

Abstract
Primary myelofibrosis is a myeloproliferative neoplasm characterized by bone marrow fibrosis, megakaryocyte atypia, extramedullary hematopoiesis, and transformation to acute myeloid leukemia. To date the stem cell that undergoes the spatial and temporal chain of events during the development of this disease has not been identified. Here we describe a CD133+ stem cell population that drives the pathogenesis of primary myelofibrosis. Patient-derived circulating CD133+ but not CD34+CD133− cells, with a variable burden for JAK2V617F mutation, had multipotent cloning capacity in vitro. CD133+ cells engrafted for up to 10 months in immunocompromised mice and differentiated into JAK2-V617F+ myeloid but not lymphoid progenitors. We observed the persistence of human, atypical JAK2-V617F+ megakaryocytes, the initiation of a prefibrotic state, bone marrow/splenic fibrosis and transition to acute myeloid leukemia. Leukemic cells arose from a subset of CD133+ cells harboring EZH2D265H but lacking a secondary JAK2V617F mutation, consistent with the hypothesis that deregulation of EZH2 activity drives clonal growth and increases the risk of acute myeloid leukemia. This is the first characterization of a patient-derived stem cell population that drives disease resembling both chronic and acute phases of primary myelofibrosis in mice. These results reveal the importance of the CD133 antigen in deciphering the neoplastic clone in primary myelofibrosis and indicate a new therapeutic target for myeloproliferative neoplasms.
Introduction
Of the four major myeloproliferative neoplasms, primary myelofibrosis (PMF) has the most complex phenotype, including persistent myeloid hyperproliferation with aberrant differentiation, extramedullary hematopoiesis associated with marked hepatosplenomegaly, anemia, gradual bone marrow (BM) fibrosis and, in 20–25% of cases, abrupt transition to acute myeloid leukemia. Megakaryocyte proliferation and atypia in the context of BM hypercellularity with or without fibrosis constitute the hallmarks of the disease.1 Mutations in JAK2 or CALR are detected in about 60% and 35% of PMF patients, respectively; however, these mutations are also found in two other myeloproliferative neoplasms, polycythemia vera (95% have a JAK2 mutation) and essential thrombocythemia (55% have mutated JAK2; 25% have a CALR mutation), which have shared but also clearly distinct pathogenic phenotypes.2–4
Transgenic and knock-in mice expressing mutant JAK2 have provided compelling evidence that mutated JAK2 (typically JAK2-V617F) is a driver in this major subset of myeloproliferative neoplasms, however these mice are poor models for PMF.5 PMF characteristics such as megakaryocyte proliferation and fibrosis have been recapitulated in mice expressing throm-bopoietin,6 the NF-E2 transcription factor,7 vascular endothelial growth factor8 or reduced levels of GATA1,9 suggesting that abnormal erythroid/megakaryocyte development and/or abnormal release of cytokines may be a key factor in the disease. Although it has been postulated that aberrant interactions between the neoplastic cells and the BM microenvironment contribute to the distinct characteristics of PMF,10 the underlying modifying mutation(s) in the context of the human neoplastic stem cell clone remain unclear.
In our studies to elucidate the sequential events in the development of human PMF, we postulated the importance of a long-term repopulating hematopoietic stem cell population in both acute and chronic phases of the disease. A long-standing hypothesis that myeloproliferative neoplasms arise from an early multipotent stem cell has been supported by evidence of clonal myelopoiesis11,12 and the presence of the JAK2V617F allele in both myeloid and lymphoid cells and in cells capable of repopulating immunodeficient mice.13–16 Previous studies to identify neoplastic stem cells of PMF have focused on CD34+ hematopoietic stem/progenitor cells (HSPC), whose high frequency in the peripheral blood (PB) is characteristic of PMF.17,18 As a potential marker for the relevant stem cell, we investigated the CD133 antigen, which can be expressed prior to CD34 within the stem cell hierarchy and has been shown to mark long-term repopulating stem cells.19,20 CD133 expression has also been used to identify cancer stem cells in a wide-range of solid cancers. Here we report the characterization of CD133+ populations isolated from patients’ PB.
Methods
Patients with primary myelofibrosis, normal donor specimens and study approval
PB was obtained from patients with late-stage PMF before stem cell transplantation. PMF stage was determined in accordance with the Lille-Cervantes scoring system.21 DNA samples from blood were analyzed for JAK2V617F mutations by polymerase chain reaction for all patients.22 Retrospectively, DNA isolated from PB or CD133+ fractions used for transplantation were also screened for common mutations in genes encoding epigenetic regulators using a IonTorrent Semiconductor Sequencer (Life Technologies) with a custom designed IonAmpliseq kit. Regions sequenced are shown in Online Supplementary Table S1. BM aspirates from a total of five normal donors were used as controls for xenotransplantation. In addition, PB was isolated from four healthy donors for analysis by fluorescence activated cell sorting (FACS). All patients and BM/PB donors agreed to donation of biological material and signed informed written consent under an approved protocol from the Ärztekammer Hamburg. The Hamburg Office of Health and Consumer Safety approved all animal experiments.
Isolation and analysis of peripheral blood mononuclear cells and bone marrow cells
Peripheral blood mononuclear cells (PBMC) and mononuclear cells from healthy BM donors were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare Life Sciences). Antibodies used to characterize the PBMC are listed in Online Supplementary Table S2. CD133+ and CD34+ cells were isolated using selection kits (Miltenyi Biotec) or an AriaIII FACS (BD Biosciences). The purity of each isolated fraction was determined by FACS to be >95%. Cells (5–10×103) from isolated fractions were assessed for clonogenic potential in Methocult H4435 and Megacult (StemCell Technologies). DNA was isolated from cell fractions or from single colonies using the DNA Microkit (Qiagen).
Mouse transplantation and human graft detection
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were obtained from Jackson Laboratories and bred at the HPI animal facilities. Isolated CD133+ cells (105 per mouse) from either PMF PB or normal BM were injected intravenously into untreated mice. Phosphate-buffered saline served as a negative control. In a second experiment, sorted cells (Lin−CD133+CD34+ or Lin−CD133−CD34+; 106 per mouse) were used. PB parameters were determined with a Hemavet (Drew Scientific). PB and BM smears were stained in accordance with the Pappenheim protocol. Spleens, livers and decalcified sterna were stained with hematoxylin & eosin (H&E) or with Gomori silver stain. PB, spleen cell suspensions, or liver leukocytes were assessed for human cells by FACS using the antibodies listed in Online Supplementary Table S2. Human cells were sorted from murine tissue and their clonogenic potential determined in Methocult H4435 (StemCell Technologies).
