

Abstract
Background
The genome of the human gastric pathogen Helicobacter pylori encodes a large number of DNA methyltransferases (MTases), some of which are shared among many strains, and others of which are unique to a given strain. The MTases have potential roles in the survival of the bacterium. In this study, we sequenced a Malaysian H. pylori clinical strain, designated UM032, by using a combination of PacBio Single Molecule, Real-Time (SMRT) and Illumina MiSeq next generation sequencing platforms, and used the SMRT data to characterize the set of methylated bases (the methylome).
Results
The N4-methylcytosine and N6-methyladenine modifications detected at single-base resolution using SMRT technology revealed 17 methylated sequence motifs corresponding to one Type I and 16 Type II restriction-modification (R-M) systems. Previously unassigned methylation motifs were now assigned to their respective MTases-coding genes. Furthermore, one gene that appears to be inactive in the H. pylori UM032 genome during normal growth was characterized by cloning.
Conclusion
Consistent with previously-studied H. pylori strains, we show that strain UM032 contains a relatively large number of R-M systems, including some MTase activities with novel specificities. Additional studies are underway to further elucidating the biological significance of the R-M systems in the physiology and pathogenesis of H. pylori.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1585-2) contains supplementary material, which is available to authorized users.
Background
The Gram-negative spiral-shaped bacterium Helicobacter pylori persistently colonizes the human stomach and is often associated with chronic gastritis and peptic ulceration. This bacterium is also implicated in more severe gastric diseases that are regarded as an early risk factor for gastric cancer. H. pylori strains are genetically diverse and the specific genotypes are associated with clinical outcomes of infection [1,2]. Previous analyses of H. pylori genomes have revealed the presence of a large number of restriction-modification (R-M) systems in several strains [3-5]. The R-M systems are often on mobile elements or associated with recombination-related genes, and divergent among different species and strains [6]. In addition to phase variation, high mutation rate and homologous recombination [7,8], the R-M system diversity has been proposed to contribute to the genetic variation of the bacteria [9,10]. Studies have suggested that R-M systems can act as geomarkers that can allow the discrimination of H. pylori populations of different geographical origins, thereby reflecting on human migration patterns [9,11].
In prokaryotes, a DNA methyltransferase (MTase) is often associated with a restriction endonuclease (REase) and forms a R-M system. R-M systems are traditionally divided into four major Types, numbered I, II, III and IV, on the basis of enzyme subunit composition, cofactor requirements and DNA specificity characteristics [12]. Type I systems are encoded by the hsdS, hsdM, and hsdR genes, whose products form multifunctional protein complexes. The HsdS subunit, which composes of two target recognition domains (TRDs), determines the specificity of DNA sequence recognition for both the methylation (HsdM) and cleavage (HsdR) activities. Methylation occurs within each half-recognition-sequence whereas cleavage occurs at a variable distance from the asymmetric recognition site or at an arrested replication fork [13]. A majority of the H. pylori R-M systems are of Type II. In contrast to Type I systems, the Type II R-M systems consist of a MTase and a REase that have enzymatic activities independent of each other, and which often, but not always, occur on independent polypeptides. When these two activities occur on the same polypeptide, the system is denoted Type IIG. Both DNA methylation and cleavage occur within or close to a defined recognition site. Type III systems have two subunits, which are products of the mod and res genes. The Mod subunit functions independently in hemi-methylation while both subunits are necessary for DNA cleavage. Specificity is determined by the Mod subunit. The Type IV systems comprise a REase that recognizes and cleaves modified DNA.
DNA methylation is an important epigenetic DNA modification in bacteria. The modified bases include 5-methylcytosine (m5C), N4-methylcytosine (m4C) and N6-methyladenine (m6A) [12]. MTases have a crucial role in bacterial biology because these enzymes affect diverse cellular and developmental processes such as gene expression and regulation, cell cycle regulation, anti-mutagenesis, DNA transposition and genome maintenance [14-17]. H. pylori is naturally competent and able to take in DNA from the environment [18] as well as being subject to bacteriophage infection [19,20]. Thus, the MTases might also serve as part of the defence mechanism that protects the genome integrity of the bacteria against transmissible DNA elements. On the other hand, strain-specific MTases are thought to influence the phenotypic traits or virulence in pathogens, host specificity and adaptability to micro-environment [21,22].
The study of MTases of H. pylori enhances our understanding of the pathogenic mechanisms of this organism. The discovery of hpyIM, which encodes a Type II MTase that recognizes CATG, revealed that the MTases may play a role in H. pylori physiology beyond the methylation function. The expression of hpyIM is growth-phase regulated and required for normal bacterial morphology [23]. It was shown that the deletion of hpyIM altered the expression of the stress-responsive dnaK operon [24]. A Type II MTase, M.HpyAIV, which recognizes GANTC, has been shown to down-regulate the expression of the katA gene that encodes for the catalase, suggesting its importance in the biology of H. pylori [25]. The expression of the modH gene, a Type III DNA MTase of H. pylori which undergoes rapid on/off switching called phase variation, was shown to regulate two proteins, FlaA and FliK, that have important roles in motility [26]. Collectively, these findings provide impetus for dissecting the roles of the DNA MTases in the cellular processes of H. pylori.
