

Significance
AMPA receptors (AMPARs) conduct the majority of excitatory synaptic transmission in the brain. Through changes in AMPAR synaptic localization or conductance the strength of a synapse can be altered. It is hypothesized that such changes underlie complex behaviors such as learning and memory. AMPAR phosphorylation is one signaling event used to alter receptor targeting and conductance. We demonstrate the neuropeptide PACAP38 stimulates AMPAR GluA1 subunit phosphorylation at S845 and dephosphorylation at T840. Investigation of PACAP38-dependent changes in AMPAR phosphorylation will help us to better understand the factors involved in regulating AMPAR function.
Keywords: PACAP38, AMPA receptor phosphorylation, synaptic transmission
Abstract
Dynamic changes in synaptic strength are thought to be critical for higher brain function such as learning and memory. Alterations in synaptic strength can result from modulation of AMPA receptor (AMPAR) function and trafficking to synaptic sites. The phosphorylation state of AMPAR subunits is one mechanism by which cells regulate receptor function and trafficking. Receptor phosphorylation is in turn regulated by extracellular signals; these include neuronal activity, neuropeptides, and neuromodulators such as dopamine and norepinephrine (NE). Although numerous studies have reported that the neuropeptide pituitary adenylate cyclase activating polypeptide 38 (PACAP38) alters hippocampal CA1 synaptic strength and GluA1 synaptic localization, its effect on AMPAR phosphorylation state has not been explored. We determined that PACAP38 stimulation of hippocampal cultures increased phosphorylation of S845, and decreased phosphorylation of T840 on the GluA1 AMPAR subunit. Increases in GluA1 S845 phosphorylation primarily occurred via PAC1 and VPAC2 receptor activation, whereas a reduction in GluA1 T840 phosphorylation was largely driven by PAC1 receptor activation and to a lesser extent by VPAC1 and VPAC2 receptor activation. GluA1 S845 phosphorylation could be blocked by a PKA inhibitor, and GluA1 T840 dephosphorylation could be blocked by a protein phosphatase 1/2A (PP1/PP2A) inhibitor and was partly blocked by a NMDA receptor (NMDAR) antagonist. These results demonstrate that the neuropeptide PACAP38 inversely regulates the phosphorylation of two distinct sites on GluA1 and may play an important role modulating AMPAR function and synaptic plasticity in the brain.
AMPA-type glutamate receptors (AMPARs) are a tetrameric assembly composed of the GluA1, 2, 3, or 4 subunits. Within the adult hippocampus, receptors consist of primarily GluA1/2 and GluA2/3 complexes (1). Because AMPARs conduct the majority of excitatory transmission in the brain, modulation of AMPAR synaptic transmission is a powerful tool by which the cell can regulate synaptic strength and cell firing. Furthermore, it is hypothesized that complex behaviors such as learning, memory, and drug addiction involve alterations in synaptic strength (2, 3).
The cell can regulate synaptic strength through changes in AMPAR conductance, trafficking, and tethering at synaptic sites. Such changes can be achieved through alterations in AMPAR expression, binding partners, and posttranslational modifications (4). A number of GluA1 and GluA2 phosphorylation sites have been proposed to play a role in AMPAR trafficking and synaptic plasticity. GluA1 S845 and T840 are two phosphorylation sites particularly relevant to this study. GluA1 S845 is phosphorylated by PKA and cGMP-dependent protein kinase II (5, 6). Its phosphorylation levels are regulated by NMDA receptors (NMDARs) (7), β-adrenergic receptors (8, 9), and muscarinic cholinergic receptors (9), and during homeostatic scaling (10), long-term depression (LTD) (11), and emotionally stressful conditions (8). Likewise, GluA1 S845 phospho-mutants show GluA1 trafficking and LTD deficits (12–14). In contrast, the GluA1 T840 site is less well characterized. PKC, calcium/calmodulin-dependent protein kinase II, protein phosphatase 1/2A (PP1/PP2A), and NMDAR activity have been reported to regulate GluA1 T840 phosphorylation (15–17). GluA1 T840 phosphorylation has also been found to enhance channel conductance (18).
