

Abstract
In the family of gas transmitters, hydrogen sulfide (H2S) is yet not adequately researched. Known for its rotten egg smell and adverse effects on the brain, lungs, and kidneys for more than 300 years, the vasorelaxant effects of H2S on blood vessel was first observed in 1997. Since then, research continued to explore the possible therapeutic effects of H2S in hypertension, inflammation, pancreatitis, different types of shock, diabetes, and heart failure. However, a considerable amount of efforts are yet needed to elucidate the mechanisms involved in the therapeutic effects of H2S, such as nitric oxide-dependent or independent vasodilation in hypertension and regression of left ventricular hypertrophy. More than a decade of good repute among researchers, H2S research has certain results that need to be clarified or reevaluated. H2S produces its response by multiple modes of action, such as opening the ATP-sensitive potassium channel, angiotensin-converting enzyme inhibition, and calcium channel blockade. H2S is endogenously produced from two sulfur-containing amino acids L-cysteine and L-methionine by the two enzymes cystathionine γ lyase and cystathionine β synthase. Recently, the third enzyme, 3-mercaptopyruvate sulfur transferase, along with cysteine aminotransferase, which is similar to aspartate aminotransferase, has been found to produce H2S in the brain. The H2S has interested researchers, and a great deal of information is being generated every year. This review aims to provide an update on the developments in the research of H2S in hypertension amid the ambiguity in defining the exact role of H2S in hypertension because of insufficient number of research results on this area. This critical review on the role of H2S in hypertension will clarify the gray areas and highlight its future prospects.
KEY WORDS: 3-3-mercaptopyruvate sulfur transferase, cystathionine γ lyase, cystathionine β synthase, cysteine aminotransferase, hydrogen sulfide, hypertension
Introduction
History and Background of Hydrogen Sulfide
A number of gases are produced endogenously in humans and have roles in the pathology of various diseases. Among these gases, nitric oxide (NO) and carbon monoxide (CO) have vital roles in the physiology of the body. Since the last decade, hydrogen sulfide (H2S) has captured the interest of scientists because of its significant role in different systems of the body. H2S has been known as a toxic gas with rotten egg smell for more than 300 years. As a toxicant, H2S mainly damages the brain, kidneys, and lungs.[1] Some studies have also reported the toxic effects of H2S on the central nervous system (CNS) and respiratory system.[2,3,4] H2S is endogenously produced from two sulfur-containing amino acids L-cysteine and L-methionine by the two enzymes cystathionine γ lyse (CSE) and cystathionine β synthase (CBS) as shown in Figure 1a and b.[5,6] Recently, a third enzyme, namely, 3-mercaptopyruvate sulfur transferase (3MST), along with cysteine aminotransferase (CAT), which is similar to aspartate amino transferase,[7,8] has been shown to produce H2S in brain. 3MST is produced by schematic chain reaction from 3-mercaptopyruvate which is also produced by CAT from cysteine and α-ketoglutarate[9,10][Figure 1].
Figure 1.
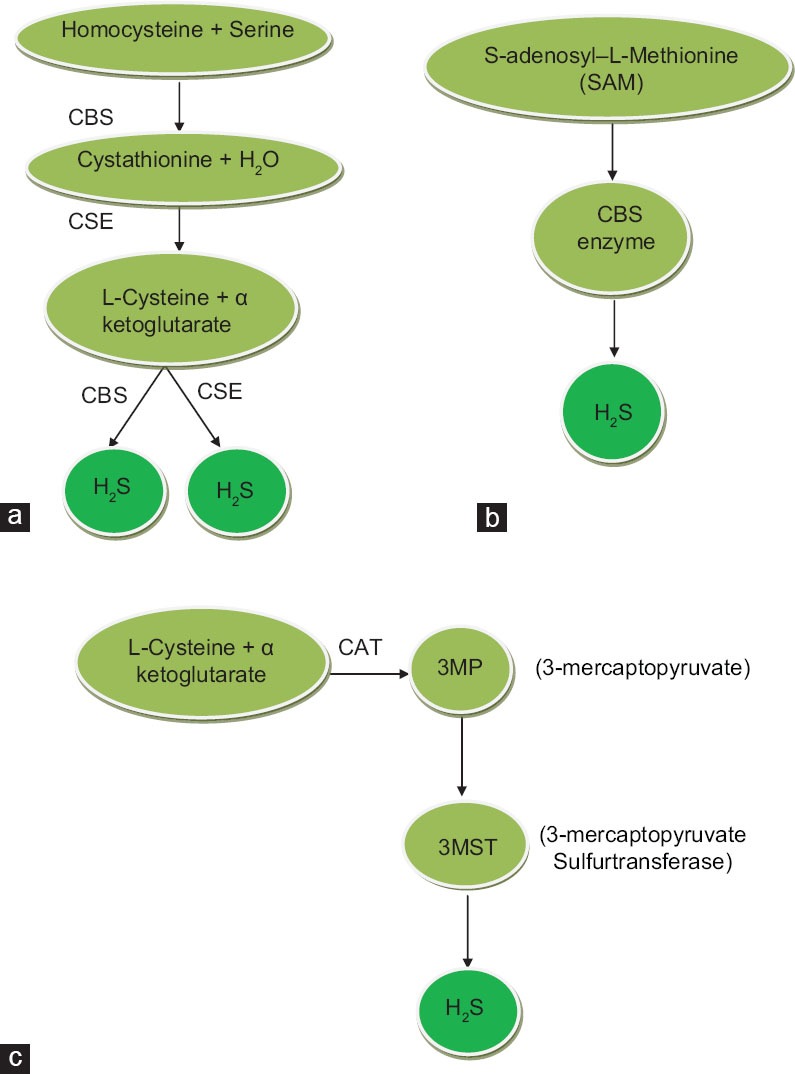
