

Abstract
The importance of factor Xa generation in thrombus formation has not been studied extensively so far. Here, we used mice deficient in either factor VIII or factor IX to determine the role of platelet-stimulated tenase activity in the formation of platelet-fibrin thrombi on collagen. With tissue factor present, deficiency in factor VIII or IX markedly suppressed thrombus growth, fibrin formation and platelet procoagulant activity (phosphatidylserine exposure). In either case, residual fibrin formation was eliminated in the absence of tissue factor. Effects of factor deficiencies were antagonized by supplementation of the missing coagulation factor. In wild-type thrombi generated under flow, phosphatidylserine-exposing platelets bound (activated) factor IX and factor X, whereas factor VIII preferentially co-localized at sites of von Willebrand factor binding. Furthermore, proteolytic activity of the generated activated factor X and thrombin was confined to the sites of phosphatidylserine exposure. With blood from a hemophilia A or B patient, the formation of platelet-fibrin thrombi was greatly delayed and reduced, even in the presence of high concentrations of tissue factor. A direct activated factor X inhibitor, rivaroxaban, added to human blood, suppressed both thrombin and fibrin formation. Together, these data point to a potent enforcement loop in thrombus formation due to factor X activation, subsequent thrombin and fibrin generation, causing activated factor X-mediated stimulation of platelet phosphatidylserine exposure. This implies that the factor VIII/factor IX-dependent stimulation of platelet procoagulant activity is a limiting factor for fibrin formation under flow conditions, even at high tissue factor concentrations.
Introduction
Platelet thrombus formation is a highly dynamic process that is tightly controlled by the rheological conditions and the activity of blood components at the vascular surface. The formation of a stable thrombus relies on interactions between activated platelets on the one hand, and the coagulation system generating thrombin and fibrin on the other hand.1–3 In vivo mouse models have shown that fibrin formation occurs hand-in-hand with platelet activation in the arterial and venous parts of the circulation.4–6 Furthermore, in vivo and in vitro flow studies have indicated that both the extrinsic coagulation pathway – triggered via tissue factor and factor (F) VIIa –7–9 and the intrinsic pathway – triggered via FXII and resulting in FXI and FIX activation –10,11 contribute to the formation of a stable thrombus. Tissue factor becomes exposed after vascular damage,12 but can also be recruited to a growing thrombus via leukocytes.13 Taken together, current evidence indicates that dual triggering of the coagulation system is required to generate sufficient amounts of thrombin and fibrin to stabilize the growing thrombus. It is recognized that thrombin and fibrin are generated in a resonance loop with platelet activation via the prothrombinase complex.14,15 However, the precise mechanism of FX activation via the tenase complex is not well understood.
Upon stimulation with strong agonists such as collagen and thrombin, platelets expose phosphatidylserine at their surface, as a consequence of Ca2+-dependent activation of phospholipid scrambling via anoctamin 6 (TMEM16F).16,17 Exposure of phosphatidylserine stimulates the procoagulant activity of platelets, by enhancing the binding and activation of coagulation factors at the platelet surface.1,2 A physiological role of phosphatidylserine exposure is evident from the observation that chelation of phosphatidylserine by annexin A5 or a genetic deficiency in anoctamin 6 greatly impairs the formation of thrombi in mouse models in vivo.4,16 Earlier studies showed that phosphatidylserine-exposing platelets stimulate prothrombinase activity, by binding FVa, FXa and prothrombin, resulting in the cleavage of prothrombin into thrombin.9,15 It has also been demonstrated that phosphatidylserine exposure is a driving force for the formation of fibrin networks on the platelet surface.18 In analogy, it has frequently been suggested,19–21 but not directly proven, that platelet phosphatidylserine exposure regulates the tenase complex, i.e. the binding of FVIIIa and FIXa to cleave FX into FXa. This is a relevant issue, since FX can also be activated by a direct interaction with the tissue factor/FVIIa complex.21
Deficiency of FVIII or FIX in humans with hemophilia A or B, respectively, causes a mild to severe bleeding disorder. Early work, particularly using animal models, indicated that in these forms of hemophilia initial platelet plug formation (primary hemostasis) is unaffected, whereas ensuing fibrin formation (secondary hemostasis) is markedly diminished.22–24 This can be explored in test-tube experiments, suggesting that certain rates of FXa as well as thrombin generation are needed to produce a stable hemostatic platelet-fibrin clot, and that both FVIII and FIX are required for sufficient levels of FXa generation.25–27 Indeed, in (treated) patients with hemophilia, residual thrombin generation appears to be linked to the remaining levels of FVIII or FIX.28 Mice deficient in FVIII have appeared to be valuable models for hemophilia A, since they show prolonged bleeding times and delayed thrombus formation under flow in vivo.29–32 Similarly, mice deficient in FIX exhibit a bleeding phenotype and show reduced thrombus formation in vivo.33–35
In the present study, we aimed to establish the role of FX activation (tenase) via FVIII and FIX in the regulation of a stable, fibrin-platelet thrombus under flow conditions. In order to do so, we measured this process using blood from F8−/− and F9−/− mice lacking these factors, and determined the binding of these on the thrombus. The data point to a prominent feed-forward role of locally generated FXa not only in fibrin-clot formation, but also in enhancement of platelet phosphatidylserine exposure.
Methods
Please see the Online Supplementary Methods for additional information.
Animals
Experiments were approved by the local animal experimental and care committee. F8−/− and F9−/− mice and matched wild-type mice came from the Jackson Laboratory and Charles River, respectively. All mice were bred on a C57Bl/6 background. Blood was obtained by aortic puncture under anesthesia (ketamine/xylazine; Eurovet) and collected into 1/10 volume of 129 mM trisodium citrate, as described elsewhere.36
Human blood preparation
Blood was taken from healthy volunteers, a patient with hemophilia A (4% FVIII) and a patient with hemophilia B (5% FIX) after their full informed consent, in accordance with the Declaration of Helsinki. Blood samples were collected by puncture of the median cubital vein into 1/10 volume of 129 mM trisodium citrate.15 In both patients, the levels of other coagulation factors were in the normal range.