Human Alu sequence-specific in situ hybridization of murine tissues
Formalin-fixed and paraffin-embedded tissue sections (~1 μm) were deparaffinized and rehydrated. For detection of human specific sequences, sections were processed with Alu Positive Control Probe and the ISH/VIEW Blue Detection kit (Roche) in an automated staining system (Benchmark XT, Ventana Medical Systems, Inc.).
Results
The prevalence of CD133+ cells is high in the peripheral blood of patients with primary myelofibrosis
To determine the existence of circulating CD133+ multipotent HSPC in PMF, we analyzed PBMC from a cohort of 36 patients (Table 1). In 75% of the patients’ samples, we detected CD133+ and/or CD34+ HSPC at levels significantly higher than those observed in PBMC from healthy donors (mean=0.07% of PBMC; n=4); the HSPC level in PMF blood was quite variable and ranged from 1% to 60% of PBMC in the positive fraction of patients (n=27) with a median value of 4.8% (Figure 1A). Furthermore, in contrast to normal BM, in which almost all CD133+ cells co-express CD34+ (and vice versa), variable levels of double positive cells were observed in PMF PBMC (Figure 1A). In one group of patients (group A; n=3), the majority of HSPC expressed CD133+ alone. In a second group (B; n=16), the majority of cells were CD133+CD34+ double positive. The third group (C; n=8) was composed primarily of CD133−CD34+ cells. No correlation between the absolute level of HSPC and the CD133/CD34 expression pattern could be discerned. FACS analysis of CD133+ cells confirmed positivity for the hematopoietic marker CD45 and the stem cell KIT tyrosine kinase (CD117) (Figure 1B); in contrast, cells were variably negative for CD38 (data not shown), consistent with studies of normal HSPC, which have shown that expression of CD38 and CD133 antigens is not mutually exclusive.20 Depletion of hematopoietic lineage specific markers (Lin−) resulted in enrichment of CD133+ cells (Figure 1B). CD133-immunofluorescent staining of isolated cells demonstrated the selective marking of plasma membrane protrusions (Online Supplementary Figure S1B). In summary, a notable expansion of circulating CD133+ HSPC, often lacking CD34+ expression, was observed in PMF PB.
Table 1.
Clinical characteristics of the PMF patients analyzed.
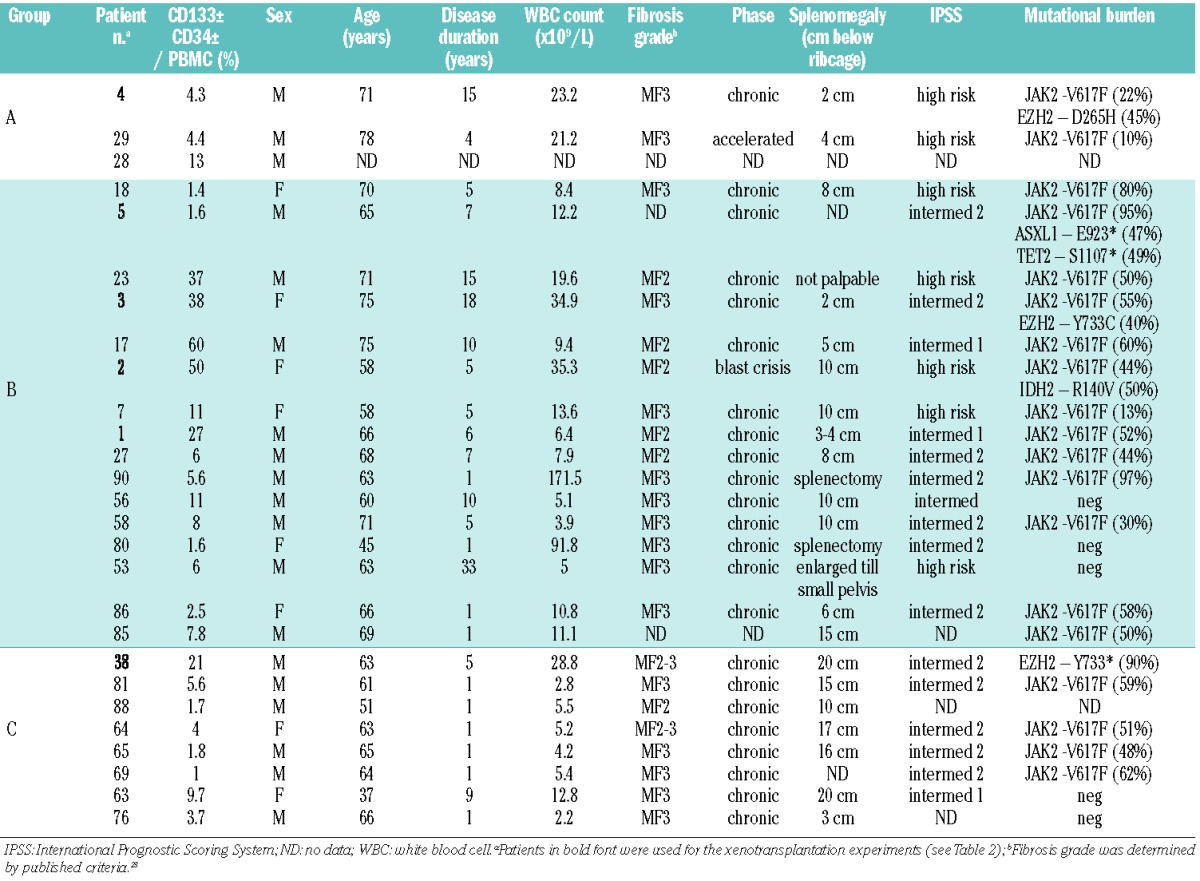
Figure 1.
CD133 marks an aberrant PMF stem cell population that gives rise to pluripotent myeloid progenitors. (A) Detection of CD133 and CD34 HSPC in PBMC from PMF patients (n=36). The incidence of HSPC (defined by CD133 and/or CD34 expression and their light scatter properties) within PBMC (excluding granulocytes and erythrocytes) was determined. Samples with <1% of PBMC were deemed not significant, whereas the remaining samples were divided into three groups based on the distribution of CD133+CD34± and CD133−CD34+ populations. Pie charts show the distribution of the three fractions and the total incidence of HSPC for each patient. As a control, normal BM cells were depleted for lineage-specific markers and examined for CD133/CD34 expression; the percentage is based on total BM cell numbers. The high proportion of double-positive cells is consistent with earlier studies.19 (B) Characterization of CD133+ cells from patients’ PB or normal BM. Patients’ PBMC were enriched for CD133+ cells to >95% using MACS microbeads. FACS analysis of the isolated fraction demonstrated co-expression of CD45 and CD117 (cKIT), characteristic of early stem cells. Alternatively, patients’ PBMC were depleted for cells expressing lineage specific antigens, and then analyzed for CD133 and CD34 expression. Normal BM cells were depleted for lineage-specific antigens by MACS and then analyzed for CD133 and CD34 antigens. Results from one of five independent samples are shown.