PACAP38 (pituitary adenylate cyclase activating polypeptide 38) is a neuropeptide that has been shown to regulate hippocampal CA1 synaptic strength (19–22). PACAP38 can bind to and activate three different G protein coupled receptors, the PAC1, VPAC1, and VPAC2 receptors, which can lead to elevated cyclic AMP and Ca2+ levels, and activation of phospholipase C and phospholipase D (23). In the hippocampus, PACAP38 stimulation has been shown to alter synaptic strength (19–22) and AMPAR excitatory postsynaptic currents (EPSCs) (24) and to reduce GluA1 synaptic localization (25). PACAP knockout mice are impaired in contextual fear conditioning and novel object recognition (26), and PAC1 receptor knockouts exhibit impaired contextual fear conditioning (27). Given the ability of PACAP38 to regulate basal synaptic transmission and AMPAR EPSCs (24), we hypothesized that PACAP38 stimulation could alter AMPAR phosphorylation levels.
We found that PACAP38 stimulation led to increased GluA1 S845 phosphorylation and decreased GluA1 T840 phosphorylation. We also demonstrated that unique signaling pathways were used to drive these phosphorylation changes. Although activation of the PAC1 and VPAC2 receptor elicited a robust increase in GluA1 S845 phosphorylation, only PAC1 receptor activity could elicit a robust decrease in GluA1 T840 phosphorylation. In addition, a PKA inhibitor blocked the increase in S845 phosphorylation, while a PP1/PP2A inhibitor blocked the decrease in T840 phosphorylation and a NMDAR antagonist partially blocked the decrease in T840 phosphorylation.
Results
To study the effect of PACAP38 on AMPAR phosphorylation, we stimulated mature [days in vitro (DIV) 14], dissociated hippocampal cultures with a low and high dose of PACAP38. Following stimulation, cells were lysed and AMPAR phosphorylation was examined by Western blot. PACAP38 stimulation resulted in elevated GluA1 S845 phosphorylation, reduced GluA1 T840 phosphorylation, and had no effect on GluA1 S831 phosphorylation (Fig. 1A). Previous reports validate the specificity of the GluA1 pS831, pS845, and pT840 antibodies (10, 16, 17). Because the GluA1 pT840 antibody detected several bands, we confirmed the specificity of our antibody using the GluA1 “penta” knock-in mouse, which harbors mutations at GluA1 S831A, T838A, S839A, T840A, and S845A (15). When WT whole brain lysate was probed with the GluA1 pT840 antibody, we observed a prominent band comigrating with GluA1 (Fig. 1B). This band diminished to negligible levels in “penta” samples. Similarly, in GluA1 immunoprecipitation experiments, this GluA1 pT840 band was present in WT but not “penta” samples. Although several nonspecific bands were observed in the input, these bands were absent following GluA1 immunoprecipitation. Thus, subsequent experiments involving the GluA1 pT840 antibody were performed exclusively upon GluA1 immunoprecipitated complexes.
Fig. 1.

Effect of PACAP38 on GluA1 phosphorylation. (A) Hippocampal cultures (DIV 14) were stimulated with different concentrations (nM) of PACAP38 for 10 min. Stimulation was followed by cell lysis and Western blot analysis. (B) GluA1 was immunoprecipitated from whole brain lysate prepared from WT and “penta” knock-in mice. Input and GluA1 IP samples were visualized by Western blot.
We next examined the dose and time sensitivity of PACAP38 effects on GluA1 phosphorylation (Fig. 2 A and B). A 0.05 nM dose of PACAP38 significantly decreased GluA1 T840 phosphorylation, and this was maximally decreased upon 1 nM dose PACAP38 application. Similarly, a significant increase in GluA1 S845 phosphorylation was observed with a 0.05 nM dose of PACAP38, reaching a maximum at 0.5 nM. To better understand the temporal regulation of these phosphorylation changes, cultures were stimulated with 1nM PACAP38 for different durations of time (Fig. 2 C and D). Two-minute stimulation with PACAP38 produced a significant reduction in GluA1 T840 phosphorylation and this was maximally reduced following 10-min stimulation. At the S845 site, a significant increase was observed at the 2-min time point, and a maximal increase was seen at the 30-min time point. Taking into account the dose response and time course data, we thereafter performed PACAP38 stimulation experiments using a 1 nM dose of PACAP38 for 10 min.