The implementation of Single Molecule, Real-Time (SMRT) DNA sequencing has allowed the direct identification of methylated bases in synthetic DNA templates, plasmids and bacterial chromosomes [27-29]. This technology monitors the real-time incorporation of fluorescently-labelled nucleotides onto growing DNA chains by individual polymerase molecules [30]. DNA methylation can be detected because the presence of certain modifications on DNA bases in the template delay the incorporation of the nucleotides by the polymerase in a characteristic manner [31]. For substrates of sufficient complexity such as genomic DNA, MTase motifs can be derived ab initio by looking for repeating patterns in sequence windows around each methylated base. Furthermore, the fraction of all instances of each motif that is modified can also be determined.
Recently, Krebes and coworkers used SMRT sequencing to analyse the methylomes of two H. pylori strains, 26695 and J99 [32]. Despite several earlier studies of the R-M systems in these strains [33-35], the SMRT-assisted analysis provided significant additional insights, including the characterization of Type I and Type III systems and the novel observation of S subunit switching between Type I systems [32]. In addition, another methylome study of five H. pylori strains (P12, F16, F30, F32 and F57) by Furuta and co-workers elucidated the relationships between each TRD sequence in S subunit of Type I systems and the corresponding half-site sequence [36]. Given the large numbers of R-M systems typical of H. pylori strains in general, it seemed likely to be fruitful to examine additional strains, particularly those isolated from more geographically diverse locations than the earlier strains. H. pylori strain UM032 was isolated from a gastroduodenal ulcer patient presenting for gastroscopy at University of Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia. It is the parental strain for the mice-adapted isolates, H. pylori 298 and H. pylori 299, and was sequenced using the PacBio platform as described in the previous study [37]. In the present study, the methylome of H. pylori UM032 was characterized using SMRT DNA sequencing and compared to those of several previously characterized H. pylori strains [32,36].
Results
Nucleotide sequence accession number
The first annotated H. pylori UM032 genome sequence was deposited in DDBJ/EMBL/GenBank with the accession number CP005490 [37]. Here, an updated version of the genome sequence was reported, where the HGAP assembled sequence was corrected by the mapping of Illumina reads. The version described in this paper is CP005490.3.
Methylome analysis of H. pylori UM032
SMRT sequencing offers the potential to study DNA methylation in H. pylori at a genome‐wide scale. Base modifications of the H. pylori UM032 genome were analysed, modified sequence motifs were determined, and the MTase responsible for each motif was deduced through a combination of prediction and characterization of cloned and isolated MTases. A total of 63,299 genomic positions were detected as methylated (m4C or m6A). Seventeen functional MTases were identified, of which 14 could be confidently assigned to their MTase sequence specificities based on formerly reported recognition sequences of highly similar examples [38]. The methylated motif GANNNNNNNTAYG, which was reported in H. pylori strain F32, was not assigned to a MTase in H. pylori UM032 genome. The remaining two systems demonstrated novel recognition motifs (GAAAG and CYANNNNNNNTRG), which were not previously described in H. pylori. The detected methylation motifs are summarized in Table 1, along with the corresponding MTase-encoding genes. All but one active R-M system was of Type II, with only one Type I R-M system and no Type III R-M systems. The analysed methylome of this isolate was deposited in REBASE [38].
Table 1.
Methylated motifs detected for H. pylori UM032
| Type of RM system | Motifs a | Type of modification | No. detected d | No. in genome | % detected | Locus tag | Nomenclature |
|---|---|---|---|---|---|---|---|
| I | GANNNNNNNTAYG | m6A | 653 | 653 | 100.00 | K747_03505 | M.HpyUM032XII |
| IIP | TCGA | m6A | 526 | 526 | 100.00 | K747_09985 | M.HpyUM032XVII |
| IIP | CA TG | m6A | 14370 | 14370 | 100.00 | K747_04980 | M.HpyUM032I |
| IIP | ACNGT | m4C | 1005 | 1104 | 91.03 | K747_10995 | M.HpyUM032II |
| IIP | GA TC | m6A | 10172 | 10172 | 100.00 | K747_09245 | M.HpyUM032III |
| IIP | GANTC | m6A | 5388 | 5388 | 100.00 | K747_12490 | M.HpyUM032IV |
| IIP | CCGG | m4C | 3396 | 3416 | 99.41 | K747_10000 | M.HpyUM032IX |
| IIP | TCNGA | m6A | 2530 | 2532 | 99.92 | K747_05140 | M.HpyUM032V |
| IIP | ATTAAT | m6A | 857 | 874 | 98.05 | K747_10980 | M.HpyUM032VII |
| IIP | TGCA | m6A | 11260 | 11270 | 99.91 | K747_12120 | M.HpyUM032VIII |
| IIS | GAGG | m6A | 4578 | 4579 | 99.98 | K747_08850 | M2.HpyUM032VI |
| IIS | CCATC | m6A | 2255 | 2255 | 100.00 | K747_03690 | M1.HpyUM032X |
| IIP | GC GCe | m5C | 774 | 2396 | 32.30 | K747_05430 | M.HpyUM032XV |
| IIG | CYANNNNNNNTRGb | m6A | 2319 | 2320 | 99.96 | K747_03825 | HpyUM032XIII |
| IIG | GAAAGb | m6A | 2514 | 4955 | 50.74 | K747_03595 | HpyUM032XIV |
| IIP | GTNNAC | m6A | 528 | 528 | 100.00 | K747_06370 | M.HpyUM032XI |
| IIP | GT AC | m6A | 174 | 174 | 100.00 | K747_06575 | M.HpyUM032XVIII |
| I | CCANNNNNNTCb,c | m6A | - | - | - | K747_10905 | M.HpyUM032XVI |
aThe methylated base within the motif is in bold while the methylated base in the complementary strand is italic.
