

Recurrent mutations are implicated in the prognosis of myeloid malignancies and are likely to be incorporated into the next WHO classification. We evaluated a pooling approach for cost-effective targeted sequencing in combination with a clinically accredited method for validating point mutations, Sequenom®. We found the pooling strategy adequate for detecting clonal mutations, but not sensitive enough to confidently detect subclonal mutations. Our data indicate that targeted sequencing adds prognostic information to existing risk scores in myelodysplastic syndromes (MDS), mixed myeloproliferative/MDS neoplasms (MDS-MPN) and acute myeloid leukemia (AML).
More than one-third of patients with MDS have higher-risk disease with poor overall survival and a high rate of progression to AML.1 Several clinical trial programs, therefore, include higher-risk MDS in current protocols for AML.2,3 Moreover, based on clinical and cytogenetic characteristics, a biological similarity between higher-risk MDS and poor prognosis AML has been suggested. Poor-risk cytogenetic abnormalities encompassing complete or partial loss of chromosome 5, 7, and 17, and complex karyotype are found in both diseases; but outside this group, the cytogenetic pattern differs markedly between MDS and AML.4 FLT3, NPM1, CEBPA and KIT mutations are integrated in the current WHO classification for AML, but are rarely mutated in MDS, or secondary or therapy-related AML.5
Consecutive adult patients were enrolled in the MDS and AML registers at the Karolinska University Hospital and diagnostic bone marrow (BM) samples were biobanked. The MDS cohort (n=100) consisted of patients with MDS and MDS-MPN (n=100) and the AML cohort (n=92) of patients with AML evolving from MDS (MDS-AML) or MPN (MPN-AML), therapy-related AML (tAML), and AML with MDS like cytogenetics (Online Supplementary Appendix and Online Supplementary Table S1). First-line treatment for both cohorts followed current therapeutic guidelines developed by the European LeukemiaNet project.6,7 Eighteen patients (7 MDS and 11 AML) underwent allogeneic stem cell transplantation. DNA samples from 20 healthy subjects were used as controls. The study was approved by the local ethics committee and samples were obtained after informed consent. Categorical data were compared using Fisher’s exact test, survival was estimated using Kaplan-Meier method and compared using the log rank test, and Cox proportional hazards model was used for uni- and multivariable analyses. Variables included in the model for MDS were mutations found to be significant in the univariable analyses (Online Supplementary Table S2) together with age, sex and IPSS-R classification. In the corresponding model for AML, IPSS-R was replaced by the cytogenetic risk group. Two-sided P values with a significance level of 0.05 were used in all analyses.8
Halogenomics target amplification technology was applied to amplify all exons of 22 selected recurrently mutated genes9,10 from BM mononuclear cell DNA (Online Supplementary Appendix). To analyze samples in a high-throughput and cost effective way we used a pooling approach with 10 samples per pool. A total of 22 pools were defined by 6 bp barcoding and samples were sequenced in 2 Illumina Hiseq2000 sequencing system using a 100 bp paired end protocol. Sequencing data were filtered based on the barcodes of each pool and aligned to the human genome reference hg19 with BWA version 0.5.9-r16.11 Only variants covered by 4 or more reads were considered for variant calling, and reads with more than 4 mismatches were disregarded. Finally, variants were annotated with ANNOVAR version 2011-11-28.12 Non-synonymous variants in protein coding regions (232 in total) were selected and analyzed by Sequenom® technology. In addition, we analyzed hotspot mutations in three splicing factor genes, SF3B1, SRSF2 and U2AF1.
The median sequencing coverage in each pool was 4658 and we detected 261 variations with functional effects in protein coding regions of which 41 variations already were reported as germ-line polymorphisms and were excluded from further analysis. Of the 232 variations analyzed by Sequenom®, 85 were identified as real mutations and were included in further analysis. As we noticed a TP53 and ASXL1 mutational frequency lower than expected in patients with del(5q) deletions and tAML, we performed re-sequencing of TP53 in 47 patients (all 20 del(5q) MDS, 18 other MDS and 9 AML samples). We discovered two additional TP53 mutations and three additional ASXL1 mutations. In 3 of these samples, the allele burden was 20% or less. Adding these mutations to the statistical model did not change outcome data, which remained based on the whole cohort.