Whole blood flow chamber experiments
Flow experiments were performed with a parallel-plate transparent flow chamber, containing a coverslip coated with spots of type I Horm collagen (Nycomed Pharma), blocked with Hepes buffer pH 7.45 (137 mM NaCl, 5 mM Hepes, 2.7 mM KCl, 2 mM MgCl2, and 0.1% glucose), containing 1% bovine serum albumin.37 Samples of citrate-anticoagulated blood (400–1500 μL) were made to flow at defined wall-shear rate, as described for murine38 and human15 blood. Coagulation was induced by co-perfusion of recalcification buffer using a dual syringe pump system (1:1 mouse: 110 mM NaCl, 13.3 mM CaCl2, 6.7 mM MgCl2, 0.1% glucose and 0.1% bovine serum albumin; 1:10 human: 10 mM Hepes, 136 mM NaCl, 2.7 mM KCl, 6.3 mM CaCl2, 3.15 mM MgCl2, 0.1% glucose and 0.1% bovine serum albumin). Where indicated, blood samples were supplemented with tissue factor (Innovin, 0.1–100 pM; Dade Behring), recombinant human FVIII (2 U/mL), recombinant human FIX (2 U/mL) or rivaroxaban (0.4 or 4 μg/mL, all f.c.). Fluorescent probes, OG488-fibrinogen or AF647-fibrinogen (16 μg/mL, f.c.), were added to the blood prior to perfusion. To detect phosphatidylserine exposure, thrombi on coverslips were post-stained with 0.5 μg/mL FITC-annexin A5. Coagulation factors were detected by post-staining with AF647-labeled antibodies against FVIII(a) or FIX(a) (1:20); control staining was performed with irrelevant AF647-labeled IgG at the same dilution. Where indicated, von Willebrand factor (VWF) was stained with FITC-labeled anti-VWF antibody (20 μg/mL), and FXa binding sites were identified with AF647-labeled FXa (16.5 μg/mL). Randomly captured images were analyzed for surface area coverage of thrombi and fluorescent labels using Metamorph software (Molecular Devices).
Real-time measurement of thrombin and activated factor X generation in flow chambers
To measure the activities of serine proteases, blood samples were pre-incubated with the fluorogenic thrombin substrate Z-GGR-AMC or FXa substrate Pefafluor, each 0.5 mM. Thrombi were generated as above on a collagen surface, after which fluorescence accumulation from cleaved substrate was recorded under stasis, by capturing microscopic images every 30 seconds.15 Regions of interest with or without thrombi images were analyzed for fluorescence intensity changes with ImageJ software (open source).
Statistical analysis
The statistical significance of differences between groups was determined with the independent samples t-test, using the Statistical Package for Social Sciences (SPSS 11.0). Data are expressed as mean ± standard error of mean. P values < 0.05 are considered statistically significant.
Results
Key roles of murine factors VIII and IX in the formation of platelet-fibrin thrombi and platelet procoagulant activity in the presence of tissue factor
The formation of platelet-fibrin thrombi, triggered via the intrinsic or extrinsic pathway, can be studied efficiently in vitro by perfusion of murine or human blood over a collagen surface.15,38 In order to investigate the role of FVIII in this process, we used citrate-anticoagulated blood from F8−/−or F8+/+ mice and made this flow during recalcification over collagen at an intermediate wall-shear rate (1000 s−1). The blood samples were supplemented with a surplus of 10 pM tissue factor to stimulate the extrinsic pathway. With blood from wild-type F8+/+ mice, this resulted in the formation of large semi-occlusive thrombi after 4 min of perfusion (Figure 1A). The thrombi were composed of aggregated platelets, large patches of phosphatidylserine-exposing platelets (annexin A5 staining) and multiple fibrin fibers (fibrinogen staining). However, blood from F8−/− mice gave only small-sized platelet thrombi with substantially fewer phosphatidylserine-exposing platelets and limited formation of fibrin (Figure 1A,B). As a consequence, the surface area coverage of F8−/− platelet-fibrin thrombi was reduced by 64%, when compared to the F8+/+ control condition (P=0.002). Fluorescent staining demonstrated a 59% reduction in phosphatidylserine-exposing platelets (annexin A5) and a 92% reduction in fibrin(ogen) accumulation (P<0.05).
Figure 1.
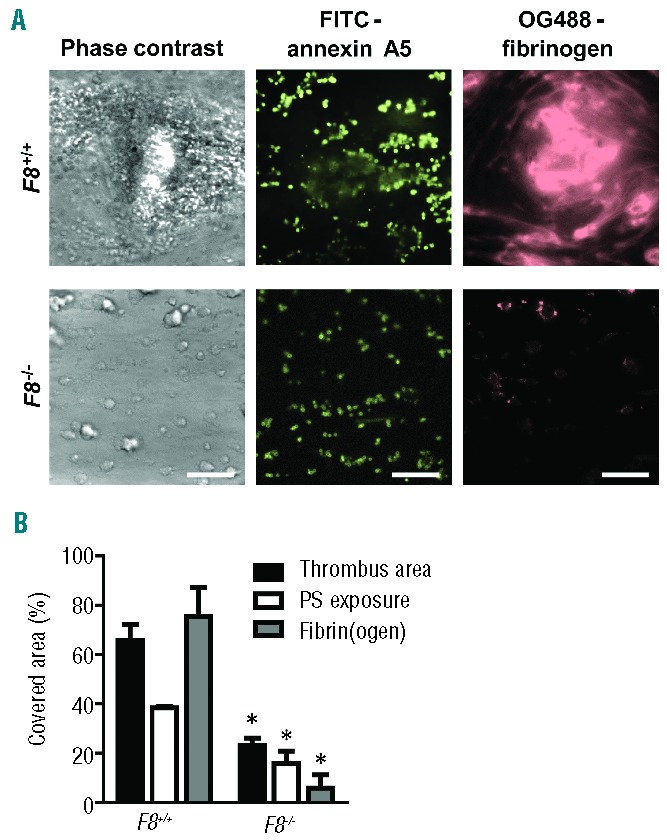
Role of murine FVIII in the formation of platelet-fibrin thrombi and procoagulant activity in the presence of tissue factor. Blood from wild-type mice or mice deficient in FVIII was perfused over collagen at a rate of shear 1000 s−1 under coagulant conditions. Blood samples were pre-incubated with tissue factor (10 pM) and OG488-fibrinogen. Thrombi were post-stained with FITC-annexin A5 to detect phosphatidylserine (PS) exposure. (A) Representative microscopic phase contrast and fluorescence images after 4 min of flow (bars, 50 μm). (B) Surface area covered by platelet-fibrin thrombi (black bars), PS-exposing platelets (white bars) and fibrin(ogen) (gray bars) as determined by fluorescence studies. Mean ± SEM; n = 8–15; *P<0.05 vs. wild-type.