Clonal analysis of CD133+ cells confirms multipotency and variable JAK2-V617F burden
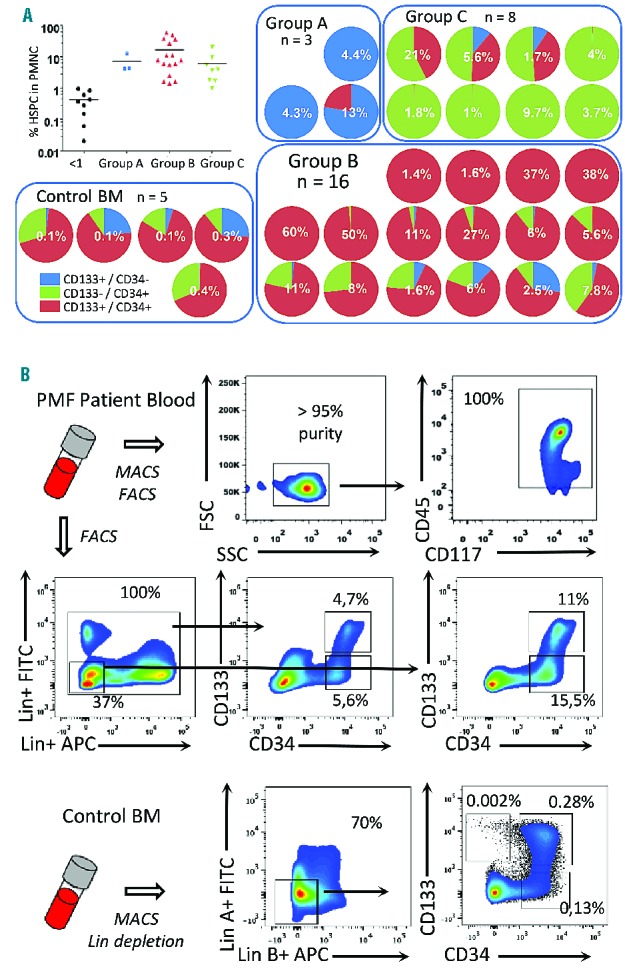
To determine the differentiation potential of the HSPC populations, CD133+CD34+, CD133−CD34+ and CD133+CD34− cells were fractionated from patients’ PBMC and assessed for clonogenic capacity (Figure 2A). Strikingly, CD133+CD34+ cells exhibited multipotent, bipotent, and unipotent myeloid (including erythroid and endothelial-like monocytes) output, whereas CD133−CD34+ cells gave rise predominantly to lineage-restricted granulocyte/monocyte progenitors or endothelial-like progenitors, previously defined as colony-forming units (CFU)-endothelial (Endo) and CFU-monocytic endothelial morphology (MEM) in assays of normal progenitors24,25 (Online Supplementary Figure S2). Gene expression analysis of single CFU-Endo and CFU-MEM colonies corroborated their granulocyte/monocyte origin (i.e. CD45+, CD14+; Online Supplementary Figure S2). Finally, we demonstrated both multipotent and restricted granulocyte/monocyte or erythroid colony differentiation capacity of CD133+CD34− cells. Thus, in contrast to circulating CD34+ cells from PMF patients which lack CD133 antigen, CD133+ cells have a broader and more robust capacity to differentiate into all myeloid cell types, including megakaryocyte/erythrocyte lineages.
Figure 2.
Determination of PMF HSPC clonogenic potential and single cell analysis for JAK2V617F allelic burden. (A) Determination of clonogenic potential of CD133+CD34−, CD133+CD34+ and CD133−CD34+ subfractions isolated from PMF patients (n=7). Due to variations in the subfraction cell numbers, not all fractions were analyzed for each patient, thus the actual number of patients analyzed for each subset is indicated. Color shading indicates individual patients, with violet bars denoting CFU-granulocyte, erythroid, megakaryocyte, monocyte (-GEMM); blue bars, CFU-GM, -G, -M or -eosinophil (Eo); green bars, CFU-Endo or -MEM; red bars, CFU-E or burst-forming unit (BFU)-E; and orange bars, CFU-megakaryocyte (-Meg). Nd: not determined. (B) The overall JAK2-V617F burden in the three cell fractions isolated from four independent PMF patients was determined by quantitative polymerase chain reaction as indicated in bar graphs. To determine the incidence of progenitors with homozygous JAK2 mutations, the JAK2 status was determined on individual colonies from the same patients. The pie graphs show the frequency of JAK2 genotypes in different colony types arising from both CD133+CD34+ and CD133-CD34+. Single colonies from the analysis in panel A were picked and the JAK2 genotype was determined by quantitative polymerase chain reaction. n represents the number of colonies analyzed (but does not reflect the distribution of colony types). Color gradient (pale to dark) denotes the percentage of negative (V/V), heterozygous (V/F) or homozygous (F/F) colonies, where V is the wild-type allele (JAK2+) and F is the mutated allele (JAK2V617F).
To determine whether the three HSPC populations varied in their JAK2-V617F status, quantitative polymerase chain reaction was performed on DNA isolated from CD133+, CD133+CD34+, and CD34+ cell fractions from four patients’ samples. This analysis showed comparatively similar mutation burdens (Figure 2B), suggesting that at least a subset of each population arose from shared stem cells. Analysis of DNA from single colonies revealed large variability in the genotypes of emerging progenitors. Homozygous JAK2V617F/V617F progenitors were detectable in all patients’ samples analyzed, even if a relatively low JAK2V617F burden (30%) was determined from the initial population. Overall the highest incidence of a homozygous JAK2V617F genotype was observed in erythroid progenitors, consistent with the profound effect of JAK2-V617F on erythropoiesis.26 Nevertheless, in two patients the incidence of JAK2V617F homozygosity was similarly high in erythroid and granulocyte/monocyte progenitors, and in one patient the highest level was observed in multipotent GEMM progenitors. We also noted JAK2V617F/V617V megakaryocytes in two independent patients’ samples. These results demonstrate the heterogeneity of the early CD133+ population with respect to JAK2V617F burden and the frequent acquisition of JAK2V617F homozygosity in a multipotent HSPC compartment in PMF, but which imparts only limited bias in differentiation potential.