(a-c) The schematic presentation of H2S production in the mammalians body by involving three different pathways
Both the enzymes CSE and CBS are present in mammalian cells and tissues. Previously, CSE has been reported to be responsible for the production of H2S in the cardiovascular system (CVS) and kidneys, whereas CBS performs the same function in CNS.[11] Few studies have supported that CSE is dominant in CVS, whereas CBS is dominant in CNS.[12] Both CBS and CSE have been reported to be present in the kidneys,[5,13] predominantly in predominantly cortical thymoma.[13,14,15] However, the mechanism of its production still remains unclear; whether or not the production of H2S is induced by CSE or CBS. Expression of both enzymes in the kidneys can lead us to conclude about the production of H2S by both enzymes. Recent studies have demonstrated that CSE is also present in endothelial cells of mice.[16] The third enzyme, 3MST, is responsible for H2S production and has also been reported to be present in endothelial cells in the thoracic aorta.[9] Recent studies have shown that greater than 90% of the total H2S is produced in the brain by 3MST.[9] Based on the literature, different enzymes are involved in the production of H2S in different parts of body; for example, H2S is predominantly produced in the heart by CSE,[12] both CBS and CSE in the kidneys,[5,13] and 3MST in the brain by.[9]
DL-propargylglycine (DL-PAG) is an inhibitor of CSE, whereas aminooxyacetic acid is an inhibitor of CBS. Both inhibitors are nonselective and may be responsible for the inhibition of other enzymes. Some selective inhibitors can elaborate the mechanisms of H2S. Some mechanisms for long-term control production of H2S are coming to light. Long-term regulation of H2S seems to be dependent on S-adenosyl-L–methionine, which activates CBS and ultimately leads to the production of H2S[9,10][Figure 1c].
The endogenous concentration of circulating H2S is 50–160 μM in rats, bovines, and humans.[12,17] Tissue level has a greater concentration than circulating level. At physiological concentration, H2S hyperpolarizes the membranes of localized cells, modulates the neuronal excitability, relaxes the smooth muscles, and controls cell apoptosis or proliferation.[12,17,18,19,20,21] In one study,[22] normal concentration of H2S in Wistar Kyoto (WKY) rats was measured at 10 μM; however, other studies[18] have demonstrated that the plasma level of H2S is 50 μM. The tissue level of H2S has been thought to be higher than its plasma level. For example, the physiological concentration of H2S in the brain has been documented at 50–160 μM.[23] Significant changes have been observed in the concentration of H2S because of various diseases. The H2S level decreased below the normal level in the body if coronary heart disease[24] spontaneously hypertensive rats (SHRs);[25] however, the level increased in diabetes and circulatory shock. In carrageenan-induced inflammation model, concentration is increased.[26] Increase in the concentration of H2S has also been observed in acute pancreatitis,[27] hemorrhagic shock,[28] and in endotoxin shock.[29,30] The vasorelaxant effect of H2S has been proven,[18] which shows that H2S relaxes the isolated aorta at a concentration as low as 18 μM and 60 μM if pretreated with 20 mM KCl or PHE.