A similar set of flow experiments over collagen was performed with blood from F9−/− and corresponding F9+/+ mice, again in the presence of tissue factor. Deficiency in FIX also resulted in a major reduction of platelet-fibrin thrombi, as well as decreased phosphatidylserine exposure and fibrin formation (Figure 2A,B). In the case of F9−/− blood, the surface area covered with thrombi and phosphatidylserine-exposing platelets decreased by 43% and 40%, respectively, compared to the coverage when wild-type blood was used. Interestingly, this reduction was less severe than that observed with FVIII-deficient blood. Regarding thrombus size (surface area coverage) and fibrin formation, the difference between F8−/− and F9−/− blood was statistically significant (P<0.005 and P<0.001, respectively). The F8 and F9 wild-type controls did not significantly differ in these parameters. Taken together, these results suggest largely overlapping functions of FVIII and FIX, but a more stringent role of FVIII, in the formation of platelet-fibrin thrombi and in the exposure of phosphatidylserine, under conditions in which exogenous tissue factor is present.
Figure 2.
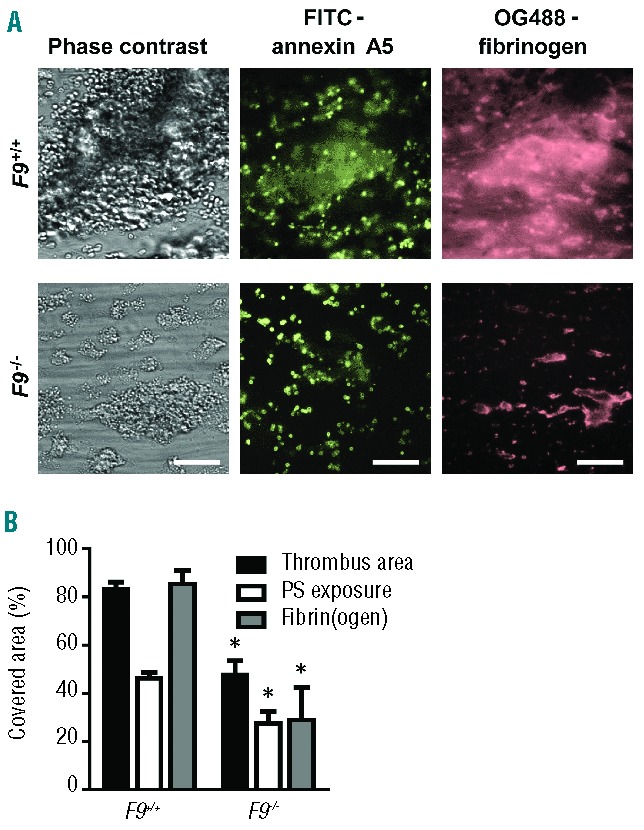
Role of murine FIX in the formation of platelet-fibrin thrombi and procoagulant activity in the presence of tissue factor. Blood from wild-type mice and mice deficient in FIX was perfused over collagen at a rate of shear 1000 s−1 under coagulant conditions. Blood samples were pre-incubated with tissue factor (10 pM) and OG488-fibrinogen. Thrombi were then stained with FITC-annexin A5, as in Figure 1. (A) Representative microscopic phase contrast and fluorescence images (bars, 50 μm). (B) Surface area covered by platelet-fibrin thrombi (black bars), PS-exposing platelets (white bars), and fibrin(ogen) (grey bars) as determined by fluorescence studies. Mean ± SEM; n = 3–6; *P< 0.05 vs. wild-type.
Maintained roles of murine factors VIII and IX in the formation of platelet-fibrin thrombi without tissue factor
Subsequent flow studies were carried out in the absence of tissue factor to eliminate the contribution of FX activation via tissue factor/FVIIa. We had previously established that in this condition the coagulation process is triggered via collagen-dependent FXII/FXI activation.38 Flow of wild-type blood resulted in a moderately delayed, but steady formation of platelet-fibrin thrombi, again containing patches of phosphatidylserine-exposing platelets and fibrin fibers (Figure 3B,C). In this condition, flow of F8−/− or F9−/− blood (again at a shear rate of 1000 s−1) gave small aggregates still containing procoagulant platelets, whereas fibrin fibers were absent (Figure 3A). More specifically, with blood from F8−/− mice, the surface area covered by thrombi was reduced by 53%, while phosphatidylserine-exposing platelets were reduced by 37% compared to the wild-type condition (P<0.001 and P<0.015) (Figure 3B,C). Similar but less stringent reductions were observed with blood from F9−/− mice. Addition of corn trypsin inhibitor (20 μg/mL), to suppress FXII activation, resulted in a further, non-significant reduction of ~20% and 40% in thrombus size for F8−/− and F9−/− blood, respectively (P>0.10).
Figure 3.