Reproduction of chronic phase primary myelofibrosis in mice transplanted with CD133+ cells
To assess the repopulating ability of CD133+ multipotent stem cells and their connection with PMF development, isolated CD133+ cells from five PMF patients positive for JAK2V617F mutations were transplanted into non-irradiated immunodeficient mice (n=2 mice per patient). Mice were examined between 4 and 10 months after transplantation (Table 2). The majority of xenotransplanted mice (8/10) exhibited one or more clinical features of chronic phase PMF, including anemia and blood pictures of anisocytosis, polychromasia and poikilocytosis of erythrocytes and abnormal, giant thrombocytes (Figure 3A,B). BM analysis of mice showed hypercellularity and increased megakaryocyte numbers in 8/10 mice in comparison to mock transplanted controls. Megakaryocytes were clustered next to sinusoids or bone trabeculae (Figure 3C). Human-specific Alu sequence in situ hybridization (HISH) of excised sterna confirmed the presence of scattered, morphologically variable, human cells in murine BM (Figure 3D). This analysis also confirmed the human origin of the megakaryocyte clusters and demonstrated the typical PMF morphology of the megakaryocytes with cloud-like nuclei and aberrant nuclear/cytoplasmic ratio (Figure 3E). All xenotransplanted animals exhibited splenomegaly. Enlarged spleen size correlated with time since transplantation (Figure 3G, Table 2). In some xenotransplanted mice (4/10) we observed a relative increase in perivascular reticulin fibers of the spleen and in one case ectopic murine bone formation with fibrosis (Online Supplementary Figure S3).
Table 2.
Disease characteristics of mice transplanted with PMF patient–derived CD133+CD34± and CD133−CD34+ cells.
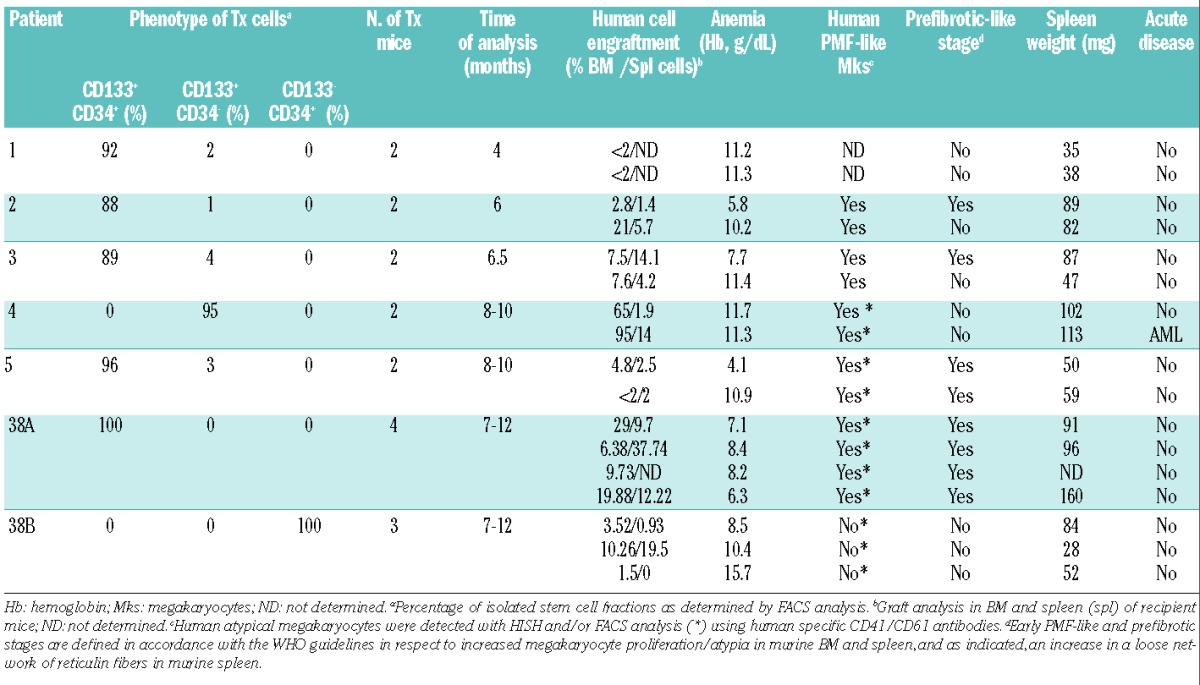
Figure 3.
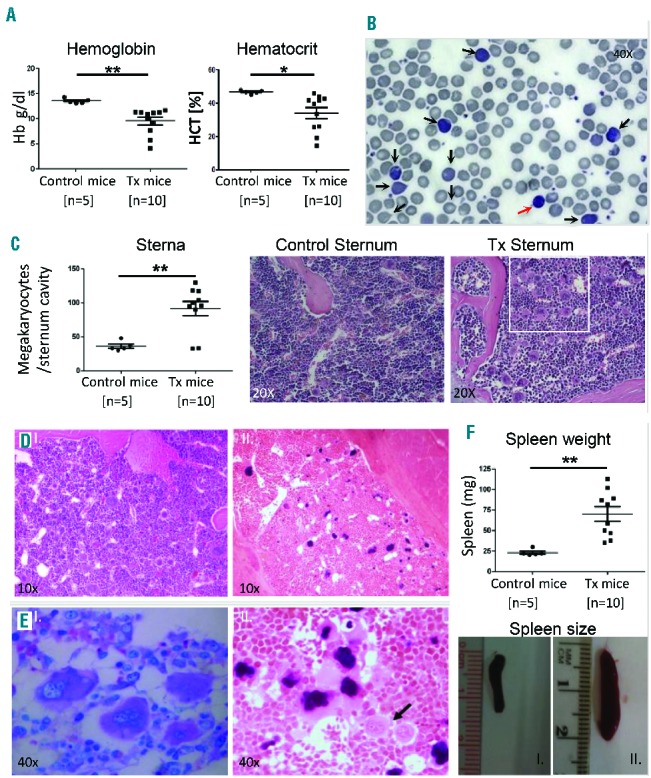
Reproduction of chronic phase PMF chronic phase in CD133+ xenograft mice. (A, B) Xenotransplanted (Tx) mice showed several PMF characteristics, including anemia, as evidenced by reduced Hb and hematocrit compared to control (transplanted with phosphate-buffered saline) mice (A) and poikilocytosis, anisocytosis, dacryocytosis, increased reticulocytes (black arrows) and giant thrombocytes (red arrow) observed in Tx blood smears (Pappenheim staining) (B). (C) Hypercellularity and increased megakaryocyte proliferation in BM of Tx mice. The mean number of megakaryocytes per sternum cavity in the indicated number of mice was determined (>6 months after transplantation) and plotted. Representative pictures of Tx BM in chronic phase PMF showing hypercellularity and increased megakaryopoiesis (right) compared to control (left). White box frames PMF-characteristic megakaryocyte clustering and atypia (H&E staining). (D) HISH confirms human cells in Tx sterna (right) as opposed to control (left). Human cell nuclei are stained black. Human cells show variable morphology and appear scattered in the whole BM space of Tx mice, 6–8 months after transplantation. (E) The prefibrotic-PMF hallmark of clustered-hypolobulated megakaryocytes of human origin (HISH, right), which morphologically resemble the atypical ones in PMF patients’ BM (Giemsa staining, left). The black arrow points to smaller unstained murine megakaryocytes. (F) Increase in spleen weight and size of all Tx mice compared to controls of the same age. Statistical significance was determined with an unpaired Student t test and is shown as mean with SEM (*P<0.05 and **P<0.01). Pictures show a normal spleen from a mock transplanted mouse (I) in comparison to a representative enlarged spleen from a Tx mouse (II) of the same age.