Fig. 2.
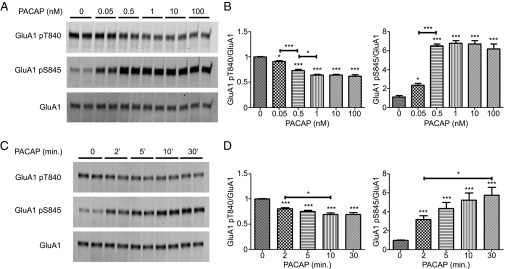
Characterization of PACAP38-dependent changes. (A) Hippocampal neurons (DIV 14) were stimulated with different concentrations (nM) of PACAP38 for 10 min. Stimulation was followed by GluA1 immunoprecipitation and Western blot. (B) Quantification of GluA1 T840 or S845 phosphorylation normalized to GluA1. (C) Hippocampal neurons (DIV 14) were stimulated for different durations of time with 1 nM PACAP38. Stimulation was followed by GluA1 immunoprecipitation and Western blot. (D) Quantification of GluA1 T840 or S845 phosphorylation normalized to GluA1. Error bars indicate ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA, Tukey posttest. n ≥ 6.
We next wanted to identify the PACAP38 receptor responsible for the AMPAR phosphorylation changes (Fig. 3 A and B). PACAP38 can bind to and activate three different GPCRs, the VPAC1, VPAC2, and PAC1 receptors. When cultures were stimulated with the VPAC1 receptor agonist, K,R,L-VIP-GRF, we observed a minor decrease in GluA1 T840 phosphorylation and a minor increase in GluA1 S845 phosphorylation (Fig. 3). Stimulation with the VPAC2 receptor agonist, Bay 55–9837, resulted in a moderate decrease in GluA1 T840 phosphorylation and a robust increase in GluA1 S845 phosphorylation (Fig. 3). Because Bay 55–9837 can weakly activate the VPAC1 receptor (28), we cannot rule out the possibility that some of the phosphorylation change is due to VPAC1 receptor activation. Application of the PAC1 receptor agonist, Maxadillan, most closely reproduced changes observed with PACAP38 stimulation, namely a strong decrease in GluA1 T840 phosphorylation and a strong increase in S845 phosphorylation (Fig. 3).
Fig. 3.
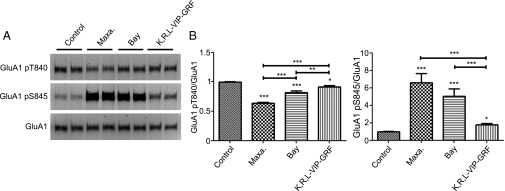
Regulation of GluA1 phosphorylation by the PACAP receptors. (A) Hippocampal neurons (DIV 14) were stimulated with the PAC1 receptor agonist (Maxadillan, 100 nM), the VPAC2 receptor agonist (Bay 55–987, 100 nM), or the VPAC1 receptor agonist (K,R,L-VIP-GRF, 1 µM) for 10 min. Stimulation was followed by GluA1 immunoprecipitation and Western blot. (B) Quantification of GluA1 T840 or S845 phosphorylation normalized to GluR1. Error bars indicate ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA, Tukey posttest. n ≥ 6.