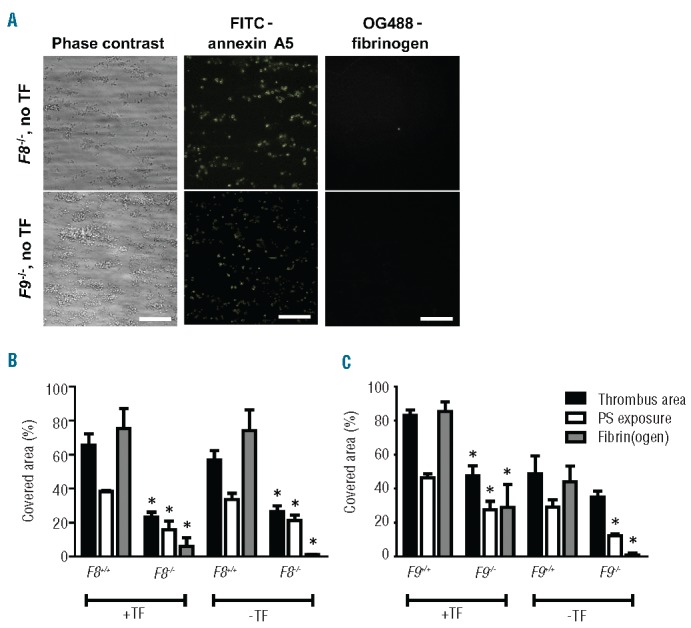
Retained roles of FVIII and FIX in the formation of platelet-fibrin thrombi and procoagulant activity without tissue factor. Blood from mice deficient in FVIII or FIX, or from corresponding wild-type mice was perfused over collagen under coagulant conditions. Where indicated, blood samples were pre-incubated with tissue factor (TF, 10 pM) and OG488-fibrinogen. Thrombi were then stained with FITC-annexin A5 as in Figure 1. (A) Representative microscopic images after 4 min of flow of F8−/− or F9−/− blood in the absence of TF (bars, 50 μm). (B) Effect of TF on parameters of thrombus formation in F8+/+ and F8−/− mice. (C) Effect of TF on parameters of thrombus formation in F9+/+ and F9−/− mice. Mean ± SEM; n = 3–7; *P<0.05 vs. corresponding wild-type.
Improved formation of platelet-fibrin thrombi and platelet procoagulant activity by addition of recombinant human factor VIII or IX
To confirm that the impaired thrombus formation with knockout mice was caused by deficiency in FVIII or FIX, blood samples from F8−/− or F9−/− mice were treated with recombinant human FVIII or FIX, respectively. Supplementing blood from F8−/− mice with recombinant human FVIII resulted in normalization of the clot-formation process and a significant increase in phosphatidylserine-exposing platelets. Similarly, addition of recombinant human FIX to F9−/− blood resulted in the formation of large platelet-fibrin clots, which stained positively with FITC-annexin A5 (Figure 4A,B).
Figure 4.
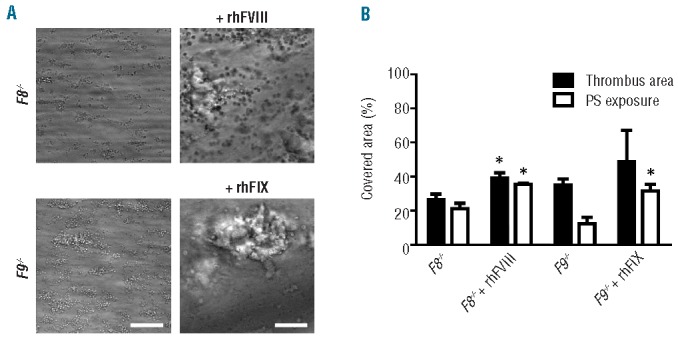
Restoration of thrombus formation in factor-deficient mice by supplementation of factor VIII or IX. Blood from F8−/− or F9−/− mice was pre-incubated for 10 min with recombinant human FVIII or FIX (2 U/mL), respectively. Samples were perfused for 4 min over collagen at a shear rate of 1000 s−1, as described for Figure 1. Thrombi were then stained with FITC-annexin A5. (A) Representative microscopic phase contrast images (bars, 50 μm). (B) Surface area covered by platelet-fibrin thrombi and PS-exposing platelets. Mean ± SEM; n = 3.
Tenase complex formation and activity on platelet-fibrin thrombi
To determine the localization of the tenase components on the murine thrombi, we first performed staining with fluorescently labeled antibodies against FVIII(a) or FIX(a). Before washing, the fluorescence of each antibody was present around the formed thrombi (Figure 5A). However, after washing, the staining for FIX(a) was exclusively detected at single platelets in a similar pattern to that seen with labeled annexin A5 staining phosphatidylserine, whereas staining for FVIII(a) was in part concentrated at fibrillar structures (Figure 5B,C). In agreement with previous results,15 staining of thrombi with the tenase product, labeled FXa, resulted in near complete co-localization with annexin A5-positive platelets. Control experiments with labeled irrelevant antibodies did not result in appreciable fluorescent staining (Figure 5D).
Figure 5.
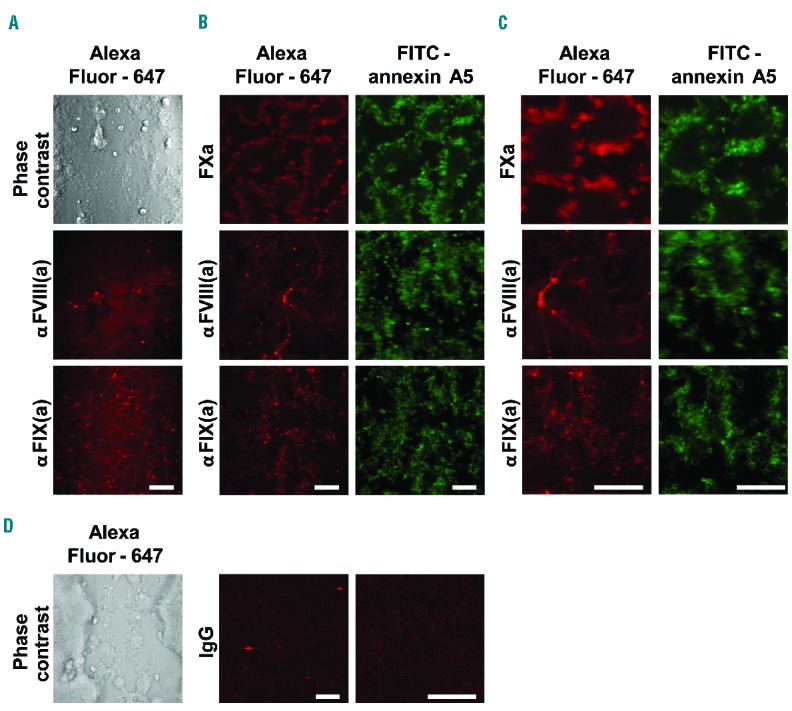
Localization of (activated) FVIII, FIX and FX on murine platelet-fibrin thrombi. Blood from wild-type mice was perfused over collagen at a shear rate of 1000 s−1 under coagulant conditions, as in Figure 1. Thrombi were then stained with FITC-annexin A5 and AF647-FXa, or AF647-labeled antibodies against FIX(a) or FVIII(a), as indicated. Representative microscopic phase contrast and fluorescence images are shown. (A) Direct antibody labeling of thrombi without rinsing. (B) Labeling of thrombi after rinsing. (C) Higher magnification images. (D) Labeling of thrombi with irrelevant AF647-antibody (bars, 20 μm).