Role of Hydrogen Sulfide in Hypertension
The role of H2S seems to be partially solved by evaluating different mechanisms; however, researchers have shifted their interest to other gaseous transmitters, such as NO and carbon monoxide. Therefore, the mechanism of H2S in hypertension is not exactly accepted.
Studying the role of H2S in hypertension chronologically, we were first informed[23] that H2S produces vasorelaxation on rat aortic tissue in vitro. In succeeding research, the vasorelaxant effect of H2S has been attributed to the opening of the ATP-sensitive potassium (KATP) channel;[18] this effect was mimicked by pinacidil (a KATP channel agonist) and antagonized by glibenclamide (a KATP channel antagonist). Blood pressure reduction by intravenous (IV) bolus injection of H2S was 12–30 mm of Hg. The study under discussion revealed that aortic rings were relaxed by 63% ± 2.2% at a dose of 180 μM, if pretreated with PHE. The vasorelaxation of H2S was partially dependent on endothelium and mostly through direct effects on the smooth muscle cells. To further exclude the involvement of NO, the vasorelaxant effects of H2S are not blocked by 1H-(1,2,4)oxadiazolo[4,3-a] quinoxalin-1-one (ODQ) (an inhibitor of guanylyl cyclase).
The KATP channel mechanism was confirmed by inducing contractions in the aortic tissue through treating the tissue with low K+ (20 mM) and high K+ (100 mM); vasorelaxation produced by H2S on aortic tissues was observed when pretreated with low K+ (20 mM) and high K+ (100 mM). The maximum vasorelaxation produced by H2S was 90% ± 8.2% and 19% ± 3.9% when pretreated with low K+ (20 mM) and high K+ (100 mM), respectively.
Hydrogen sulfide produces more relaxation with low K+ (20 mM) because of K+ conductance. The K+ channel opener effect was further verified using 10 mM tetraethylammonium and 100 nM of charybdotoxin or 100 nM iberiotoxin (specific inhibitors of Kca channel), completely inhibiting H2S-induced relaxation.
In upcoming years, the vasorelaxation produced by H2S was proven to be through a different mechanism than that of NO and CO.[19] Furthermore, the vasorelaxation effect on the vascular tissue by NO is reduced when pretreated with H2S. However, the presence of NO does not alter the H2S vascular response. This study also suggests that an additive response can be achieved using NO and H2S. A study[31] elaborated that the cardioprotective action of H2S mediated by KATP channel opening is caused by the physiological production of H2S in the heart. This study has proven the negative inotropic effects of H2S both in vivo and in vitro,[32] and concluded that H2S can effectively prevent hypertension in rats when induced by L-NG-nitro arginine methyl ester.
Nitric oxide and CO play important roles in the pathogenesis of hypertension.[33,34] This mystery was solved.[35] In this study, deficiency in NO and CO has been proven to contribute to the pathogenesis of hypertension.[36,37] H2S has also been reported to play the same role in hypertension because of similar biological activities.[12] In subsequent studies, the level of H2S in the plasma and its production rate in seriously hypertensive rats (SHR) was lower than that in WKY rats. Therefore, CSE is a specific enzyme for H2S production in the thoracic aorta and its decreased activity in hypertension may lead to less production of H2S, resulting in decreased circulating level of H2S.[35] The above-mentioned theory proposes that less activity and decreased transcription of CSE results in decreased circulating aortic H2S level and that vasoconstriction is a dominant phenomenon over vasorelaxant one. Sodium hydrogen sulfide (NaHS) has been selected as an exogenous source ofH2S because of to four reasons. (1) Na+ dissociates from HS_ in a NaHS solution, then HS associates with H and produces H2S, regardless if the H2S solution was prepared by bubbling H2S gas or dissolving NaHS. In physiological saline, about one-third of H2S exists in the undissociated form (H2S), and the remaining two-thirds is HS - at equilibrium with H2S. (2) NaHS enables a more accurate and reproducible measurement of H2S concentrations in a solution than by bubbling H2S gas. (3) The influence of 1 mM or less sodium ion on the physiological experiments is negligible. (4) NaHS at concentrations used in the current study does not change the pH of the medium.