bNovel recognition sequences.
cActivity identified only after cloning. No methylation activity was observed in H. pylori UM032.
dThe total number includes motifs occurring on the “+” and “–” strands.
eLow percentage detected, due to m5C modification.
Characterization of DNA MTases with unknown specificities
To identify the MTases that recognize and methylate the three unassigned recognition motifs, candidate MTase genes, and their associated S subunits where necessary, were cloned into pRRS and overexpressed in Escherichia coli ER2796. Genomic DNA was then isolated from each recombinant strain and subjected to SMRT sequencing to confirm the enzymatic activity of the MTase candidate and to identify the modified motif. Those MTases that were active either in the H. pylori UM032 genome or as clones are shown in Table 1, while all MTases not responsible for any activity in the genome or shown to be inactive as clones are shown in Additional file 1: Table S3.
K747_03505
This Type I MTase would require association with an S subunit for activity, and the most likely candidate was encoded by the adjacent ORF (K747_03510). Concomitant overexpression of K747_03505 and K747_03510 revealed methylation of the recognition motif GANNNNNNNTAYG. This MTase was designated as M.HpyUM032XII.
K747_03595
This Type IIG gene belonging to the CjeFIII/Eco57-like MTase family was cloned, and SMRT sequencing of genomic DNA from the recombinant E. coli strain revealed hemi-methylation of the target sequence GAAAG. This MTase was named HpyUM032XIV.
K747_03825
This is a BcgI-like Type IIG R-M system, comprising two S subunit genes (S1 and S2) and a hybrid gene (RM) encoding both MTase and REase domains (Figure 1). The two S subunit genes (K747_11950 and K747_11945) are separated by a homopolymeric G repeat, which may have resulted in a previously intact single S subunit becoming split as a result of a frameshift mutation. When the RM, S1, and S2 genes were overexpressed together in E. coli, the palindromic motif CYANNNNNNNTRG was found to be methylated just as in the genome. This R-M system was named HpyUM032XIII. Interestingly, when the S1 and S2 were artificially fused by “correcting” the frameshift and overexpressed with the RM, a change of methylation pattern was observed leading to recognition of CYANNNNNNNTTC. This is a new specificity that was not detected in the methylome of H. pylori UM032 during normal growth. It was named as HpyUM032XIII-mut1, indicating its artificially derived sequence (Figure 1). Expressing S2, but not S1, with the RM gene gave no activity. On the basis of these results S1, which only encodes one TRD, must be responsible for recognition of the CYA half-site. The second TRD would then recognize the half-site GAA. The sequences of HpyUM032XIII and HpyUM032XIII-mut1 were deposited in DDBJ/EMBL/GenBank with the accession number KM875507 and KM875508 respectively.
Figure 1.
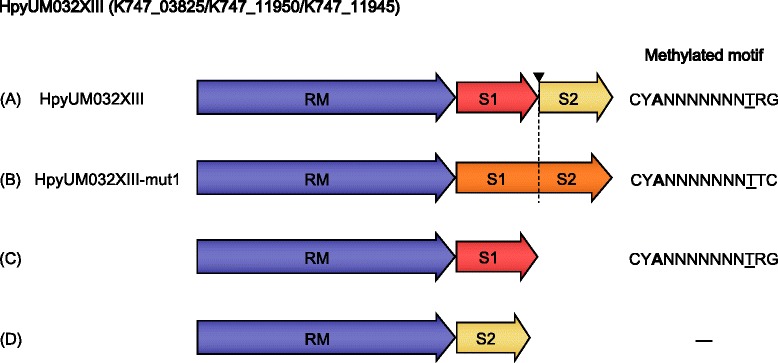
Schematic representation of the specificity switching of the Type IIG MTase HpyUM032XIII. (A) The S1 and S2 subunits are separated by a homopolymeric tract of 12 G residues at the location shown by ▼, which appears to create a natural frameshift. (B) Reducing the tract length from 12 to 11 corrects the frameshift at ▼, thereby fusing the S1 and S2 subunits. This ‘corrected’ sequence is denoted by ‘mut’, to create fusion of S1 and S2 subunits. (C–D) Expression of individual S subunits with the RM gene shows that the S1 subunit is active in the absence of S2.