PB, BM, spleen and liver from xenotransplanted mice were analyzed for human cells by flow cytometry using a broad panel of markers. We detected early human HSPC (CD133+CD34±; ~1%), myeloid progenitors (CD45+CD33+/CD14±; ~0.3–23% or CD36+CD61+CD31+; ~0.1–30%), and endothelial-like monocytic progenitors (VE-Cadherin+CD45dim; ~0.1–15%) in murine BM and PB (Figure 4A,B). Human HSPC isolated from xenotransplanted mice formed colonies in semisolid media and were JAK2-V617F+ (Figure 4A). The CD36+CD61+CD31+ cells are uncharacterized progenitor/precursors that warrant further investigation. In addition, we detected mature cells of several myeloid lineages: erythroblasts, megakaryocytes and monocytes (Figure 4A,B), while no lymphoid cells were observed. Our analysis also confirmed a high JAK2V617F burden of the transplanted patients’ cells, with a mean mutated allele status >50% in erythroid (CD71+GlycophorinA+), megakaryocytes (CD41+CD61+) and endothelial-phenotype monocytic cells (VE-Cadherin+), as well as in the CD36+/CD61+/CD31+ myeloid progenitors (Figure 4D). Endothelial-like JAK2-V617F+ progenitor cells in PMF patients’ PB have been reported previously,27,28 but our study confirms their origin in a CD133+ stem cell. Despite diversity of patients as an independent variable, our CD133+ xenotransplantation model demonstrated striking reproducibility in the human cell types arising and their persistence at different time points after transplantation (Figure 4C, Table 2, and Online Supplementary Figure S4).
Figure 4.

PMF patient-derived CD133+ stem cells exhibit long-term engraftment and myeloid/endothelial-like cell differentiation in vivo. (A) Detection, isolation and assessment of clonogenic capacity of human progenitors in mouse xenotransplanted (Tx) BM: early stem (I), myeloid (II, IV) and endothelial-like (III) progenitors. Indicated cell fractions from Tx mice were isolated by cell sorting and the cells were subjected to colony assays. The typical colony morphology arising from each fraction is shown. Detection of terminally differentiated human cells of all myeloid lineages: erythroid (CD71+ GlyA+), megakaryocytic (CD41+CD61+), granulocytic-monocytic (CD14+) in all Tx mice (V). Initial JAK2 burden ranged from 20% to 95% in the CD133+CD34+ fraction. (B) Characterization of circulating human endothelial-like cells (I), myeloid progenitors (II) and monocytes (III) in peripheral blood of Tx mice in chronic phase of PMF. (C) Graphs show the distribution of human cell phenotypes in Tx mice with chronic phase PMF (4–10 months after transplantation) in (I) BM, (II) PB. Analysis of mouse 4.2 is not shown, as it developed leukemia at 10 months (see Figure 6). (D) JAK2-V617F mutation status of various human stem, precursor and terminally differentiated myeloid cells in murine BM (4–10 months after transplantation). Data are shown as mean with SD and n≥3.
CD133 marks the neoplastic stem cell compartment in primary myelofibrosis
To compare both differentiation potential and PMF reproduction of disparate HSPC fractions that vary in CD133 expression, we performed xenotransplantation of lineage depleted CD133+/CD34+ (n=4 mice) and CD133−/CD34+ (n=3 mice) HSPC from PB of one patient with adequate cells (patient 38, Tables 1 and 2). All mice were sacrificed 7–12 months after transplantation. Strikingly, all CD133+ transplanted mice had anemia and enlarged spleens (hematocrit <25, spleen size >90 mg) with accumulation of atypical megakaryocytes in their BM (Figure 4A, Table 2). This is in contrast to CD133−/CD34+ transplanted mice, in which no pathological features were observed (Table 2). Human cell engraftment was greater in CD133+/CD34+ (~20%) than in CD133−/CD34+ (<1%) stem cell transplanted recipients. Human graft analysis revealed that only CD133+ stem cells gave rise to a constant number of CD45+ cells (1%), which included both primitive (CD133+/CD34+, ~0.2%) and committed myeloid progenitors (CD34+ and/or CD33+, ~0.5%) (Figure 5B,C). CD133+/CD34+ HSPC gave rise to all classes of myeloid (granulocytic, monocytic, endothelial-like), erythroid and megakaryocyte cells, in contrast to CD133−/CD34+ stem cells that exhibited limited myeloid differentiation potential towards the granulocyte/monocyte lineage (Figure 5B,C). Stem cell fractionation and xenotransplantation thus provided substantial evidence that the neoplastic clone in PMF is derived from the CD133+ compartment.
Figure 5.

CD133+/CD34+ hematopoietic stem cells (HSC) exhibit long-term engraftment and greater multilineage myeloid differentiation with features of PMF, as compared to CD133−/CD34+ HSC. (A) Histological analysis of sterna from Mock, Lin−CD133−CD34+ and Lin−CD133+/CD34+ HSC transplanted mice. Extended atypical megakaryocyte proliferation and initiation of fibrosis was observed only in Lin−CD133+/CD34+ recipients (H&E stain, 20×). (B) Comparison of emerging human populations in bone marrow from Lin−CD133+/CD34+ and Lin−CD133−/CD34+ transplanted mice. Various human cell classes (myeloid progenitors and megakaryocytes/erythrocytes) were exclusively observed in mice which received Lin−CD133+/CD34+ PMF cells and were analyzed 10 months after transplantation. (C) Human graft analysis of individual animals transplanted with Lin−CD133+/CD34+ (n=4), Lin−CD133−/CD34+ (n=3) from the same PMF patient and normal bone marrow CD133+ cells (n=2) (analysis 10–12 months after transplantation; number of transplanted cells 106/mouse).