PACAP38 could modulate phosphorylation at the GluA1 T840 or the S845 sites through the regulation of kinase or phosphatase activity. Because PACAP38 has been shown to increase PKA activity (23) and PKA can phosphorylate GluA1 at S845 (5), we investigated the role of PKA in PACAP38-dependent phosphorylation changes. The PKA inhibitor, H89, blocked the PACAP38-dependent increase in GluA1 S845 phosphorylation but had no effect on the PACAP38-dependent reduction in GluA1 T840 phosphorylation (Fig. 4 A and E). Previously, activation of PKC has been demonstrated to regulate phosphorylation of the T840 site (15, 17). It is possible the reduction in GluA1 T840 phosphorylation is caused by a down-regulation of PKC activity. Application of the PKC inhibitor, Go6983, resulted in a significant decrease in GluA1 T840 phosphorylation. Despite the basal effect of Go6983, Go6983 did not inhibit the ability of PACAP38 to stimulate phosphorylation changes at the GluA1 T840 or S845 site (Fig. 4 B and F). These data suggest that although PACAP38 can modulate PKA to effect changes specific to S845 phosphorylation state, PACAP38 does not modulate S845 and T840 phosphorylation by altering PKC activity. Lastly we sought to determine whether phosphatases might play a role in PACAP38 regulation of GluA1 phosphorylation. We first investigated the ability of protein phosphatase 2B (PP2B) to regulate PACAP38-dependent phosphorylation changes. We found the PP2B inhibitor, cyclosporine A, led to a significant decrease in basal levels of GluA1 T840 phosphorylation. However, cyclosporine A was unable to block PACAP38-dependent phosphorylation changes at the GluA1 T840 and S845 sites (Fig. 4 D and H). Consistent with published data (15), the PP1/PP2A inhibitor, okadaic acid, led to a significant increase in GluA1 T840 phosphorylation (Fig. 4 C and G). We also found okadaic acid blocks the PACAP38-dependent GluA1 T840 dephosphorylation, but had no effect on the PACAP38-dependent GluA1 S845 phosphorylation.
Fig. 4.

Identification of kinases or phosphatases responsible for PACAP38-dependent GluA1 phosphorylation changes. Hippocampal neurons (DIV 14) were preincubated with 10 µM H89 for 10 min (A), with 1 μM Go6983 for 10 min (B), with 2 µM okadaic acid (OA) for 10 min (C), or with 2 µM cyclosporine A (CsA) for 15 min (D), and then stimulated with PACAP38 (1 nM) for 10 min. Cells were lysed, GluA1 immunoprecipitated, and samples visualized by Western blot. (E–H) Quantification of GluA1 T840 or S845 phosphorylation normalized to GluA1. Error bars indicate ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA, Bonferroni posttest. n ≥ 6.
It has been reported that a low dose of PACAP38 may influence synaptic transmission through the regulation of NMDARs (20). NMDAR activation has also been shown to result in GluA1 T840 dephosphorylation (16, 17). Thus, we wanted to investigate whether PACAP38 might act through the NMDAR to modulate AMPAR phosphorylation. We found the NMDAR antagonist, D-APV, partially blocked the GluA1 pT840 reduction but had no affect on changes at the S845 site (Fig. 5 A and B).
Fig. 5.
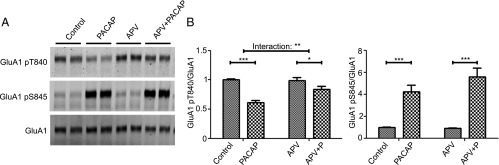
NMDA receptor involvement in GluA1 phosphorylation changes. (A) Hippocampal neurons (DIV 14) were preincubated with D-APV (50 µM) for 45 min and then stimulated with PACAP38 (1 nM) for 10 min. Cells were lysed, GluA1 was immunoprecipitated, and samples were examined by Western blot. (B) Quantification of GluA1 T840 or GluA1 S845 phosphorylation normalized to GluA1. Error bars indicate ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA, Bonferroni posttest. n ≥ 6.
Discussion
A number of studies have shown that PACAP38 regulates CA1 synaptic transmission, AMPAR EPSCs, and GluA1 synaptic clustering (19–22, 24, 25). In humans, a sex-specific association between a single-nucleotide polymorphism in a PACAP38 receptor, the PAC1 receptor, and posttraumatic stress disorder (PTSD) has been reported (29). Moreover, the PAC1 receptor knockout exhibits impaired contextual fear conditioning (27), and the PACAP38 knockout exhibit impaired contextual fear and novel object recognition (26).