K747_04185
This is a putative Type III MTase that showed no activity in either H. pylori UM032 or when the mod gene was cloned into E. coli ER2796. A frameshift mutation was identified in the REase gene upstream of the MTase and this may disrupt the functional expression of the MTase if it is transcribed as an operon. Since the cloned MTase was also inactive, the prolonged absence of expression may have allowed the accumulation of less obvious inactivating mutations in the MTase gene itself.
K747_05620
This ORF shares 92.8% amino acid identity with the functional M.Hpy99XVIII of H. pylori J99 that methylates TCNNGA. However, when cloned into E. coli it did not confer methylation, nor is it active in the genome, assuming that it would have the same recognition specificity as M.Hpy99XVIII.
K747_08715
This is an orphan Type II MTase located within a putative Type III R-M system (between the REase gene K747_08710 and the corresponding unannotated MTase gene). As the two MTases are located adjacent to each other in the genome, both of the genes were cloned and overexpressed both individually and together in E. coli. Nevertheless, the overexpressed gene products showed no methylation activity.
K747_10905
Overexpression of this Type I MTase along with its S subunit (K747_10900) in E. coli resulted in adenine methylation of the recognition site CCANNNNNNTC. Despite having a functional MTase, no methylation of this motif was detected in the H. pylori UM032 genome which could be due to the frameshift in the upstream REase gene that may have disrupted the transcription of this Type I R-M operon.
Discussion
The complete genome sequence of the Malaysian H. pylori clinical strain UM032 was obtained using PacBio sequencer as described in the previous study [37]. However, despite the long read length, error rates of single-molecule reads can be as high as 13% [39,40]. To address this limitation, the strain was sequenced with Illumina Miseq platform in this study to increase genome coverage thereby improved error-correction in single-molecule sequences.
This study describes a methylome analysis of the Malaysian H. pylori clinical strain UM032 using SMRT DNA sequencing technology, which can detect m6A and m4C methylation with high precision. The kinetic signatures of m5C bases may not have been strong enough to properly study. Nonetheless, because of the relatively high sequence coverage [41], one native m5C methylated motif in the H. pylori UM032 genomic DNA was identified, GCGC. The specificity of the m5C MTases was predicted based on high similarity with homologous examples in other H. pylori strains, and so the GCGC motif has been tentatively assigned to the remaining MTase (Table 1). TET treatment of the DNA and cloning of the m5C MTases may reveal additional m5C modification in this genome.
Seventeen R-M systems were identified, of which 16 are of Type II, which is in agreement with previous findings that H. pylori encodes mostly Type II R-M systems [11]. H. pylori genomes encode unusually high numbers of R-M systems, in particular the Type II R-M systems are highly diverse between strains. However, it is not clear why H. pylori possesses this unique characteristic. Three of the recognition motifs (CATG, TCGA and ATTAAT) present in H. pylori UM032 were also detected in other H. pylori strains with characterized methylome shown in Additional file 1: Table S2, suggesting that they may be essential for the survival and/or maintenance of the genome integrity of H. pylori strains in general. The specificity CATG is shared by a previously characterized MTase, M.HpyI, which associates with the epithelial-responsive REase IceAI [24]. The hpyIM gene, which encodes M.HpyI, is highly conserved in the genomes of H. pylori clinical strains of different geographical origins [23,35]. Strain UM032 encodes a putative REase, HpyUM032IP, that is 88% identical to IceAI, and is located adjacent to the MTase responsible for methylation of CATG, suggesting this system may have similar epithelial-responsive properties. Two novel methylation motifs were detected in the current study: 1, GAAAG, methylated by a Type IIG R-M system designated HpyUM032XIV and; 2, CYANNNNNNNTRG, methylated by another Type IIG R-M system designated HpyUM032XIII. On the other hand, HpyUM032XII, which recognizes GANNNNNNNTAYG, was the only active Type I R-M system identified in H. pylori UM032 genome.
HpyUM032XIII, which resembles the BcgI system in that it consists of a fused RM protein and a separate S protein, also differs from BcgI in that the genetic system encodes two S genes, each of which is one half of the typical length of such genes. It seemed likely that these “half-genes” resulted from a frameshift that had occurred in an ancestral, full-length S gene. Although such frameshift often abolish activity, the cloned system, including RM, S1 and S2 demonstrated MTase activity recognizing the palindromic site CYANNNNNNNTRG. Identical activity was observed when the S2 subunit was omitted, and no activity was observed when S1 was omitted, suggesting the activity resulted from a complex of RM and S1 alone. Surprisingly, when S1 and S2 were artificially fused, the recognition sequence had changed and was now CYANNNNNNNTTC (Figure 1). These observations indicate that S1.HpyUM032XIII must contain a TRD capable of recognizing the half-site CYA. Active BcgI, which also recognizes a palindromic sequence, has a stoichiometry of [(RM)2S]2 [42], and HpyUM032XIII would have a similar stoichiometry, where S is replaced by S1. S.BcgI and S1.HpyUM032XIII must each recognize only a single half-site and therefore require dimerization for functionality. By fusing S1 and S2 into a single protein, two TRDs would be present, and dimerization of S would no longer be required. HpyUM032XIII-mut1 should exhibit a stoichiometry of (RM)2S. A similar phenomenon has been observed in Type I systems such as M.NgoAV [43] and M.Hpy99XVI [32], but to our knowledge this is the first example of this phenomenon in the context of a Type IIG systems, in which the MTase and REase activities are fused into a single protein. Further studies are required to verify these hypotheses.