The concentrated staining for FVIII(a) seemed to coincide with earlier staining patterns for VWF, binding to collagen fibers.39 To investigate this further, we compared the localization of FVIII(a) with VWF and fibrinogen by triple color high-resolution confocal microscopy using fluorescently labeled antibodies against FVIII(a) and VWF as well as labeled fibrinogen (Online Supplementary Figure S1). Image inspection and overlap analysis indicated marked co-localization of FVIII(a) with VWF in part on collagen fibers and furthermore on platelet thrombi in a similar labeling pattern as fibrin(ogen). Together, this points to FVIII(a) bound to VWF as a reservoir for tenase complex formation, in contrast to FIX(a) which seems to be bound in particular to phosphatidylserine-exposing platelets in a thrombus.
To assess the main sites of tenase activity further, murine blood was pre-incubated with fluorescence peptide substrates for thrombin or FXa and, once platelet-fibrin thrombi had been formed, fluorescence microscopic images were taken to determine the generation of thrombin and FXa. Image analysis under stasis indicated consistent activity of both thrombin (Figure 6A) and FXa (Figure 6B), occurring with highly similar kinetics, and being most prominent at areas where platelet thrombi were present. With either substrate, areas in between thrombi showed limited accumulation of fluorescence, most likely due to substrate diffusion from neighboring areas. No fluorescence was seen at microscopic spots where no platelets (no collagen) were present (data not shown).
Figure 6.
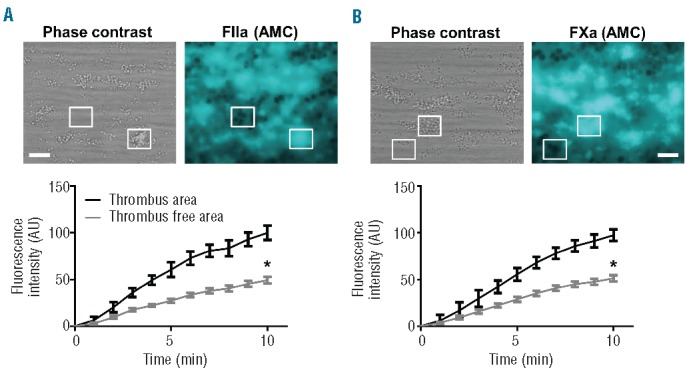
Generation of FXa and thrombin on preformed thrombi. Blood from wild-type mice was pre-incubated with (A) thrombin substrate peptide Z-GGR-AMC or (B) FXa substrate peptide Pefafluor, and perfused over collagen at a shear rate of 1000 s−1, as in Figure 1. After 4 min of thrombus formation, a time-series of microscopic fluorescence images was taken under stasis. Representative phase contrast and fluorescence images after 10 min (bars, 20 μm); and quantification of fluorescence intensity over time of regions-of-interest containing or not containing thrombi. Mean ± SEM; n = 8–12, *P<0.05 vs. thrombi.
Roles of human intrinsic and extrinsic coagulation pathways in the formation of platelet-fibrin thrombi under flow
Also with human blood, in the absence of added tissue factor, fibrin clot formation on collagen under flow is known to be initiated by the intrinsic coagulation pathway.38 To determine the role of the extrinsic pathway herein, we performed perfusion experiments over collagen using whole blood with a range of tissue factor concentrations. Notably, platelet deposition over time was not affected by increasing the tissue factor concentration (Figure 7A), indicating that this primary process is thrombin independent. However, the extent of fibrin formation was significantly enhanced after 4 minutes of perfusion with 1–100 pM tissue factor (Figure 7B). In agreement with this, time to first fibrin formation decreased dose-dependently with 1, 10 and 100 pM of tissue factor (P<0.05, P<0.001 and P<0.0001, respectively) (Figure 7C).
Figure 7.
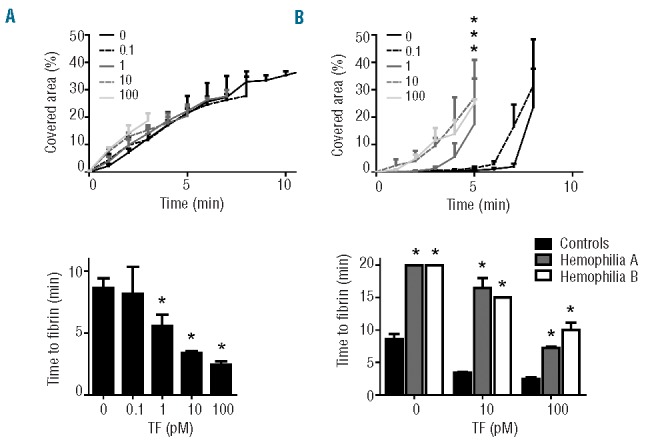
Maintained defective formation of platelet-fibrin thrombi with hemophilic blood at high tissue factor concentrations. Blood from normal control donors or a patient with hemophilia A (HA) or hemophilia B (HB) was perfused over collagen at a shear rate of 1000 s−1 for up to 20 min. Blood samples were pre-incubated with DiOC6 to label platelets, AF647-fibrinogen and different concentrations of tissue factor (TF, 0–100 pM), as indicated. (A, B) Effect of TF concentration on surface area covered by (A) platelets and (B) fibrin(ogen) with normal blood samples. (C) Effect of TF concentration on time to first fibrin formation. (D) Prolonged time to first fibrin formation with blood from a HA or HB patient. Mean ± SEM; n = 4–6; *P<0.05 compared to normal controls.