Despite the accumulating evidence that PACAP38 can regulate CA1 synaptic transmission and AMPAR EPSCs, very little is known about how this regulation occurs. A number of groups have demonstrated that AMPAR phosphorylation affects receptor recycling (4, 30). Therefore, we hypothesized that PACAP38 may regulate AMPAR phosphorylation. In our study we demonstrated that PACAP38 stimulation of mature, hippocampal cultures results in an up-regulation of GluA1 S845 phosphorylation and a down-regulation of GluA1 T840 phosphorylation. We found that phosphorylation changes at the GluA1 T840 and S845 site result from PACAP38 dose applications as low as 0.05 nM. Furthermore, the reduction in GluA1 T840 phosphorylation and increase in GluA1 S845 phosphorylation could be observed as early as 2 min following stimulation. Phosphorylation increases at the S845 site were robustly driven by VPAC2 and PAC1 receptor activation, and phosphorylation decreases at the T840 site were most robustly driven by PAC1 receptor activation. Downstream of the PACAP38 receptors, we found that PKA activity was necessary for the GluA1 S845 phosphorylation increase, and PP1/PP2A activity was necessary for the GluA1 T840 phosphorylation decrease. We also found that GluA1 T840 dephosphorylation was partially blocked by a NMDAR antagonist. Interestingly, previous reports have shown that NMDA stimulation results in GluA1 T840 and S845 dephosphorylation and that phosphorylation changes were blocked by a PP1/PP2A inhibitor (11, 16, 17). Our antagonist experiment along with these studies suggests there is crosstalk between PACAP38 and NMDAR signaling pathways to regulate GluA1 T840 dephosphorylation but not S845 phosphorylation. Thus, it is conceivable that during NMDAR-dependent processes such as LTD or LTP, PACAP38 may act to modulate NMDAR-dependent changes in AMPAR phosphorylation. Further study is needed to determine if and how crosstalk between PACAP- and NMDAR-dependent AMPAR regulation affect AMPAR phosphorylation, trafficking and synaptic plasticity.
These findings offer a potential mechanism by which PACAP38 may regulate CA1 synaptic transmission. PACAP38 has been found to have a dose-dependent effect on CA1 synaptic transmission, where lower doses of PACAP38 enhance synaptic transmission and AMPAR EPSCs (20, 24), and high doses reduce synaptic transmission and AMPAR EPSCs (20, 24). Although it is unclear how this dose-dependent effect would occur, our data indicates that PACAP38-dependent changes in GluA1 phosphorylation could be a contributing factor that modulates synaptic transmission.
GluA1 T840 phosphorylation has been shown to increase AMPAR conductance (18). It is possible the PACAP38-dependent T840 dephosphorylation reduces AMPAR conductance and synaptic transmission. The reduction in GluA1 T840 phosphorylation could also alter AMPAR trafficking. There is evidence that GluA1 S845 phosphorylation results in increased GluA1 membrane insertion (12) and there is a correlation between increased GluA1 S845 phosphorylation and elevated surface GluA1 levels (12, 31). The PACAP38-dependent increase in GluA1 phosphorylation may increase surface GluA1 levels. Another potential function for PACAP38 regulation of GluA1 phosphorylation may be to facilitate the synaptic delivery of GluA1. For example, the neuromodulator norepinephrine (NE) has been shown to increase GluA1 S845 phosphorylation and to lower the threshold for long-term potentiation (LTP) (8). In a GluA1 S831, 845A knock-in mouse, NE-facilitated LTP is impaired (8). It is possible that PACAP38-dependent changes in AMPAR phosphorylation may also alter the LTP threshold. In future studies, it will be important to demonstrate that PACAP38’s ability to regulate synaptic strength is impaired by GluA1 T840 or S845 phospho-mutants. Likewise it will be interesting to see if changes in synaptic strength occur through alterations in AMPAR trafficking or conductance. Finally, these findings suggest that deficits in AMPAR phosphorylation may underlie the role of PACAP38 and the PAC1 receptor in PTSD and fear memory (26, 27, 29).