Several MTases exhibited different behaviour in various contexts. There was one MTase (K747_10905) of Type I R-M system that was not functional in the genome of H. pylori UM032, but was shown to be active when cloned and overexpressed in E. coli. Similar examples of apparent activation when cloned have been noted previously and presumably reflect some silencing mechanism in the genome [32]. Transcriptional silencing [44,45] or antisense RNA [46] could have been involved in switching off the genes in H. pylori, while the lack of such regulation(s) in E. coli would result in the expression of this gene. On the other hand, the Type II MTase, K747_08715, and the MTase of a putative Type III R-M system that located adjacent to K747_08715 were both non-functional. This phenomenon could be explained by Nobusato et al. [47]. As the R-M systems are often linked with the mobile genetic elements, K747_08715 could have been inserted to this putative Type III R-M system, resulting in inactivation of both systems. A different MTase, K747_05620, which has strong sequence similarity to M.Hpy99XVIII from H. pylori J99, was shown to be inactive in both native and cloned contexts. Pairwise alignment revealed that K747_05620 was missing ten amino acid residues from the C-terminus compared to that of M.Hpy99XVIII, which could be the cause of inactivation of the MTase.
Conclusions
This analysis provides yet another illustration of the variability in methylation patterns and MTases that is a hallmark of H. pylori biology. Because of its very restricted habitat, it seems unlikely that the large number of potential R-M systems in H. pylori strains is needed to protect against bacteriophages. In looking for alternative functions for this extreme methylation it is tempting to speculate that the MTases are involved in the regulation of gene expression that might facilitate rapid adaptation of H. pylori to changes in the host environment and thus successful gastric colonisation. They may also play a pivotal role in maintaining genome and strain identity in this naturally competent organism: since multiple strains are often present in the same niche, DNA methylation may act to limit recombination between strains and thus preserve diversity.
Methods
Bacterial cultivation and preparation of genomic DNA
H. pylori strain UM032 was inoculated onto non-selective lysed blood agar and incubated for three days in humidified air with 10% CO2 at 37°C. The genomic DNA was extracted from H. pylori UM032 using an RTP® Bacteria DNA Mini Kit (Stratec, Germany).
E. coli strains ER2683 [48] and ER2796 [27] were used as hosts for the preparation of plasmid DNA while E. coli ER2796 was used to express MTases. All the E. coli strains were cultured aerobically overnight at 37°C on Luria-Bertani (LB) agar or in LB broth supplemented with ampicillin (100 μg/ml) when necessary. Genomic DNA from E. coli was purified using phenol:methylene chloride extraction as described [49] and resuspended in TE buffer.
Genomic DNA sequencing
The genome of H. pylori UM032 was sequenced using a combination of next-generation sequencing platforms. Genomic DNA sequencing was first performed on the Pacific Biosciences (PacBio) RS instrument (Menlo Park, CA) using 10-kb libraries prepared by the manufacturer’s kits with C2 chemistry. H. pylori UM032 was sequenced on eight SMRT Cells, with one 120-minute movie per Cell, yielding >300× average genome coverage. To improve the quality of the sequence, the genomic DNA was subjected to additional sequencing on an Illumina MiSeq platform. Preparation of the MiSeq library was performed according to the Nextera XT protocol (Ver. May 2012) using Illumina Nextera XT chemistry (Illumina, San Diego, CA, USA) as previously described with minor modifications [50]. The final libraries were instead normalized by quantification with bioanalyzer (Agilent) and the concentration was adjusted to 4 nM as required by the MiSeq loading protocol. Libraries were sequenced using MiSeq reagent kit v3 (Illumina Inc., San Diego, CA, USA) for a 300-bp paired-end sequencing run using the MiSeq Personal Sequencer (Illumina Inc., San Diego, CA, USA), yielding 135× genome coverage.
E. coli genomes were sequenced using a PacBio RS II instrument (PacBio, Menlo Park, CA, USA). The genomic DNA was treated for 1-hr at 37°C with RNase If (New England Biolabs, Ipswich, MA, USA), sheared to an average size of 5-kb using g-TUBEs (Covaris Inc., Woburn, MA, USA) and purified using the PowerClean DNA Clean-Up Kit (MoBio Laboratories Inc., Carlsbad, CA, USA). PacBio SMRTbell™ template libraries were prepared according to the manufacturer’s instructions. SMRT sequencing was performed using Sequencing Reagent 2.0 with DNA polymerase P4. Typically, samples were sequenced with two SMRT Cells using one 120-min movies per Cells, and this typically resulted in coverage of 70-fold across the ER2796 reference. In some cases where methylation levels were low, additional SMRT Cells were employed.
