand J. P.; Purington L. C.; Pogozheva I. D.; Traynor J. R.; Mosberg H. I. Modulation of opioid receptor ligand affinity and efficacy using active and inactive state receptor models. Chem. Biol. Drug Des. 2012, 80, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor J. R.; Nahorski S. R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-S-35 thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [PubMed] [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X.-P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. W.; Husbands S. M. The orvinols and related opioids—high affinity ligands with diverse efficacy profiles. Curr. Pharm. Des. 2004, 10, 717–732. [DOI] [PubMed] [Google Scholar]
- Husbands S. M.; Lewis J. W. Structural determinants of efficacy for kappa opioid receptors in the orvinol series: 7,7-spiro analogues of buprenorphine. J. Med. Chem. 2000, 43, 139–141. [DOI] [PubMed] [Google Scholar]
- Zaveri N.; Polgar W. E.; Olsen C. M.; Kelson A. B.; Grundt P.; Lewis J. W.; Toll L. Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur. J. Pharmacol. 2001, 428, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K.; Eitan S.; Bryant C. D.; Yang Y. C.; Saliminejad N.; Walwyn W.; Kieffer B. L.; Takeshima H.; Carroll F. I.; Maidment N. T.; Evans C. J. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J. Neurosci. 2003, 23, 10331–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K.; Cowan A. Buprenorphine: a unique drug with complex pharmacology. Curr. Neuropharmacol. 2004, 2, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cami-Kobeci G.; Polgar W. E.; Khroyan T. V.; Toll L.; Husbands S. M. Structural determinants of opioid and NOP receptor activity in derivatives of buprenorphine. J. Med. Chem. 2011, 54, 6531–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan T. V.; Polgar W. E.; Cami-Kobeci G.; Husbands S. M.; Zaveri N. T.; Toll L. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J. Pharmacol. Exp. Ther. 2011, 336, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Yan L.-D.; Li Y.-L.; Wen Q.; Dong H.-J.; Gong Z.-H. TH-030418: a potent long-acting opioid analgesic with low dependence liability. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 125–131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.