Materials and Methods
Reagents and Antibodies.
Maxadillan and (Lys15, Arg16,Leu27)-VIP(1-7)-GRF (8–27), abbreviated as K,R,L-VIP-GRF, were purchased from Bachem. PACAP38, Bay 55–9837, Go6983, D-APV, and H89 were purchased from Tocris. Okadaic acid was purchased from LC Laboratories and cyclosporine A was purchased from Sigma-Aldrich. Commercial antibodies GluA1 pT840 (Abcam), GluA1 pS845 (Millipore), and GluA1 pS831 (Millipore) were used. Antibodies against the GluA1 N terminus (JH4296, 4.9D) were generated in house.
Preparation of Whole Brain Lysate.
Animals were handled in accordance with guidelines set by the Johns Hopkins University Animal Care and Use Committee. Whole brains from WT or “penta” knock-in mice were lysed with NL buffer (1% SDS, 150 mM NaCl, 50 mM Tris pH 7.4, 2 mM EGTA, 50 mM NaF, 10 mM NaPPi, PICA+B, 1 µM okadaic acid). Samples were sonicated and incubated at 95 °C for 5 min. The protein concentration of each sample was measured using the BCA Protein Assay (Thermo Scientific) and samples were diluted to equivalent concentrations. Cell lysate was diluted 10-fold with dilution buffer (final concentration: 1% Triton X-100, 150 mM NaCl, 50 mM Tris pH 7.4, 2 mM EGTA, 50 mM NaF, 10 mM NaPPi) and GluA1 was immunoprecipitated. Input and IP samples were visualized by Western blot.
Cell Culture.
E18 rat pup hippocampal neurons were plated onto poly-l-lysine coated plates containing NM5: Neurobasal growth medium (Invitrogen) supplemented with 5% (vol/vol) FBS (HyClone), 2% (vol/vol) B27 (Invitrogen), 50 U/mL PenStrep (GIBCO), and 2 mM Glutamax (GIBCO). One day after plating, this media was completely replaced with NM0: Neurobasal growth medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 50 U/mL PenStrep (GIBCO), and 2 mM Glutamax. Every 3–4 d thereafter, half of the media was replaced with fresh NM0.
PACAP38 Stimulation and Immunoprecipitation in Hippocampal Neurons.
At DIV 14 hippocampal cells were stimulated with NM0 containing 1nM PACAP38 for 10 min, unless otherwise noted. Cells were then rinsed with ACSF and lysed with NL buffer (1% SDS, 150 mM NaCl, 50 mM Tris pH 7.4, 2 mM EGTA, 50 mM NaF, 10 mM NaPPi, PICA+B, 1 µM okadaic acid), incubated at 95 °C for 5 min, sonicated, and spun down at 16,000 × g for 10 min. Cell lysate was diluted 10 fold with dilution buffer (Final concentration: 1% Triton X-100, 150 mM NaCl, 50 mM Tris pH 7.4, 2 mM EGTA, 50 mM NaF, 10 mM NaPPi) and incubated with a GluA1 antibody (JH4296) and protein A Sepharose beads overnight. The following day, beads were washed three times with Wash buffer (0.1% SDS, 1% Triton X-100, 150 mM NaCl, 50 mM Tris pH 7.4, 2 mM EGTA, 50 mM NaF, 10 mM NaPPi) and eluted in 2× SDS sample buffer. Samples were separated on a SDS/PAGE gel, transferred to nitrocellulose membrane, blocked with Odyssey Blocking Buffer, and incubated with antibody. Blots were then washed, incubated with Alexa Fluor 680 and 800 secondary antibodies, washed, and imaged using an Odyssey Imaging System. Data were analyzed using Odyssey software.
Acknowledgments
We thank Dr. Natasha Hussain, Dr. Graham Diering, Dr. Nicolas Arbez, Kitty Xu, and Dr. Sachin Deshmukh for their helpful advice.
Footnotes
The authors declare no conflict of interest.
