

T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy of thymocytes resulting from the transformation of T-cell progenitors. Around half of T-ALL patients harbor recurrent cytogenetic alterations, including juxtaposition of strong promoters and enhancers located in the TCRB (chr. 7q34) or TCRA-TCRD (chr. 14q11) loci with a variety of oncogenic transcription factors, such as LIM-only domain (LMO) genes, LMO1 and LMO2 resulting in their aberrant expression.1 The LIM-only gene LMO2 encodes a protein that participates in a transcription factor complex, which includes E2A, GATA1, and LDB1, TAL1. LMO2 was reportedly activated in 4 cases of T-ALL arising via retroviral insertion mutagenesis in a gene therapy trial for X-linked severe combined immunodeficiency.2–4 In addition, aberrant expression of LMO2 has been found in 9% of pediatric T-ALL cases,5 though higher figures have also been reported.6 LMO2 is activated via chromosomal translocations t(11;14)(p13;q11) or t(7;11)(q35;p13) and del(11)(p12p13) in T-ALL patients.5 Interestingly, high LMO2 expression levels have also been reported in many T-ALL patients without these changes, suggesting that cryptic LMO2 rearrangements may exist in T-ALL. Recently, we identified LMO2 rearrangements in 5 of 26 (19.2%) T-ALL cell lines including two novel cryptic non-TCR chromosome translocations t(3;11)(q25;p13) and t(X;11)(q25;p13), respectively activating LMO2 by juxtaposition with MBNL1 and STAG2.7 This prompted us to investigate novel rearrangements involving LMO2 in primary samples from T-ALL patients using fluorescence in situ hybridization (FISH) with tilepath BAC/fosmid clones, array-comparative genomic hybridization (CGH), and next generation sequencing (NGS) techniques.
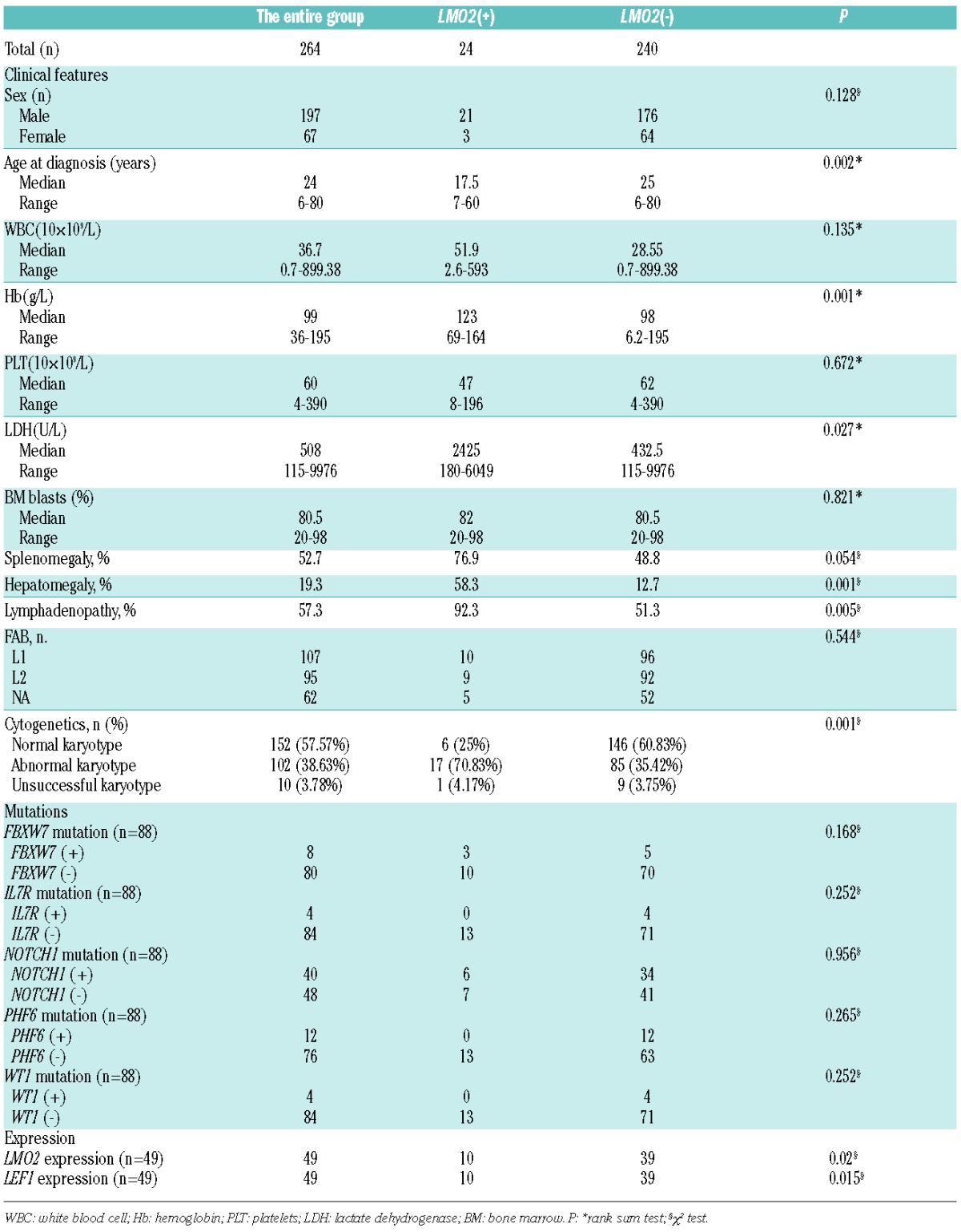
Between July 1997 and April 2013, 409 T-ALL patients were identified following admission to JIH. A total of 264 patients’ samples were included in the present study. The median age of the case series was 24 years (range 6–80 years); the majority were male (74.6%). T-cell phenotype was defined according to the EGIL criteria. Conventional R-banding was used for karyotypic analysis on bone marrow (BM) cells at diagnosis. Clonal karyotypic abnormalities were described according to ISCN.8
We screened all 264 T-ALL patients by FISH using BAC clones on methanol/acetic acid-fixed cells obtained from the BM cultures, as previously described.7 For detection of LMO2 rearrangements, dual color FISH experiments were performed with two contiguous BAC clones: RP11-646J21 and RP11-278N12, respectively labeled with Spectrum Green-dUTPand Spectrum Red-dUTP. LMO2 rearrangements were identified in 9.1% (24 of 264) of patients. Characteristics of the 24 patients are listed in Table 1. The clinical features of LMO2 rearranged versus LMO2 unre-arranged patients are compared in Online Supplementary Table S1. LMO2 rearrangements were significantly associated with younger age, higher hemoglobin concentrations, higher lactate dehydrogenase serum levels, higher frequency of hepatomegaly or lymphadenopathy, and higher frequency of abnormal karyotype.
Table 1.
Clinical and biological characteristics of T-ALL patients.