To assess the amplification role of FVIII and FIX at different trigger strengths, we perfused blood samples from patients with hemophilia A (4% FVIII) or hemophilia B (5% FIX) in the presence of increasing doses of tissue factor. With blood from either patient, no fibrin was formed in the absence of tissue factor over a period of 20 minutes, in comparison to the lag time of 8.6±1.9 minutes for blood from control subjects (P<0.0001). Strikingly, with blood samples from both hemophilic patients, time to fibrin formation shortened at 10 and 100 pM tissue factor, but still remained delayed compared to that of control blood (Figure 5D). These results indicate that the direct pathway of tissue factor/FVII(a)-induced coagulation can contribute to platelet-fibrin thrombus formation under flow, and that both FVIII and FIX enhance this process even at considerably high levels of tissue factor.
Finally, the sensitivity of this process to anticoagulant treatment was investigated by making human blood, treated with the direct FXa inhibitor rivaroxaban,40 flow over a collagen/tissue factor surface under coagulant conditions. Rivaroxaban treatment, at 4 μg/mL, did not affect platelet deposition at the collagen surface, but greatly suppressed fibrin accumulation (Figure 8A,B). In agreement with this, rivaroxaban strongly suppressed the cleavage of fluorescence thrombin substrate at the site of platelet thrombi (Figure 8C). This confirmed, also for human thrombi, the presence of a potent enforcement loop of FX activation, thrombin and fibrin generation.
Figure 8.
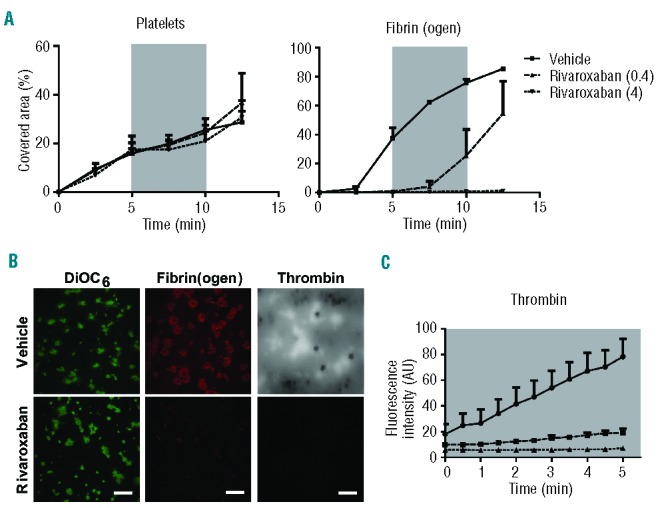
Effect of the FXa inhibitor rivaroxaban on the formation of platelet-fibrin thrombi. Human blood was pre-incubated with (A, B) OG488-fibrinogen or (B, C) thrombin substrate peptide Z-GGR-AMC and indicated concentrations of rivaroxaban (μg/mL), and then perfused under coagulant conditions over a collagen/tissue factor surface at a shear rate of 1000 s−1. After 5 min of flow, the shear rate was reduced to 150 s−1, as indicated by the gray areas. Flow was continued at 1000 s−1 after 5 min. (A) Microscopic images taken at indicated times were quantified for platelet deposition and fibrin(ogen) staining. (B) Representative fluorescence images of DiOC6-labeled thrombi, and fibrin(ogen) and accumulated AMC staining. (C) Fluorescence images of AMC fluorescence due to thrombin activity were quantified during 5 min of stasis after an initial 5 min of thrombus formation. Mean ± SEM; n = 3.
Discussion
The present data point to key roles of FVIII and FIX in FX activation at the site of a platelet thrombus by supporting: (i) thrombin generation, (ii) thrombus growth and platelet phosphatidylserine exposure, and (iii) fibrin formation at the platelet surface. The likely mechanism is that tenase activity via FVIIIa and FIXa, which is confined to the sites of platelet thrombi, generates FXa that directly catalyzes the conversion of prothrombin into thrombin. The locally produced thrombin fulfills its multiple roles, including fibrinogen cleavage and evoking persistent intracellular signaling in platelets to stimulate phosphatidylserine exposure and enhancement of procoagulant activity.41
Early evidence for the assembly of the tenase complex of FVIIIa and FIXa came from experiments using only procoagulant phospholipid membranes.42,43 Our localization studies of the (activated) coagulation factors on platelet thrombi indicate that both FIX(a) and FXa concentrate on phosphatidylserine-exposing platelets, as expected, because both contain Gla-domains. However, we observed only limited and probably weak binding of FVIII(a) to phosphatidylserine-exposing platelets. Instead, FVIII(a) immuno-activity accumulated at sites of VWF localization, coinciding with either collagen or fibrin(ogen)-labeled platelets in the thrombus. This is in agreement with the established role of VWF as a carrier of FVIII. Given that FVIII is one of the least abundant coagulation factors in plasma, these results suggest that a steady supply of (VWF-bound) FVIII to the procoagulant platelets is needed to ensure continued FXa generation. This suggestion is supported by recent evidence that locally immobilized FVIII increases fibrin clot formation under flow,44 and that platelet-targeted FVIII can restore hemostasis in hemophilic dogs.45 Interestingly, engineered FIX variants that stimulate the coagulation process independently of FVIII appeared to reduce bleeding and normalize in vivo thrombus formation in hemophilic mice lacking FVIII.46 This latter observation is compatible with a primary role of FVIII in (soluble?) FIX activation, and the concept that thrombus-localized active FIX is sufficient for FX activation and ensuing thrombin generation.
Based on the observation of low FVIII(a) staining of thrombi before rinsing, and relatively high, concentrated FVIII(a) staining at sites of VWF localization after rinsing, we came to the following hypothesis. Assuming a high affinity of the FVIII(a) antibody, there appears to be low or reversible FVIII(a) binding to procoagulant platelets, i.e. at sites where FIXa is present. Likely, the FVIII(a) can be delivered to those sites from a reservoir of FVIII bound to the VWF on collagen and in the growing platelet thrombus. Earlier studies indicated that binding to VWF has no effect on the kinetics of heavy-chain FVIII activation, but that the cleavage of its light chain enhances the dissociation of FVIII from VWF.47,48 This is also an attractive mechanism for the presentation of FVIII from its VWF reservoir sites to procoagulant platelets during the thrombus-forming process.