De novo assembly of the H. pylori UM032 genome
The results of both sequencing platforms were used to perform de novo assembly. The de novo assembly of 10-kb insert reads by PacBio sequencing was conducted using the hierarchical genome assembly process (HGAP) version 2.0 [51]. This resulted in a single, complete contig. The raw reads generated from the Illumina platform were aligned to the H. pylori UM032 contig using the Geneious R7 in-house read mapper with medium sensitivity option [52]. Gene prediction was conducted using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP).
Analysis of methylated bases from SMRT® sequencing data
DNA methylation detection was carried out using the kinetic data collected during the genome sequencing process. Genome‐wide detection of base modification and the affected motifs were performed using the “RS_Modification_and_Motif_Analysis.1” protocol from PacBio. Motifs were determined using the default quality value (QV) score of 30. While the coverage levels were high enough to warrant raising the QV threshold to a more stringent level, the lower (default) value was chosen to minimize the false negatives. Despite the low threshold, the mean modification QVs of all of the motifs in Table 1 were between 80 and 350. Furthermore, all of the m4C and m6A motifs identified were methylated in 98-100% of the instances of each motif (Table 1), suggesting that none of these were false positives generated by an inappropriately low threshold.
Identification and assignment of MTase genes
The assembled genome was scanned for homologs of R-M system genes using in-house, BLAST-based software (E-value < 1e-11) to identify putative MTases as previously described [53]. Predicted specificities were assigned to candidate MTases based on specificities of previously characterized homologs. The presence of functional motifs, syntenic information, and known characteristics of different MTase types were also used to support or reject those assignments. As examples of characteristic information, Type III and most Type IIG MTases methylate only one strand of their recognition sequence, whereas Type I systems have bipartite recognition sequences consisting of two “half-sites.” MTase candidates with predicted specificities were matched where possible with observed motifs found in our motif analyses. If a single candidate MTase existed for an observed motif, then that gene was assumed to be responsible for that particular specificity. If multiple candidates existed for a single motif, no automatic assignment was made. When assigning a novel specificity to a given MTase, the MTase gene sequence was cross-checked against other similar genes in REBASE, and the novel specificity against unassigned SMRT-derived motif data in REBASE. In many cases, the same motif occurred in a different genome with an essentially identical MTase or specificity subunit protein sequence, adding weight to the strength of the assignment. MTase information and sequences were deposited in REBASE (http://rebase.neb.com/rebase/rebase.html) [12].
Cloning and over-expression of MTases
Putative MTase and specificity (S) subunit genes were amplified from H. pylori UM032 with Q5® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA) using gene-specific oligonucleotide primers and cloned into PCR-amplified pRRS plasmid vector using the Gibson Assembly® Cloning kit (New England Biolabs, Ipswich, MA, USA). Mutations to correct the frameshift in the S subunit of K747_03825 and silent mutations to stabilize polynucleotide repeat sequences were likewise introduced using Gibson Assembly. For example, in K747_03825, the 12-bp repeat sequence GGGGGGGGGGGG was changed to GGAGGAGGCGG, which simultaneously introduced silent mutations to prevent replication slippage and shortened the length to 11, bringing S2 in frame with S1. The expression of all MTase genes was under the regulation of the same E. coli Plac promoter present in the pRRS vector. Primer sequences are shown in Additional file 1: Table S1.
Recombinant constructs were used to transform E. coli ER2683. Restriction analysis was performed to confirm that the bacterial transformants carried the desired plasmid construct. The plasmid constructs were then used to transform E. coli strain ER2796, which lacks endogenous MTase activity. The genomic DNA of the E. coli ER2796 recombinant strain was subjected to SMRT sequencing to determine the resulting methylation pattern. Plasmid sequences were confirmed by re-sequencing the PacBio reads against the plasmid reference.
Acknowledgement
This work was supported by University of Malaya-Ministry of Education (UM-MoE) High Impact Research (HIR) Grant UM.C/625/1/HIR/MoE/CHAN/13/2 (Account No. H-50001-A000032) to KLG and the Small Business Innovation Research Program (NIGMS) of the National Institutes of Health under award number R44GM105125 to RJR. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Additional file
Modification analysis for H. pylori UM032 at QV 30 and QV 50. Table S1. Oligonucleotide primers used for H. pylori putative MTase expression in E. coli. Table S2. Comparison of methylation patterns among H. pylori UM032 and various H. pylori strains. Table S3. Other MTase genes in H. pylori UM032 not responsible for observed activities.
Footnotes
Competing interests
BPA and RJR work for New England Biolabs, a company that commercializes REases, MTases and other enzymes. SS is a full-time employee at PacBio Singapore whereas SW, PB and MA are full-time employees at Pacific Biosciences, a company that commercializes the SMRT DNA sequencing technology.