[23] The results of one study[35] suggest that upregulation of H2S results in the reduction of SBP in the SHR + NaHS group (158.13 ± 12.52 mm vs. 183.57 ± 11.8 mm of Hg). A study[18] demonstrated that an IV bolus injection of H2S at 2.8 μmol/kg and 14 μmol/kg body weight results in a decrease in the mean arterial blood pressure of rats by 12.5 ± 2.1 mmHg and 29.8 ± 7.6 mmHg, respectively. Therefore, the physiological concentration of H2S is responsible for maintaining the mean arterial blood pressure. However, the expression of CSE activity decreases with the onset of disease. Based on the findings,[18] the finding that the physiological concentration of H2S is responsible for the normal function of CVS is a matter of great interest. The concentration of H2S in the body is a predictor of disease. Diseases like hypertension occur when the concentration of H2S decreases; however, the opposite occurs in hemorrhagic shock. In a study,[28] induction of hemorrhagic shock results in a prolonged decrease in the mean arterial pressure (MAP) and heart rate (HR). However, data suggest that vasoconstriction is responsible for a hemorrhagic shock because of vasopressin, noradrenaline, and angiotensin II.[38] A reasonable number of previous studies have shown that excessive formation of inducible nitric oxide synthase (iNOS) in hypotension is responsible for hemorrhagic shock;[38,39] thus, the concentration of H2S increases in the induction of hemorrhagic shock. Treatment with inhibitors of H2S-producing enzymes CSE and DL-PAG, a suicidal inhibitor, resulted in the rapid and partial restoration of MAP and HR. Previously, glibenclamide (a K+ channel blocker) has been proven to perform the same partial but equally effective function after hemorrhagic shock in a rat.[40] A comparative study was conducted[28] to compare the effect of H2S with PAG and β-cyano-L-alanine, a reversible inhibitor of CSE, on the blood pressure of rats subjected to hemorrhagic shock.
After many years of earning good repute among researchers, H2S research started to intermingle with each other. As a result, few discrepancies that need solutions arise. Two different schools of thought exist between interchangeable production of H2S and NO.
In 1997, a study[23] has proven that a low concentration of H2S increases the relaxation of smooth muscles by 13-folds. This study also explained that low concentration (30 μM) of H2S enhances the vasodilator effect of NO. Therefore, a synergistic response between H2S and NO can be the therapeutic outcome in hypertension. This study further elaborated that NO-induced vasorelaxation is specifically for H2S, but NO cannot potentiate the vasorelaxant effect of H2S.

Another study[19] has proven that NO is responsible for the upregulated production of H2S in rats in a dose-dependent manner (1–100 μM), as shown in the following figure.

This study claims that production of H2S by NO is narrated by two mechanisms: (1) NO increases CSE activity, which ultimately leads to the production of H2S, and (2) NO increases the activity of protein kinase, which is dependent on cyclic guanosine monophosphate, thereby increasing CSE protein. Another study[41] also supported the concept.[18] Another study[12] elaborated on the release of H2S by NO.
A growing body of evidence[42] has proven that H2S as a cofactor is responsible for the generation of NO from nitrite.

In his previous studies in 2002,[43] documented the interaction between hydrogen sulfide and NO resulting in the formation of the intermediate compound nitrosothiol, which releases NO through hemolysis. In his next experimentation,[44] proposed the mechanism of NO production from sodium nitroprusside. In 2006,[45] has proven that the interaction between two gasotransmitters results in the formation of nitrosothiol and the release of only a small portion of NO unless an antioxidant is involved. These findings contradict those of other studies,[19,43,44] which demonstrate that the interaction between NO and H2S enhances vasodilatation.
In the battle of the interchangeable production of H2S and NO, another finding cropped up, supporting the claim that H2S is responsible for the direct inhibition of endothelial nitric oxide synthase (eNOS).[46]

Endothelial nitric oxide synthase is responsible for the production of NO in endothelial cells. This theory is born out of the result of the first school of thought, which proposed that NO can potentiate the response of H2S, but NO can do this.[46] has elaborated that H2S is responsible for the inhibition of eNOS, as well as iNOS and nNOS. They further strengthened the results of their study by explaining that H2S and tetrahydrobiopterin (BH4) reverse the H2S inhibition of eNOS and nNOS, but not iNOS.
A study in 2006[47] elaborated that the intermediate complex formed from the interaction between H2S and NO has no vasorelaxant effect; H2S was responsible for the regulation of NO. This study concluded that H2S inhibits the vasorelaxant effect of NO.