Whereas prothrombinase activity under flow conditions has been studied for 20 years,49 little has been reported on the regulation of FX activation, i.e. tenase activity, during thrombus formation under flow. Our data indicate that complete deficiency of murine FVIII or FIX leads to a marked impairment of the formation of fibrin-containing thrombi under flow, effects that can be restored by supplementation with human recombinant FVIII or FIX, respectively. Overall, the residual surface area covered by thrombi and fibrin(ogen) appeared to be higher for F9−/− than for F8−/− blood in the presence of tissue factor, suggesting that the remaining coagulant activity is higher in the absence of FIX than of FVIII. On the other hand, the data also indicate that without FIX or FVIII, procoagulant platelets and fibrin can still be formed, implying that residual amounts of thrombin are generated with tissue factor present.
In human hemophilia A, the amount of (platelet- or phospholipid-dependent) thrombin generation is considered to be a parameter related to the patient’s bleeding phenotype.28,50 In this context, our findings with hemophilia A and B blood may suggest that local tenase activity at a platelet thrombus is a major regulator of fibrin clot formation and, hence, bleeding cessation. Indeed, even at high tissue factor concentrations, we still observed a prolongation of platelet-dependent fibrin formation under flow conditions. This is even more striking given the residual concentrations of FVIII (4%) and FIX (5%) present in the patients’ blood. Jointly, these data point to a limited, supplementary role of tissue factor that still relies on both FVIII and FIX in platelet-fibrin thrombus formation under flow, likely via direct activation of FX.
Other authors have demonstrated that, in mouse models of arterial thrombosis, thrombus formation and fibrin accumulation are greatly impaired in animals deficient in either FVIII or FIX.30, 35 Flow chamber studies in vitro also pointed to diminished fibrin clot formation with blood from mice lacking FVIII.32,51 Similar observations have been made in flow assays using blood from patients with FVIII or FIX deficiency under coagulant conditions. It was concluded that platelet deposition and fibrin formation under flow diminished depending on the residual factor activity, with fibrin formation being more sensitive to factor deficiency than platelet deposition.52–54 The present results extend these findings significantly, by indicating for the first time an enforcing role in the thrombus-forming process of procoagulant, phosphatidylserine-exposing platelets capable of FIX(a) and FXa binding.
We have unraveled a potent enforcement loop in thrombus formation, involving FX activation, subsequent thrombin and fibrin generation, and FXa-mediated stimulation of platelet phosphatidylserine exposure. The FVIII/FIX-dependent stimulation of platelet procoagulant activity seems to be a limiting factor in fibrin clotting under flow conditions.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This research was supported by the Dutch Landsteiner Foundation for Blood Transfusion Research (1006) and the Centre for Translational Molecular Medicine (INCOAG).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Heemskerk JW, Mattheij N, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. 2013;11(1):2–11. [DOI] [PubMed] [Google Scholar]
- 2.Mazepa M, Hoffman M, Monroe D. Superactivated platelets: thrombus regulators, thrombin generators, and potential clinical targets. Arterioscler Thromb Vasc Biol. 2013;33(8):1747–1752. [DOI] [PubMed] [Google Scholar]
- 3.Jackson SP. Arterial thrombosis: insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. [DOI] [PubMed] [Google Scholar]
- 4.Kuijpers MJ, Munnix IC, Cosemans JM, et al. Key role of platelet procoagulant activity in tissue factor- and collagen-dependent thrombus formation in arterioles and venules in vivo. Differential sensitivity to thrombin inhibition. Microcirculation. 2008;15(4):269–282. [DOI] [PubMed] [Google Scholar]
- 5.Furie B, Furie BC. In vivo thrombus formation. J Thromb Haemost. 2007;5(Suppl 1):12–7. [DOI] [PubMed] [Google Scholar]
- 6.Von Brühl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils and platelets cooperate to inititate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood. 2006;107(10):3902–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mangin P, Yap CL, Nonne C, et al. Thrombin overcomes the thrombosis defect associated with platelet GPVI/FcRgamma deficiency. Blood. 2006;107(11):4346–4353. [DOI] [PubMed] [Google Scholar]
- 9.Munnix IC, Kuijpers MJ, Auger J, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27(11):2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renné T, Pozgajova M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuijpers MJ, van der Meijden PE, Feijge MA, et al. Factor XIIa regulates the pathological process of thrombus formation on ruptured plaques. Arterioscler Thromb Vasc Biol. 2014;34(8):1674–1680. [DOI] [PubMed] [Google Scholar]
- 12.Day SM, Reeve JL, Pedersen B, et al. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105(1):192–198. [DOI] [PubMed] [Google Scholar]
- 13.Gross PL, Furie BC, Merrill-Skoloff G, Chou J, Furie B. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J Leukoc Biol. 2005;78(6):1318–1326. [DOI] [PubMed] [Google Scholar]
- 14.Béguin S, Kumar R. Thrombin, fibrin and platelets, a resonance loop in which von Willebrand factor is a necessary link. Thromb Haemost. 1997;78(1):590–594. [PubMed] [Google Scholar]
- 15.Berny MA, Munnix IC, Auger JM, et al. Spatial distribution of factor Xa, thrombin, and fibrin(ogen) on thrombi at venous shear. PloS One. 2010;5(4):e10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Kim A, David T, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151(1): 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Kruchten R, Mattheij NJ, Saunders C, et al. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood. 2013;121(10):1850–1857. [DOI] [PubMed] [Google Scholar]
- 18.Cosemans JM, Schols SE, Stefanini L, et al. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood. 2011;117(2):651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zwaal RF, Schroit AJ. Pathophysiological implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89(4): 1121–1132. [PubMed] [Google Scholar]
- 20.Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol. 2006;26(1):41–48. [DOI] [PubMed] [Google Scholar]