Authors’ contributions
MFL, KLG, BJM, RJR and JV designed the experiments. SW, PB, SS and MA involved in the PacBio sequencing for H. pylori and interpretation of the data. EGC, CYT and FT involved in the Illumina MiSeq sequencing for H. pylori and interpretation of the data. MFL analysed and assembled the data. WCL and BPA performed the experiments involving methylation and analysed the data. WCL, BPA and RJR drafted the manuscript. All authors were involved in revising the manuscript, read and approved the final manuscript.
Contributor Information
Woon Ching Lee, Email: woonching.lee@gmail.com.
Brian P Anton, Email: anton@neb.com.
Susana Wang, Email: swang@pacificbiosciences.com.
Primo Baybayan, Email: pbaybayan@pacificbiosciences.com.
Siddarth Singh, Email: ssingh@pacificbiosciences.com.
Meredith Ashby, Email: mashby@pacbiosciences.com.
Eng Guan Chua, Email: eng.chua@uwa.edu.au.
Chin Yen Tay, Email: alfred.tay@uwa.edu.au.
Fanny Thirriot, Email: fanny.thirriot@uwa.edu.au.
Mun Fai Loke, Email: lokemunfai@um.edu.my.
Khean Lee Goh, Email: gohkl@um.edu.my.
Barry J Marshall, Email: barry.marshall@uwa.edu.au.
Richard J Roberts, Email: roberts@neb.com.
Jamuna Vadivelu, Email: jamuna@um.edu.my.
References
- 1.Go M, Graham D. How does Helicobacter pylori cause duodenal ulcer disease: the bug, the host, or both? J Gastroenterol Hepatol. 1994;9:S8–10. doi: 10.1111/j.1440-1746.1994.tb01294.x. [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay AK, Kersulyte D, Jeong J-Y, Datta S, Ito Y, Chowdhury A, et al. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J Bacteriol. 2000;182:3219–26. doi: 10.1128/JB.182.11.3219-3227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, et al. The complete genome sequence of Helicobacter pylori strain G27. J Bacteriol. 2009;191:447–8. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh JD, Kling-Bäckhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc Natl Acad Sci USA. 2006;103:9999–10004. doi: 10.1073/pnas.0603784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alm RA, Ling LL, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–80. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 6.Furuta Y, Kobayashi I, Diversity I. DNA methyltransferases suggests epigenetics-driven adaptive evolution. Mob Genet Elem. 2012;2:292–6. doi: 10.4161/mge.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, et al. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci USA. 2011;108:5033–8. doi: 10.1073/pnas.1018444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Björkholm B, Sjölund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci USA. 2001;98:14607–12. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vale FF, Mégraud F, Vítor JMB. Geographic distribution of methyltransferases of Helicobacter pylori: evidence of human host population isolation and migration. BMC Microbiol. 2009;9:193. doi: 10.1186/1471-2180-9-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001;29:3742–56. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Q, Morgan RD, Roberts RJ, Blaser MJ. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc Natl Acad Sci USA. 2000;97:9671–6. doi: 10.1073/pnas.97.17.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–12. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loenen WAM, Dryden DTF, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42:20–44. doi: 10.1093/nar/gkt847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahramanoglou C, Prieto AI, Khedkar S, Haase B, Gupta A, Benes V, et al. Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat Commun. 2012;3:886. doi: 10.1038/ncomms1878. [DOI] [PubMed] [Google Scholar]
- 15.Wion D, Casadesús J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol. 2006;4:183–92. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa K, Handa N, Sears L, Raleigh EA, Kobayashi I. Cleavage of a model DNA replication fork by a methyl-specific endonuclease. Nucleic Acids Res. 2011;39:5489–98. doi: 10.1093/nar/gkr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donczew R, Zakrzewska-Czerwińska J, Zawilak-Pawlik A. Beyond DnaA: The role of DNA topology and DNA methylation in bacterial replication initiation. J Mol Biol. 2014;426:2269–82. doi: 10.1016/j.jmb.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Hofreuter D, Odenbreit S, Haas R. Natural transformation competence in Helicobacter pylori is mediated by the basic components of a type IV secretion system. Mol Microbiol. 2001;41:379–91. doi: 10.1046/j.1365-2958.2001.02502.x. [DOI] [PubMed] [Google Scholar]
- 19.Luo C-H, Chiou P-Y, Yang C-Y, Lin N-T. Genome, integration, and transduction of a novel temperate phage of Helicobacter pylori. J Virol. 2012;86:8781–92. doi: 10.1128/JVI.00446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchiyama J, Takeuchi H, Kato S, Gamoh K, Takemura-Uchiyama I, Ujihara T, et al. Characterization of Helicobacter pylori bacteriophage KHP30. Appl Environ Microbiol. 2013;79:3176–84. doi: 10.1128/AEM.03530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heithoff DM. An essential role for DNA adenine methylation in bacterial virulence. Science. 1999;284:967–70. doi: 10.1126/science.284.5416.967. [DOI] [PubMed] [Google Scholar]