Sixty-one percent of MDS and 50% of AML patients had mutations in at least one gene, and 22% and 15%, respectively, had mutations in more than one gene. In the MDS cohort, the most frequently mutated genes were SF3B1, TET2, SRSF2 and IDH2 (Figure 1), confirming the results of two recent large studies of MDS.9,10 In the AML cohort, TET2, SRSF2, U2AF1 and IDH2 were the most frequently mutated genes (Figure 1 and Online Supplementary S3). SF3B1 mutations dominated in RARS/RCMD-RS, while mutations in epigenetic regulators dominated in other MDS subtypes. As expected, patients with 5q- syndrome had the lowest frequency of gene mutations, 21%. TET2 mutations were observed more frequently in CMML than in MDS (60% vs. 9%; P=0.001) (Figures 1 and 3A), with SRSF2 being the second most common mutation.13
Figure 1.
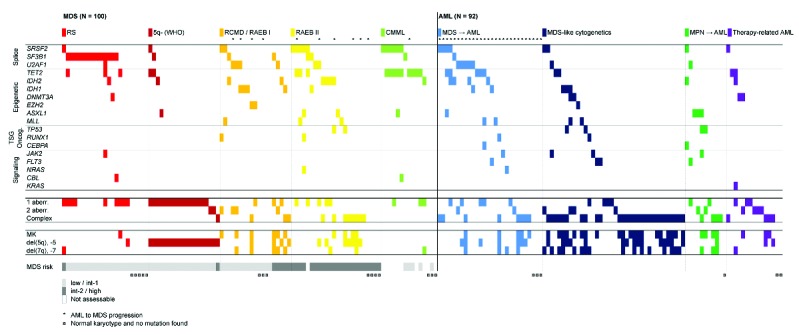
Distribution of mutations and cytogenetic aberrations in 100 patient samples with myelodysplastic syndromes (MDS) and 92 samples with acute myeloid leukemia (AML). Each column represents one patient. The colors represent different subgroups of the diseases. *Denotes patients with MDS who subsequently progressed to AML and AML with a morphologically identified preceding MDS phase. ¤Denotes patients with normal karyotype and no mutation. aberr.: aberration; MK: monosomal karyotype; TSG: tumor suppressor gene.
Figure 3.
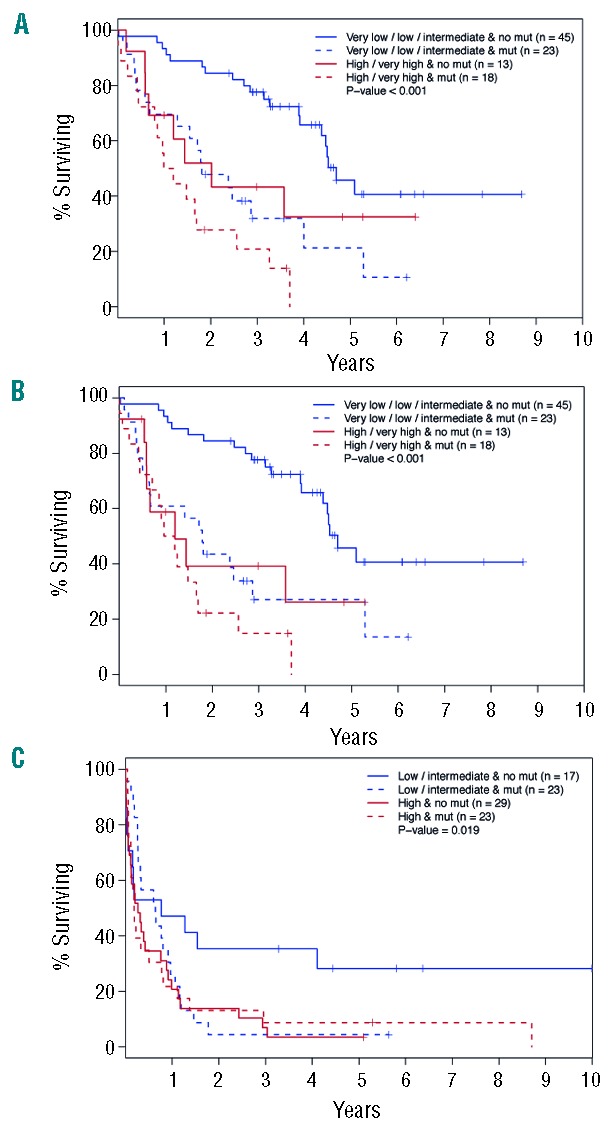
Survival according to risk classification combined with mutational status. (A) Survival in IPSS-R very low+intermediate risk MDS versus high+very high risk myelodysplastic syndromes (MDS) with or without the presence of any mutation other than SF3B1. (B) Acute myeloid leukemia (AML)-free survival in IPSS-R very low+intermediate risk myelodysplastic syndromes (MDS) versus high+very high risk myelodysplastic syndromes (MDS) with or without the presence of any mutation other than SF3B1. (C) Survival in low+intermediate cytogenetic risk acute myeloid leukemia (AML) versus high cytogenetic risk acute myeloid leukemia (AML) with or without the presence of any mutation.