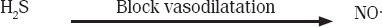
A decrease in H2S concentration results in over stimulation of β adrenoreceptor. H2S inhibits the activity of β adrenoreceptor by inhibiting adenylyl cyclase.[48] The H2S has also been reported to inhibit the L-type Ca++ channels in cardiomyocyte, which leads to a decrease in contractility, thereby revealing the Ca++ channel-blocking mechanism of H2S.[49] In the same year, H2S has been found to have the ability to block the angiotensin-converting enzyme and responsible for additive vasorelaxant response leading to the inhibition of angiotensin II production, ultimately resulting in reduced degradation of bradykinin.
In a related study in 2010,[50] reported that eNOS and neuronal nitric oxide synthase (NOS) activation is important to produce a cardioprotective response. The cardioprotective response of the NOS family mentioned in[50,51] is contrary to the results by.[46]
Another area of interest is the formation of the intermediate complex nitrosothiol, which is responsible for the vasorelaxant effect in the blood vessels through interaction between two gasotransmitters, H2S and NO.[23,45] The presence of this intermediate molecule, nitrosothiol, has not been fully confirmed and has been considered as a possible molecule for the said action. A previously mentioned study[47] has stated that the intermediate molecule is nitrosothiol but has no vasorelaxant effect in vivo and in vitro. Another school of thought[50] has reported that the interaction between NO and H2S leads to the formation of the nitroxyl group; nitroxyl has been reported to be involved in positive inotropic and vasodilation activities.[52] These results support the evidence that HNO/NO− produce positive inotropic and lusitropic effects (independent of β-adrenergic stimulation) in a failing heart.[31]
Most recently, a group of researchers studied the role of H2S in hypertension along with diabetes[53] and emphasized that H2S improves the renal blood flow by reducing renal vasculature resistance through vasodilation. The same group of researchers extended their study to investigate the role of H2S in cardiac protection in dihydroxycortisone acetate-induced hypertension and SHR combined with diabetes.[54] They showed the cardiac protection role of H2S by reducing HR and vasodilation.
Future Prospects
In view of the above-mentioned literature review, many things remain to be explored and many questions still to be answered. One of the major ambiguities is the interchangeable production of H2S and NO.

Future studies must verify whether the intermediate molecule formed as a result of interaction between NO and H2S is nitroxyl or nitrosthiol or both. If both molecules are present; then, which one is responsible for the vasorelaxant effect?
Concentration of H2S decreased in hypertension. Is it a cause or consequence of the disease?
H2S acts by vasodilatation, so α blocking effect can be investigated
BH4 and H2S effects in hypertension can be studied
The molecular mechanism of H2S can be studied using reverse transcription polymerase chain reaction
Some selective inhibitors can be introduced to avoid inhibition of unwanted enzymes
Releasers of H2S introduced as present releasers are inconsistent and have sustained-release property.
Footnotes
Source of Support: AA is a recipient of an USM fellowship (Teaching) (No. PF-D 0067/11 (R) from the Institute of Postgraduate Students, Universiti Sains Malaysia and is thankfully acknowledged. The Institute of Postgraduate Studies (IPS) is acknowledged for providing USM fellowship (Teaching) to Ashfaq Ahmad (No. PF-D 0067/11 (R)) and Universiti Sains Malaysia and Ministry of Science Technology & Innovation (MOSTI) Malaysia for providing grant no. 203/PFARMASI/6711452 to Dr. Hassaan A. Rathore for this work.
Conflict of Interest: No.