As a final control, we transplanted two mice with CD133+/CD34+ cells isolated from BM from healthy donors. Levels of all classes of myeloid cells, including erythroid cells, were comparable to those observed in PMF-donor mice erythroid (Figure 5C). However, consistent with the hypothesis that the high level and aberrant megakaryopoiesis in PMF-transplanted mice is inherent to the PMF phenotype, no human megakaryocytes were detectable. Furthermore, in contrast to mice receiving PMF cells, control mice contained human lymphoid cells (CD19+ or CD3+), consistent with findings of previous studies.29 In summary, these results demonstrate that PMF CD133+ long-term repopulating stem cells are skewed toward myeloid differentiation with robust megakaryopoiesis.
CD133+ cells contain leukemia-initiating stem cells
In one of two mice transplanted with JAK2-V617F+ CD133+ cells lacking CD34 expression (patient 4, group A in Figure 1A, Tables 1 and 2), we observed transition to acute myeloid leukemia 9 months after transplantation. The animal’s disease was characterized by splenomegaly and the accumulation of myeloblasts in the blood (Figure 6A, Table 2). The BM cavity was hypercellular with >50% blasts showing a limited degree of differentiation (Figure 6B). The human origin of the proliferating cells in the BM was demonstrated by HISH and confirmed by FACS analysis, indicating >90% human cells positive for either human CD45+ or erythroid antigens (human CD71 and human glycophorin A) (Figure 6C). Leukemic cells in the BM represented a population expressing CD133+CD34+CD45hi,dimCD33+ myeloid markers (Figure 6C). FACS and histological analysis of the spleen confirmed invasion of myeloid blasts, although leukemic involvement was limited and splenomegaly was mostly attributable to extramedullary erythropoiesis. Notably, JAK2V617F mutations were not detected in leukemic cells, but a missense mutation (D265H) in EZH2, which was present in the original CD133+ cell fraction, was readily detectable. Due to the high burden of the EZH2-D265H mutation (45%) as compared to the JAK2-V617F burden (22%) in the original CD133+ fraction, we predict that the EZH2 mutation preceded the JAK2 mutation, which occurred in a subset of CD133+ cells (Figure 6F). This result demonstrates that transformation to acute disease originated from human CD133+ cells lacking JAK2 mutations but with an underlying epigenetic mutation, consistent with findings in patients’ samples.30–32
Figure 6.

Development of acute myeloid leukemia (AML) lacking JAK2V617Fmutation from transplanted human PMF CD133+CD34− stem cells. (A) Blood films showing increased circulating blasts (Pappenheim staining) in one mouse receiving cells from patient 4. (B) Bone marrow smear (Giemsa staining) shows massive myeloproliferation, as indicated by elevated numbers of erythroid/granulocyte progenitors and significantly increased numbers of immature myeloblasts (black arrows) (I). HISH reveals complete occupancy of bone marrow space by human cells. The red arrow points to an unstained murine megakaryocyte (II). (C) Immunophenotyping of human cells and detection of the leukemic clone. (I) Analysis of total BM cells shows massive myeloproliferation of human origin (92% human CD45+ cells). (II, and III) Analysis of BM cells depleted of murine cells (CD45+) showed high levels of human erythropoiesis marked by Glycophorin A (7%) as well as a large fraction of proliferating CD71+ cells, which may either represent early erythroid progenitors or myeloblasts (14%). The predominant surface phenotype of leukemic cells were CD45hi,dimCD133+CD33+CD34±. (D) HISH depicts the infiltration of human cells in murine spleen at leukemic transition. (E) Detection of myeloid blasts (5%), human megakaryocytes (7%) and myeloid progenitors (1%) in the spleen of the xenotransplanted mouse with human AML. (F) Detection of the EZH2-D265H mutation both in the CD133+ fraction isolated from PB of patient 4 and in the PB of the AML transition mouse. The JAK2-V617F mutation burden for the same sample was determined by polymerase chain reaction.
Discussion
We have characterized an expanded human CD133+ HSPC population in the PBMC of PMF patients. Our results support the hypothesis that these cells embody the neoplastic stem cell clone harboring collaborating mutations that both maintain and drive the distinct PMF chronic disease, and underscore the heterogeneity of the stem cell pool in PMF patients, in whom the spectrum of phenotypic and genotypic differences likely reflect the evolving disease.
Colony assays and/or xenotransplantation demonstrated that the CD133+ but not the single CD34+ fraction is multipotent but with a high megakaryocytic differentiation capacity and long-term repopulating potential in immunodeficient mice (up to 12 months). Multipotent differentiation capacity of normal CD133+CD34+ cells, in particular megakaryocyte differentiation, has been noted previously in vitro.20,33 However, in contrast to normal CD133+CD34+ cells isolated from BM, circulating CD133+CD34+ PMF cells showed a greater contribution to megakaryocytic differentiation in vivo. A reduced lymphoid repopulation capacity was also observed, although we cannot rule out that the apparent myeloid skewing was partly due to normal age-related reduction of lymphoid potential, as our BM controls were not aged-matched.
In contrast to previous studies using PMF-derived CD34+ cells, we showed that engraftment of CD133+ cell reproduced many disease hallmarks – including increased and atypical megakaryopoiesis in BM and spleen, high levels of myeloid progenitors (CD36+) in blood, as well as anemia and splenomegaly. Fibrosis was more frequently observed in the spleen than in the BM, perhaps reflecting the unique hematopoietic microenvironment of the mouse spleen. In view of the overall modest level of human cell engraftment, it is likely that some disease parameters, such as splenomegaly and anemia, are an indirect consequence of the human PMF-cell graft. Human megakaryocytes, which were observed at high levels in our engrafted animals, are a potential source of cytokines that stimulate proliferation or skewed differentiation of mouse hematopoietic cells.10 This was highlighted in an independent cohort of mice, reported separately, in which we showed that transplanted PMF cells conferred susceptibility to mouse myeloid progenitors for retroviral transformation.34
Long-term repopulating potential and myeloid-restricted differentiation of CD34+ PBMC from PMF patients has been reported previously in immunodeficient mice;15,16,18,35 however, these studies did not report high or aberrant levels of megakaryopoiesis and/or erythropoiesis. None of these studies investigated CD133+ expression, so it is possible that CD133+CD34+ double-positive cells were underrepresented in these transplants and thus gave rise to mice with similar phenotypes to those receiving CD133−/CD34+ cells in our study. Nevertheless, we cannot ignore technical differences in our experimental setup that may have contributed to the manifestation of PMF parameters not observed by others. In addition to using NSG mice, in which the absence of T, B and NK cells permits high human cell engraftment,36 the mice were not subjected to irradiation before transplantation. Irradiation is known to impair the SDF1/CXCR4 axis essential for homing of quiescent stem cells in the BM.37 CD34+ HSPC from PMF patients have also been reported to express low levels of CXCR4,38,39 thus we argue that compromising this important gradient further by irradiation may be preferentially detrimental to PMF versus normal cells. Another important parameter is the time point of analysis. Most PMF xenograft studies have only considered short-term engraftment (up to 20 weeks); however, it is well established that the CD34+ HSPC compartment is composed of a wide spectrum of HSPC with divergent self-renewal and differentiation capacities. Accordingly, different output kinetics are observed in transplanted mice, with different progenitor fractions contributing to sequential waves of early repopulation, with the more primitive cells responsible for repopulation after 4 months.40,41 Thus it is likely that primitive hematopoietic stem cells contained within the CD133+CD34+ cell fraction became the dominant clone in transplanted mice only at later stages, at which time the atypical megakaryopoiesis and aberrant erythropoiesis were observed. Indeed, a pronounced phenotype was only observed in mice examined after 6 months in our study. We admit that a rigorous “self-renewal” potential of the neoplastic stem cell was not assayed by performing secondary transplantation; however, we argue that sustainment of myelopoiesis for up to a year after transplantation confirms long-term repopulation capacity in a mouse model.