Within the AML cohort, mutational frequencies varied significantly between different subgroups, which indicates that these patients are more heterogeneous than previously thought (Figure 1). MDS-AML shared features with higher risk MDS with mutations predominantly in epigenetic and splicing genes, and with a minor proportion of mutations in signaling genes and oncogenes. By contrast, AML with MDS-like cytogenetics showed lower frequency of splice factor and epigenetic mutations (Figure 1). The mutational pattern of MPN-AML differed from the other AML by a higher incidence of signaling and oncogenic mutations. Interestingly, 87% of patients with tAML showed cytogenetic abnormalities, while the mutational rate was relatively low (33%), indicating that chromosomal aberrations constitute the main driver of disease in these patients. However, albeit only 13% of tAML carried del(5q), an underestimation of TP53 mutations cannot be excluded. In order to discover mutations associated with transformation from MDS to AML in MDS, we grouped MDS patients with subsequent progression to AML with AML patients with morphologically identified preceding MDS and compared them with MDS without AML progression (Figure 2). The only gene that was positively associated with progression was U2AF1 (3 of 11 MDS with AML progression and 5 of 28 MDS-AML, compared to 4 of 89 MDS without transformation; P=0.008). No other mutation was significantly associated with transformation and our data point towards a distinct prognostic role of U2AF1. We also confirm the previously described negative correlation between SF3B1 mutation and risk of AML progression (20 of 89 MDS without transformation versus 1 of 34 with AML transformation; P=0.004).9,10
Figure 2.
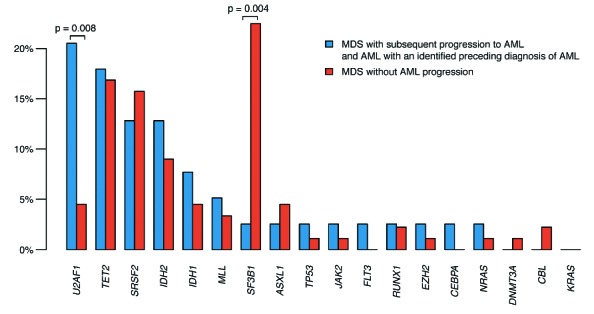
Proportion of patients with a certain gene mutation that did or did not progress from myelodysplastic syndromes to acute myeloid leukemia. Blue bars consist of MDS with subsequent progression to AML and AML with an identified preceding diagnosis of MDS (n=39). Red bars consist of all cases from the myelodysplastic syndromes cohort without a known progression to acute myeloid leukemia (n=89). Median follow up was 57.3 months (9.7–113, SD±27.0) for MDS and 88.5 months (27.6–190, SD ±40.5) for acute myeloid leukemia patients.