References
- 1.Hydrogen sulfide. Baltimore, MD: University Park Press; 1979. US National Research Council, Subcommittee on Hydrogen Sulfide, Division of Medical Sciences. [Google Scholar]
- 2.Beauchamp RO, Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- 3.Guidotti TL. Hydrogen sulphide. Occup Med (Lond) 1996;46:367–71. doi: 10.1093/occmed/46.5.367. [DOI] [PubMed] [Google Scholar]
- 4.Warenycia MW, Goodwin LR, Benishin CG, Reiffenstein RJ, Francom DM, Taylor JD, et al. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem Pharmacol. 1989;38:973–81. doi: 10.1016/0006-2952(89)90288-8. [DOI] [PubMed] [Google Scholar]
- 5.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–77. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaroop M, Bradley K, Ohura T, Tahara T, Roper MD, Rosenberg LE, et al. Rat cystathionine beta-synthase. Gene organization and alternative splicing. J Biol Chem. 1992;267:11455–61. [PubMed] [Google Scholar]
- 7.Akagi R. Purification and characterization of cysteine aminotransferase from rat liver cytosol. Acta Med Okayama. 1982;36:187–97. doi: 10.18926/AMO/30697. [DOI] [PubMed] [Google Scholar]
- 8.Ubuka T, Umemura S, Yuasa S, Kinuta M, Watanabe K. Purification and characterization of mitochondrial cysteine aminotransferase from rat liver. Physiol Chem Phys. 1978;10:483–500. [PubMed] [Google Scholar]
- 9.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–14. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 10.Kuo SM, Lea TC, Stipanuk MH. Developmental pattern, tissue distribution, and subcellular distribution of cysteine: Alpha-ketoglutarate aminotransferase and 3-mercaptopyruvate sulfurtransferase activities in the rat. Biol Neonate. 1983;43:23–32. doi: 10.1159/000241634. [DOI] [PubMed] [Google Scholar]
- 11.Moore PK, Bhatia M, Moochhala S. Hydrogen sulfide: From the smell of the past to the mediator of the future? Trends Pharmacol Sci. 2003;24:609–11. doi: 10.1016/j.tips.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Wang R. Two's company, three's a crowd: Can H 2 S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–8. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 13.House JD, Brosnan ME, Brosnan JT. Characterization of homocysteine metabolism in the rat kidney. Biochem J. 1997;328:287–92. doi: 10.1042/bj3280287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, et al. Murine cystathionine gamma-lyase: Complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J. 2004;381:113–23. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li N, Chen L, Muh RW, Li PL. Hyperhomocysteinemia associated with decreased renal transsulfuration activity in Dahl S rats. Hypertension. 2006;47:1094–100. doi: 10.1161/01.HYP.0000219634.83928.6e. [DOI] [PubMed] [Google Scholar]
- 16.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H 2 S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–90. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- 18.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H 2 S as a novel endogenous gaseous K (ATP) channel opener. EMBO J. 2001;20:6008–16. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao W, Ndisang JF, Wang R. Modulation of endogenous production of H 2 S in rat tissues. Can J Physiol Pharmacol. 2003;81:848–53. doi: 10.1139/y03-077. [DOI] [PubMed] [Google Scholar]
- 20.Yang G, Sun X, Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004;18:1782–4. doi: 10.1096/fj.04-2279fje. [DOI] [PubMed] [Google Scholar]
- 21.Siegel LM. A direct microdetermination for sulfide. Anal Biochem. 1965;11:126–32. doi: 10.1016/0003-2697(65)90051-5. [DOI] [PubMed] [Google Scholar]
- 22.Mason J, Cardin CJ, Dennehy A. The role of sulphide and sulphide oxidation in the copper molybdenum antagonism in rats and guinea pigs. Res Vet Sci. 1978;24:104–8. [PubMed] [Google Scholar]
- 23.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–31. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 24.Jiang HL, Wu HC, Li ZL, Geng B, Tang CS. Changes of the new gaseous transmitter H 2 S in patients with coronary heart disease. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:951–4. [PubMed] [Google Scholar]
- 25.Du J, Yan H, Tang C. Endogenous H 2 S is involved in the development of spontaneous hypertension. Beijing Da Xue Xue Bao. 2003;35:102. [PubMed] [Google Scholar]
- 26.Bhatia M, Sidhapuriwala J, Moochhala SM, Moore PK. Hydrogen sulphide is a mediator of carrageenan-induced hindpaw oedema in the rat. Br J Pharmacol. 2005;145:141–4. doi: 10.1038/sj.bjp.0706186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM, Moore PK. Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 2005;19:623–5. doi: 10.1096/fj.04-3023fje. [DOI] [PubMed] [Google Scholar]