In agreement with the hypothesis that the observed heterogeneity of the stem cell compartment in PMF is due to collaborating mutations, there is growing evidence that JAK2 mutations are insufficient to promote clonal myelopoiesis, to expand an early hematopoietic stem cell compartment, or to explain the distinctive phenotypes of the various myeloproliferative neoplasms.5,42,43 Although we noted a striking heterogeneity in the burden of JAK2-V617F in circulating HSPC, in agreement with a previous study of three PMF patients,14 we confirmed a major contribution of JAK2-V617F+ CD133+ cells in the engrafted animals. Thus our results are consistent with earlier studies suggesting that JAK2-V617F in the context of PMF does not hinder self-renewal or proliferation of immunodeficient repopulating cells,15,16 which contrasts with studies of polycythemia vera and chronic myeloid leukemia, in which Scid-repopulating cells expressing the “driver” mutations, JAK2-V617F and BCR/ABL, respectively, are detected at relatively low levels.16,26,44 To determine whether or not these differences reflect the target cell of mutations within the stem cell hierarchy and/or the interplay with secondary mutations will require much larger cohorts of patients.
Importantly, in a retrospective analysis of the patients’ samples used for transplantation in this study, we found that all samples but one used for xenografts carried common mutations in epigenetic regulators (Online Supplementary Table S1).45,46 It is likely that these mutations may have facilitated clonal selection, expansion of the stem cell compartment, and progression of acute myeloid leukemia. Indeed in CD133+ cells from patient 4, which induced acute myeloid leukemia in a recipient mouse, we observed the persistence of the EZH2-D265H mutation in the leukemic clone (but loss of the JAK2 mutation). EZH2 encodes the catalytic component of the polycomb-repressor complex 2 (PRC2), which facilitates transcriptional repression and chromatin condensation of gene loci implicated in cell fate decisions.47 The observed EZH2 mutation is within Domain 2, which is known to interact with SUZ12, another member of PRC2 that is essential for its catalytic activity, and in which other mutations have been found in PMF and chronic myeloid leukemia.48–50 EZH2 mutations are relatively rare in acute myeloid leukemia, but have been shown to confer a poor prognosis in PMF, in which the incidence of mutations is about 7%.48,49 Patient 4 is currently in remission after successful stem cell transplantation. Although no obvious correlation between the expansion of the CD133+ HSPC population and the presence or type of cooperating epigenetic mutation was observed in this study, further investigation is warranted.
The novelty of our study is based on the identification of CD133 as a marker for a distinct class of circulating HSPC in PMF and the demonstration that these cells have neoplastic stem cell activity. This work sets the stage for the further characterization and quantification of these disease stem cells, which would be instrumental for unraveling the genetic and epigenetic events that lead to the manifestation of the distinct PMF disease and that underlie progression to acute myeloid leukemia.
Acknowledgments
The authors would like to thank U. Bacher for providing hematological evaluation expertise, S. Zeschke and A. Badbaran for genetic analysis of patients’ material, and M. Lachmann, S. Roscher, and C. Siggel of the UKE FACS Core Facility for their assistance in sorting experiments. This work was initiated at the Aristotle University of Thessaloniki, Greece and submitted to the Faculty of the Department of Pharmaceutical Sciences as a requirement for the Doctorate Degree awarded to IT. The manuscript is dedicated to the memory of Dr. Ursula Gehling.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This study was supported by grants from Deutsche Akademische Austauschdienst (DAAD) to IT, Erich and Gertrud Roggenbuck Foundation to IT and NK, Hamburger Krebsgesellschaft to NK, Else-Kröner-Fresenius-Stiftung to CS and Deutsche Forschungsgemeinschaft to BF (FE 568/11-2). The Heinrich-Pette-Institute is financed by the German Federal Ministry of Health and the Freie und Hansestadt Hamburg.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 2.Klampfl T, Gisslinger H, Harutyunyan A, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 3.Levine R, Pardanani A, Tefferi A, Gilliland D. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673–683. [DOI] [PubMed] [Google Scholar]
- 4.Nangalia J, Massie C, Baxter E, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullally A, Lane S, Brumme K, Ebert B. Myeloproliferative neoplasm animal models. Hematol Oncol Clin North Am. 2012;26(5):1065–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villeval J, Cohen-Solal K, Tulliez M, et al. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90(11):4369–4383. [PubMed] [Google Scholar]
- 7.Kaufmann K, Gründer A, Hadlich T, et al. A novel murine model of myeloproliferative disorders generated by overexpression of the transcription factor NF-E2. J Exp Med. 2012;209(1):35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maes C, Goossens S, Bartunkova S, et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 2010;29(2):424–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vannucchi A, Bianchi L, Cellai C, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood. 2002;100(4): 1123–1132. [DOI] [PubMed] [Google Scholar]
- 10.Lataillade J-J, Pierre-Louis O, Hasselbalch H, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112(8):3026–3035. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson R, Salo A, Fialkow P. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978;51(2):189–194. [PubMed] [Google Scholar]
- 12.Levine R, Belisle C, Wadleigh M, et al. X-inactivation-based clonality analysis and quantitative JAK2V617F assessment reveal a strong association between clonality and JAK2V617F in PV but not ET/MMM, and identifies a subset of JAK2V617F-negative ET and MMM patients with clonal hematopoiesis. Blood. 2006;107(10):4139–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bogani C, Guglielmelli P, Antonioli E, et al. B-, T-, and NK-cell lineage involvement in JAK2V617F-positive patients with idiopathic myelofibrosis. Haematologica. 2007;92(2):258–259. [DOI] [PubMed] [Google Scholar]