We assessed the additive prognostic value of recurrent mutations (Figure 3). As SF3B1 mutations alone were associated with a favorable prognosis both in the univariable analysis and in previous studies,14 patients were grouped as no mutations or SF3B1 mutation only versus any mutation except SF3B1. Furthermore, MDS patients were grouped according to the Revised International Prognostic Score System (IPSS-R) in IPSS-R very low, low and intermediate risk versus high and very high risk. Overall survival (OS) was better in patients with no or SF3B1 mutations than in patients with other mutations, and the difference was more pronounced in the low/intermediate risk group (P<0.001) (Figure 3A). Similar results were obtained when OS was replaced by progression-free survival (P<0.001) (Figure 3B). This strongly supports the additional prognostic value of mutational screening in addition to IPSS-R in MDS.
Acute myeloid leukemia patients were grouped based on cytogenetic risk assessment15 in low risk + intermediate risk (n=40) versus high risk (n=52) and then further divided into mutated and non-mutated patients. No AML patient had SF3B1 mutation only. As illustrated in Figure 3C, the mutational status did not contribute to OS in high-risk AML patients. However, the OS of low + intermediate risk patients with mutations was lower than that of patients without mutations, indicating that gene mutations per se may influence disease pathogenesis (Figure 3C).
Finally, a Cox proportional hazards model was used to analyze OS in the MDS cohort (Online Supplementary Table S2). In univariable analysis, SF3B1 significantly correlated with better, and SRSF2, IDH2, U2AF1 and RUNX1 with worse survival. After adjusting for sex, age and IPSS-R, only SF3B1 and U2AF1 remained independent prognostic factors [mutated vs. non-mutated (HR 0.34, 95%CI: 0.14–0.82) and (HR 2.78, 95%CI: 1.07–7.25)]. In the corresponding model for AML, TP53 and NRAS showed a significant correlation with worse survival, even after adjusting for sex, age and cytogenetic risk, although the number of patients were very few entailing wide confidence intervals.
In conclusion, mutational screening by targeted sequencing provides important clinical information for MDS and AML and will develop to become a prognostic tool in centers managing these disorders. Targeted sequencing will constitute a cornerstone in patient management during the next years. While the pooling approach is adequate for detecting clonal mutations, important subclonal mutations, such as TP53 and ASXL1 may be missed by this strategy. Sequenom® methodology, or equivalent quick genotyping tests, may prove to be a rapid and cost-effective method to detect hotspot mutations.
Acknowledgments
We would like to thank SciLifeLab and MAF core facilities at Karolinska Institutet for their generous help with high-throughput sequencing and mutation analysis services.
Footnotes
Funding: this study was supported by research grants from the Swedish Research Council, Cancerfonden and Karolinska Institutet to EHL, SL and JK.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012; 120(12):2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Manero G, Tambaro FP, Bekele NB, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol. 2012; 30(18):2204–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim ZY, Ingram W, Brand R, et al. Impact of pretransplant comorbidities on alemtuzumab-based reduced-intensity conditioning allogeneic hematopoietic SCT for patients with high-risk myelodysplastic syndrome and AML. Bone Marrow Transplant. 2010;45(4):633–639. [DOI] [PubMed] [Google Scholar]
- 4.Lubbert M, Muller-Tidow C, Hofmann WK, Koeffler HP. Advances in the treatment of acute myeloid leukemia: from chromosomal aberrations to biologically targeted therapy. J Cell Biochem. 2008; 104(6):2059–2070. [DOI] [PubMed] [Google Scholar]
- 5.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008. [Google Scholar]
- 6.Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 7.Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013; 122(17):2943–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.R Core Team. R: A language and environment for statistical computing. 3.0.3 ed. Vienna, Austria: R Foundation for Statistical Computing, 2014. [Google Scholar]
- 9.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627; quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. [DOI] [PubMed] [Google Scholar]