References
- 1.Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22(3):461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: The last 25 years. Neuron. 2013;80(3):704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61(3):340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol. 2012;22(3):470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 6.Serulle Y, et al. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56(4):670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 8.Hu H, et al. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131(1):160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 9.Seol GH, et al. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55(6):919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diering GH, Gustina AS, Huganir RL. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron. 2014;84(4):790–805. doi: 10.1016/j.neuron.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405(6789):955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 12.Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci USA. 2007;104(9):3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HK, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112(5):631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 14.Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103(1):479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HK, et al. Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol Cell Neurosci. 2007;36(1):86–94. doi: 10.1016/j.mcn.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delgado JY, et al. NMDA receptor activation dephosphorylates AMPA receptor glutamate receptor 1 subunits at threonine 840. J Neurosci. 2007;27(48):13210–13221. doi: 10.1523/JNEUROSCI.3056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray EE, Guglietta R, Khakh BS, O’Dell TJ. Inhibitory interactions between phosphorylation sites in the C terminus of α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor GluA1 subunits. J Biol Chem. 2014;289(21):14600–14611. doi: 10.1074/jbc.M114.553537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins MA, et al. Regulation of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol Pharmacol. 2014;85(4):618–629. doi: 10.1124/mol.113.091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberto M, Brunelli M. PACAP-38 enhances excitatory synaptic transmission in the rat hippocampal CA1 region. Learn Mem. 2000;7(5):303–311. doi: 10.1101/lm.34200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberto M, Scuri R, Brunelli M. Differential effects of PACAP-38 on synaptic responses in rat hippocampal CA1 region. Learn Mem. 2001;8(5):265–271. doi: 10.1101/lm.40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo T, Tominaga T, Ichikawa M, Iijima T. Differential alteration of hippocampal synaptic strength induced by pituitary adenylate cyclase activating polypeptide-38 (PACAP-38) Neurosci Lett. 1997;221(2-3):189–192. doi: 10.1016/s0304-3940(96)13323-1. [DOI] [PubMed] [Google Scholar]
- 22.Ciranna L, Cavallaro S. Opposing effects by pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal peptide on hippocampal synaptic transmission. Exp Neurol. 2003;184(2):778–784. doi: 10.1016/S0014-4886(03)00300-5. [DOI] [PubMed] [Google Scholar]
- 23.Dickson L, Finlayson K. VPAC and PAC receptors: From ligands to function. Pharmacol Ther. 2009;121(3):294–316. doi: 10.1016/j.pharmthera.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Costa L, Santangelo F, Li Volsi G, Ciranna L. Modulation of AMPA receptor-mediated ion current by pituitary adenylate cyclase-activating polypeptide (PACAP) in CA1 pyramidal neurons from rat hippocampus. Hippocampus. 2009;19(1):99–109. doi: 10.1002/hipo.20488. [DOI] [PubMed] [Google Scholar]
- 25.Gardoni F, et al. The neuropeptide PACAP38 induces dendritic spine remodeling through ADAM10-N-cadherin signaling pathway. J Cell Sci. 2012;125(Pt 6):1401–1406. doi: 10.1242/jcs.097576. [DOI] [PubMed] [Google Scholar]
- 26.Takuma K, et al. An enriched environment ameliorates memory impairments in PACAP-deficient mice. Behav Brain Res. 2014;272:269–278. doi: 10.1016/j.bbr.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Otto C, et al. Impairment of mossy fiber long-term potentiation and associative learning in pituitary adenylate cyclase activating polypeptide type I receptor-deficient mice. J Neurosci. 2001;21(15):5520–5527. doi: 10.1523/JNEUROSCI.21-15-05520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsumi M, et al. A potent and highly selective VPAC2 agonist enhances glucose-induced insulin release and glucose disposal: A potential therapy for type 2 diabetes. Diabetes. 2002;51(5):1453–1460. doi: 10.2337/diabetes.51.5.1453. [DOI] [PubMed] [Google Scholar]
- 29.Ressler KJ, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25(11):578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 31.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281(2):752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]