- 14.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. [DOI] [PubMed] [Google Scholar]
- 15.Ishii T, Zhao Y, Sozer S, et al. Behavior of CD34+ cells isolated from patients with polycythemia vera in NOD/SCID mice. Exp Hematol. 2007;35(11):1633–1640. [DOI] [PubMed] [Google Scholar]
- 16.James C, Mazurier F, Dupont S, et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood. 2008;112(6):2429–2438. [DOI] [PubMed] [Google Scholar]
- 17.Barosi G, Viarengo G, Pecci A, et al. Diagnostic and clinical relevance of the number of circulating CD34(+) cells in myelofibrosis with myeloid metaplasia. Blood. 2001;98(12):3249–3255. [DOI] [PubMed] [Google Scholar]
- 18.Xu M, Bruno E, Chao J, et al. The constitutive mobilization of bone marrow-repopulating cells into the peripheral blood in idiopathic myelofibrosis. Blood. 2005;105(4): 1699–1705. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia M. AC133 expression in human stem cells. Leukemia. 2001;15(11):1685–1688. [DOI] [PubMed] [Google Scholar]
- 20.Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90(12):5002–5012. [PubMed] [Google Scholar]
- 21.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. [DOI] [PubMed] [Google Scholar]
- 22.Kröger N, Badbaran A, Holler E, et al. Monitoring of the JAK2-V617F mutation by highly sensitive quantitative real-time PCR after allogeneic stem cell transplantation in patients with myelofibrosis. Blood. 2007;109(3):1316–1321. [DOI] [PubMed] [Google Scholar]
- 23.Thiele J, Kvasnicka H, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128–1132. [PubMed] [Google Scholar]
- 24.Gehling U, Ergün S, Schumacher U, et al. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood. 2000;95(10):3106–3112. [PubMed] [Google Scholar]
- 25.Yoder M, Mead L, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jamieson C, Gotlib J, Durocher J, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci USA. 2006;103(16):6224–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sozer S, Ishii T, Fiel M, et al. Human CD34+ cells are capable of generating normal and JAK2V617F positive endothelial like cells in vivo. Blood Cells Mol Dis. 2009;43(3):304–312. [DOI] [PubMed] [Google Scholar]
- 28.Teofili L, Martini M, Iachininoto M, et al. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood. 2011;117(9):2700–2707. [DOI] [PubMed] [Google Scholar]
- 29.Sloma I, Beer P, Saw K, et al. Genotypic and functional diversity of phenotypically defined primitive hematopoietic cells in patients with chronic myeloid leukemia. Exp Hematol. 2013;41(10):837–847. [DOI] [PubMed] [Google Scholar]
- 30.Beer P, Delhommeau F, LeCouédic J-P, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115(14):2891–2900. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen-Khac F, Lesty C, Eclache V, et al. Chromosomal abnormalities in transformed Ph-negative myeloproliferative neoplasms are associated to the transformation subtype and independent of JAK2 and the TET2 mutations. Genes Chromosomes Cancer 2010;49(10):919–927. [DOI] [PubMed] [Google Scholar]
- 32.Theocharides A, Boissinot M, Girodon F, et al. Leukemic blasts in transformed JAK2-V617F–positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2006;110(1):375–379. [DOI] [PubMed] [Google Scholar]
- 33.Charrier S, Boiret N, Fouassier M, et al. Normal human bone marrow CD34(+)CD133(+) cells contain primitive cells able to produce different categories of colony-forming unit megakaryocytes in vitro. Exp Hematol. 2002;30(9):1051–1060. [DOI] [PubMed] [Google Scholar]
- 34.Triviai I, Ziegler M, Bergholz U, et al. Endogenous retrovirus induces leukemia in a xenograft mouse model for primary myelofibrosis. Proc Natl Acad Sci USA. 2014;111(23):8595–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Prakash S, Lu M, et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J Clin Invest. 2012;122(11):3888–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shultz L, Lyons B, Burzenski L, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10): 6477–6489. [DOI] [PubMed] [Google Scholar]
- 37.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. [DOI] [PubMed] [Google Scholar]
- 38.Bogani C, Ponziani V, Guglielmelli P, et al. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells. 2008;26(8):1920–1930. [DOI] [PubMed] [Google Scholar]
- 39.Migliaccio A, Martelli F, Verrucci M, et al. Altered SDF-1/CXCR4 axis in patients with primary myelofibrosis and in the Gata1 low mouse model of the disease. Exp Hematol. 2008;36(2):158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung A, Leung D, Rostamirad S, et al. Distinct but phenotypically heterogeneous human cell populations produce rapid recovery of platelets and neutrophils after transplantation. Blood. 2012;119(15):3431–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glimm H, Eisterer W, Lee K, et al. Previously undetected human hematopoietic cell populations with short-term repopulating activity selectively engraft NOD/SCID-beta2 microglobulinnull mice. J Clin Invest. 2001;107(2):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.James C. The JAK2V617F mutation in polycythemia vera and other myeloproliferative disorders: one mutation for three diseases? Hematology Am Soc Hematol Educ Program. 2008:69–75. [DOI] [PubMed] [Google Scholar]
- 43.Kent D, Li J, Tanna H, et al. Self-renewal of single mouse hematopoietic stem cells is reduced by JAK2V617F without compromising progenitor cell expansion. PLoS Biol 2013;11(6) e1001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sirard C, Lapidot T, Vormoor J, et al. Normal and leukemic SCID-repopulating cells (SRC) coexist in the bone marrow and peripheral blood from CML patients in chronic phase, whereas leukemic SRC are detected in blast crisis. Blood. 1996;87(4):1539–1548. [PubMed] [Google Scholar]
- 45.Shih A, Abdel-Wahab O, Patel J, Levine R. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. [DOI] [PubMed] [Google Scholar]
- 46.Vainchenker W, Delhommeau F, Constantinescu S, Bernard O. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118(7):1723–1735. [DOI] [PubMed] [Google Scholar]
- 47.Simon JA, Kingston RE. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell. 2013;49(5):808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. [DOI] [PubMed] [Google Scholar]
- 49.Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118(19):5227–5234. [DOI] [PubMed] [Google Scholar]
- 50.Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220–2228. [DOI] [PubMed] [Google Scholar]