- 21.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451(7181):914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hovig T, Dodds WJ, Rowsell HC, Mustard JF. The transformation of hemostatic platelet plugs in normal and factor IX deficient dogs. Am J Pathol. 1968;53(3):355–373. [PMC free article] [PubMed] [Google Scholar]
- 23.Sixma JJ, van den Berg A. The haemostatic plug in haemophilia A: a morphological study of haemostatic plug formation in bleeding time skin wounds of patients with severe haemophilia A. Br J Haematol. 1984;58(4):741–753. [DOI] [PubMed] [Google Scholar]
- 24.Van der Velden P, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol. 1988;70(3):345–355. [DOI] [PubMed] [Google Scholar]
- 25.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annu Rev Biochem. 1988;57:915–956. [DOI] [PubMed] [Google Scholar]
- 26.Hemker HC. Thrombin generation, an essential step in haemostasis and thrombosis. In: Haemostasis and Thrombosis, Vol I (Forbes CD, Thomas DP, Tuddenham EGD, Eds) Churchill Livingstone, UK: 1993:477–490. [Google Scholar]
- 27.Orfeo T, Brummel-Ziedins KE, Gissel M, Butenas S, Mann KG. The nature of the stable blood clot procoagulant activities. J Biol Chem. 2008;283(15):9776–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dargaud Y, Beguin S, Lienhart A, et al. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost. 2005;93(3):475–480. [DOI] [PubMed] [Google Scholar]
- 29.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–121. [DOI] [PubMed] [Google Scholar]
- 30.Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. Von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109(6):2424–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neyman M, Gewirtz J, Poncz M. Analysis of the spatial and temporal characteristics of platelet-delivered factor VIII-based clots. Blood. 2008;112(4):1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogawa S, Szlam F, Dunn AL, et al. Evaluation of a novel flow chamber system to assess clot formation in factor VIII-deficient mouse and anti-factor IXa-treated human blood. Haemophilia. 2012;18(6):926–932. [DOI] [PubMed] [Google Scholar]
- 33.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90(10):3962–3966. [PubMed] [Google Scholar]
- 34.Kundu RK, Sangiorgi F, Wu LY, et al. Targeted inactivation of the coagulation factor IX gene causes hemophilia B in mice. Blood. 1998;92(1):168–174. [PubMed] [Google Scholar]
- 35.Wang X, Cheng Q, Xu L, et al. Effects of factor IX and factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3(4):695–702. [DOI] [PubMed] [Google Scholar]
- 36.Munnix IC, Cosemans JM, Auger JM, Heemskerk JW. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102(6):1149–1156. [DOI] [PubMed] [Google Scholar]
- 37.De Witt SM, Swieringa F, Cavill R, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van der Meijden PE, Munnix IC, Auger JM, et al. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114(4):881–890. [DOI] [PubMed] [Google Scholar]
- 39.Kuijpers MJ, Schulte V, Oury C, et al. Facilitating roles of murine platelet glycoprotein Ib and aIIbb3 in phosphatidylserine exposure during vWF-collagen-induced thrombus formation. J Physiol. 2004;558(2): 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer KA. Recent progress in anticoagulant therapy: oral direct inhibitors of thrombin and factor Xa. J Thromb Haemost. 2011;9(Suppl. 1):12–19. [DOI] [PubMed] [Google Scholar]
- 41.Van der Meijden PE, Feijge MA, Swieringa F, et al. Key role of integrin αIIbβ3 signaling to Syk kinase in tissue factor-induced thrombin generation. Cell Mol Life Sci. 2012;69(20): 3481–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curtis JE, Helgerson SL, Parker ET, Lollar P. Isolation and characterization of thrombin-activated human factor VIII. J Biol Chem. 1994;269(8):6246–6251. [PubMed] [Google Scholar]
- 43.Lamphear BJ, Fay PJ. Factor IXa enhances reconstitution of factor VIIIa from isolated A2 subunit and A1/A3-C1-C2 dimer. J Biol Chem. 1992;267(6):3725–3730. [PubMed] [Google Scholar]
- 44.Doi M, Sugimoto M, Matsui H, Matsunari Y, Shima M. Coagulation potential of immobilised factor VIII in flow-dependent fibrin generation on platelet surfaces. Thromb Haemost. 2013;110(2):316–322. [DOI] [PubMed] [Google Scholar]
- 45.Du LM, Nurden P, Nurden AT, et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milanov P, Ivanciu L, Abriss D, et al. Engineered factor IX variants bypass FVIII and correct hemophilia A phenoype in mice. Blood. 2014;119(2):602–611. [DOI] [PubMed] [Google Scholar]
- 47.Hill-Eubanks DC, Lollar P. von Willebrand factor is a cofactor for thrombin-catalyzed cleavage of the factor VIII light chain. J Biol Chem. 1990;265(29):17854–17858. [PubMed] [Google Scholar]
- 48.Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN. Hemostasis and Thrombosis. Philadelphia: Lippincott Williams & Wilkins; 2001. [Google Scholar]
- 49.Billy D, Speijer H, Willems G, Hemker HC, Lindhout T. Prothrombin activation by prothrombinase in a tubular flow reactor. J Biol Chem. 1995;270(3):1029–1034. [DOI] [PubMed] [Google Scholar]
- 50.Wartiovaara-Kautto U, Joutsi-Korhonen L, Ilveskero S, Armstrong E, Lassila R. Platelets significantly modify procoagulant activities in haemophilia A. Haemophilia. 2011;17(5):743–751. [DOI] [PubMed] [Google Scholar]
- 51.Sugita C, Yamashita A, Moriguchi-Goto S, et al. Factor VIII contributes to platelet-fibrin thrombus formation via thrombin generation under low shear conditions. Thromb Res. 2009;124(5):601–607. [DOI] [PubMed] [Google Scholar]
- 52.Weiss HJ, Turitto VT, Vicic WJ, Baumgartner HR. Fibrin formation, fibrinopeptide A release, and platelet thrombus dimensions on subendothelium exposed to flowing native blood: greater in factor XII and XI than in factor VIII and IX deficiency. Blood. 1984;63(5):1004–1014. [PubMed] [Google Scholar]
- 53.Onasoga-Jarvis AA, Leiderman K, Fogelson AL, et al. The effect of factor VIII deficiencies and replacement and bypass therapies on thrombus formation under venous flow conditions in microfluidic and computational models. PloS One. 2013;8(11):e78732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colace TV, Fogarty PF, Panckeri KA, Diamond SL. Microfluidic assay of hemophilic blood clotting: distinct deficits in platelet and fibrin deposition at low factor levels. J Thromb Haemost. 2014;12(2):147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]