- 28.Mok YY, Atan MS, Yoke Ping C, Zhong Jing W, Bhatia M, Moochhala S, et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: Protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol. 2004;143:881–9. doi: 10.1038/sj.bjp.0706014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collin M, Anuar FB, Murch O, Bhatia M, Moore PK, Thiemermann C. Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br J Pharmacol. 2005;146:498–505. doi: 10.1038/sj.bjp.0706367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–8. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 31.Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM, et al. Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: Independence from beta-adrenergic signaling. Proc Natl Acad Sci U S A. 2003;100:5537–42. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens. 2003;21:1879–85. doi: 10.1097/00004872-200310000-00015. [DOI] [PubMed] [Google Scholar]
- 33.Raij L. Workshop: Hypertension and cardiovascular risk factors: Role of the angiotensin II-nitric oxide interaction. Hypertension. 2001;37:767–73. doi: 10.1161/01.hyp.37.2.767. [DOI] [PubMed] [Google Scholar]
- 34.Sabaawy HE, Zhang F, Nguyen X, ElHosseiny A, Nasjletti A, Schwartzman M, et al. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension. 2001;38:210–5. doi: 10.1161/01.hyp.38.2.210. [DOI] [PubMed] [Google Scholar]
- 35.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–7. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 36.Lyons D. Impairment and restoration of nitric oxide-dependent vasodilation in cardiovascular disease. Int J Cardiol. 1997;62(Suppl 2):S101–9. doi: 10.1016/s0167-5273(97)00247-7. [DOI] [PubMed] [Google Scholar]
- 37.Ndisang JF, Zhao W, Wang R. Selective regulation of blood pressure by heme oxygenase-1 in hypertension. Hypertension. 2002;40:315–21. doi: 10.1161/01.hyp.0000028488.71068.16. [DOI] [PubMed] [Google Scholar]
- 38.Thiemermann C, Szabó C, Mitchell JA, Vane JR. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci U S A. 1993;90:267–71. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ungureanu-Longrois D, Balligand JL, Kelly RA, Smith TW. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: Role of a cytokine inducible nitric oxide synthase in cardiac myocytes. J Mol Cell Cardiol. 1995;17:155–67. doi: 10.1016/s0022-2828(08)80015-6. [DOI] [PubMed] [Google Scholar]
- 40.Salzman AL, Vromen A, Denenberg A, Szabó C. K(ATP)-channel inhibition improves hemodynamics and cellular energetics in hemorrhagic shock. Am J Physiol. 1997;272:H688–94. doi: 10.1152/ajpheart.1997.272.2.H688. [DOI] [PubMed] [Google Scholar]
- 41.Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: Gaseous messengers in cerebrovascular circulation. J Appl Physiol. 2006;100:1065–76. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grossi L. Hydrogen sulfide induces nitric oxide release from nitrite. Bioorg Med Chem Lett. 2009;19:6092–4. doi: 10.1016/j.bmcl.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 43.Grossi L, Montevecchi PC. A kinetic study of S-nitrosothiol decomposition. Chem Eur J. 2002;8(Suppl 2):380–7. doi: 10.1002/1521-3765(20020118)8:2<380::AID-CHEM380>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 44.Grossi L, d’Angelo SJ. Sodium nitroprusside: Mechanism of no release mediated by sulfhydryl-containing molecules. Med Chem. 2005;48:2622–6. doi: 10.1021/jm049857n. [DOI] [PubMed] [Google Scholar]
- 45.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun. 2006;343:303–10. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 46.Kubo S, Doe I, Kurokawa Y, Nishikawa H, Kawabata A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: Contribution to dual modulation of vascular tension. Toxicology. 2007;232:138–46. doi: 10.1016/j.tox.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 47.Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol. 2006;149:625–34. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yong QC, Pan TT, Hu LF, Bian JS. Negative regulation of beta-adrenergic function by hydrogen sulphide in the rat hearts. J Mol Cell Cardiol. 2008;44:701–10. doi: 10.1016/j.yjmcc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. 2008;79:632–41. doi: 10.1093/cvr/cvn140. [DOI] [PubMed] [Google Scholar]
- 50.Yong QC, Hu LF, Wang S, Huang D, Bian JS. Hydrogen sulfide interacts with nitric oxide in the heart: Possible involvement of nitroxyl. Cardiovasc Res. 2010;88:482–91. doi: 10.1093/cvr/cvq248. [DOI] [PubMed] [Google Scholar]
- 51.Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–96. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Nitroxyl (HNO): The Cinderella of the nitric oxide story. Trends Pharmacol Sci. 2008;29:601–8. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Hassaan AR, Din F, Mohammed HA, Munavvar AS, Norazizan A, Edward JJ. Hydrogen sulfide improves the renal function in a combined state of hypertension and diabetes. FASEB J. 2012;26:1131–212. [Google Scholar]
- 54.Hassaan AR, Din F, Mohammad HA, Ayaz AK, Munavvar AS, Norazizan A, et al. Effect of exogenously administered hydrogen sulfide on cardiac and renal function in hypertensive diabetic rats. FASEB J. 2012;26:876–7. [Google Scholar]