- 22.Takata T, Aras R, Tavakoli D, Ando T, Olivares AZ, Blaser MJ. Phenotypic and genotypic variation in methylases involved in Type II restriction-modification systems in Helicobacter pylori. Nucleic Acids Res. 2002;30:2444–52. doi: 10.1093/nar/30.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Q, Peek RM, Miller GG, Blaser MJ. The Helicobacter pylori genome is modified at CATG by the product of hpyIM. J Bacteriol. 1997;179:6807–15. doi: 10.1128/jb.179.21.6807-6815.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donahue JP, Israel DA, Torres VJ, Necheva AS, Miller GG. Inactivation of a Helicobacter pylori DNA methyltransferase alters dnaK operon expression following host-cell adherence. FEMS Microbiol Lett. 2002;208:295–301. doi: 10.1111/j.1574-6968.2002.tb11097.x. [DOI] [PubMed] [Google Scholar]
- 25.Skoglund A, Björkholm B, Nilsson C, Andersson AF, Jernberg C, Schirwitz K, et al. Functional analysis of the M.HpyAIV DNA methyltransferase of Helicobacter pylori. J Bacteriol. 2007;189:8914–21. doi: 10.1128/JB.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srikhanta YN, Gorrell RJ, Steen JA, Gawthorne JA, Kwok T, Grimmond SM, et al. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori. PLoS One. 2011;6:e27569. doi: 10.1371/journal.pone.0027569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, et al. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012;40:e29. doi: 10.1093/nar/gkr1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–5. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol. 2012;30:1232–9. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 31.Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, et al. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 2013;23:129–41. doi: 10.1101/gr.136739.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krebes J, Morgan RD, Bunk B, Spröer C, Luong K, Parusel R, et al. The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 2014;42:2415–32. [DOI] [PMC free article] [PubMed]
- 33.Kong H, Lin L-F, Porter N, Stickel S, Byrd D, Posfai J, et al. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28:3216–23. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin L-F, Posfai J, Roberts RJ, Kong H. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc Natl Acad Sci USA. 2001;98:2740–5. doi: 10.1073/pnas.051612298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitkute J, Stankevicius K, Tamulaitiene G, Maneliene Z, Timinskas A, Berg DE, et al. Specificities of eleven different DNA methyltransferases of Helicobacter pylori strain 26695. J Bacteriol. 2001;183:443–50. doi: 10.1128/JB.183.2.443-450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furuta Y, Namba-Fukuyo H, Shibata TF, Nishiyama T, Shigenobu S, Suzuki Y, et al. Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet. 2014;10:e1004272. doi: 10.1371/journal.pgen.1004272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khosravi Y, Rehvathy V, Wee WY, Wang S, Baybayan P, Singh S, et al. Comparing the genomes of Helicobacter pylori clinical strain UM032 and mice-adapted derivatives. Gut Pathogens. 2013;5:25. doi: 10.1186/1757-4749-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–6. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quail M, Smith ME, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:1. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Detecting DNA Base Modifications. [www.pacb.com/pdf/TN_Detecting_DNA_Base_Modifications.pdf]
- 42.Kong H. Analyzing the functional organization of a novel restriction modification system, the BcgI system. J Mol Biol. 1998;279:823–32. doi: 10.1006/jmbi.1998.1821. [DOI] [PubMed] [Google Scholar]
- 43.Adamczyk-Poplawska M, Lower M, Piekarowicz A. Deletion of one nucleotide within the homonucleotide tract present in the hsdS gene alters the DNA sequence specificity of Type I restriction-modification system NgoAV. J Bacteriol. 2011;193:6750–9. doi: 10.1128/JB.05672-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casadesús J, Low DA. Programmed heterogeneity: epigenetic mechanisms in bacteria. J Biol Chem. 2013;288:13929–35. doi: 10.1074/jbc.R113.472274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bikard D, Marraffini LA. Innate and adaptive immunity in bacteria: mechanisms of programmed genetic variation to fight bacteriophages. Curr Opin Immunol. 2012;24:15–20. doi: 10.1016/j.coi.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Mruk I, Kobayashi I. To be or not to be: regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014;42:70–86. doi: 10.1093/nar/gkt711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nobusato A, Uchiyama I, Ohashi S, Kobayashi I. Insertion with long target duplication: A mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene. 2000;259:99–108. doi: 10.1016/S0378-1119(00)00456-X. [DOI] [PubMed] [Google Scholar]
- 48.Sibley MH, Raleigh EA. Cassette-like variation of restriction enzyme genes in Escherichia coli C and relatives. Nucleic Acids Res. 2004;32:522–34. doi: 10.1093/nar/gkh194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb Protoc. 2006;2006:pdb.prot4455. doi: 10.1101/pdb.prot4455. [DOI] [Google Scholar]
- 50.Perkins TT, Tay CY, Thirriot F, Marshall B. Choosing a benchtop sequencing machine to characterise Helicobacter pylori genomes. PLoS One. 2013;8:e67539. doi: 10.1371/journal.pone.0067539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 52.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, et al. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–62. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]