Among the 24 positive patients, karyotypic analysis revealed chromosomal aberrations involving 11p12-13 in 12 patients: 10 with t(11;14)(p13;q11); 2 with del(11p12). Overall, half of the LMO2 rearrangements were cryptic by routine karyotypic analysis at a rate of 4.5% (12 of 264). LMO2 mRNA expression levels were measured on 10 TALL patients with and 39 without LMO2 rearrangements. The qRT-PCR results showed that LMO2 transcripts were significantly higher in cases with LMO2 rearrangements (P=0.02) than without (Figure 1A). Meanwhile, the mRNA expression levels of LEF1, LYL1, MEF2C, STAG2, SEPT1, TLX1, and TLX3 were also measured by qRT-PCR in these patients, showing no differences between patients with and without LMO2 rearrangements, with the exception of LEF1. Our findings show that patients with LMO2 rearrangements had higher LEF1 transcripts (P=0.015) (Figure 1A), which raises the possibility that LMO2 interactome includes LEF1 in T-ALL, as reported in B-cell lymphomas.9
Figure 1.

Integrative genomic and transcriptional analyses of LMO2 rearrangements in T-ALL patients. (A) The q(uantitative)R(everse)Transcription-PCR results (left) showed that LMO2 transcripts were significantly higher in cases with LMO2 rearrangements (P=0.02) than without. Meanwhile, LEF1 transcripts (right) were significantly higher in cases with LMO2 rearrangements (P=0.015) than without. (B) FISH with RP11-646J21 (green) and RP11-278N12 (red) probes and array-CGH analysis revealed a 475 Kbp of del(11p13p13) including the upstream LMO2 region in T-ALL samples. (C) Whole genome sequencing (WGS) was performed using the Illumina Hiseq 2500 system (Agilent, Santa Clara, CA, USA) according to the manufacturer’s protocol. We identified a fusion between 11p13 (33,856,828 bp) with 7q34 (142,494,025 bp) in case 6 with normal karyotype (left). Dual color FISH experiments with probes RP11-114L10 (TCRB, red) and RP11-646J21 (LMO2, green) confirmed the balanced translocation between 7q34 and 11p13 (right). (D) WGS identified a fusion between 11p13 (33,957,035 bp) and 14q32 (98,842,615 bp) in case 10 with complex karyotype (left). Dual color FISH experiments with RP11-646J21 (green) and RP11-278N12 (red) probes confirmed the involvement of LMO2 in this patient (right). (E) LMO2 and BCL11B breakpoints in T-ALL. Diagram shows distribution of translocation breakpoints at chromosome 11p13 and 14q32.2 previously reported in T-ALL.7,13 Arrows indicate patient breakpoints above and cell lines below co-ordinate plots. The t(7;11)(q35;q13) and t(11;14)(q13;q32) breakpoints mapped in this report are indicated by a diamond and asterisk, respectively. The black wedge (“E”) shows a remote downstream enhancer region characterized by us previously,13 which coincides with the distal BCL11B breakpoint cluster region boundary. Note placement (right figure) of the LMO2-BCL11B breakpoint amid other BCL11B partners, TLX3 and NKX2-5, consistent with analogous activation mechanisms for all three oncogene targets. Note also contrasting non-canonical placement (left figure) of the LMO2-BCL11B patient breakpoint upstream of LMO2 breakpoints all of which involved TCR loci. The same patient breakpoint lay instead amid other non-TCR cell-line breakpoints (bullets), all located more distally upstream of LMO2, implying mechanistic differences between the oncogene activation mechanisms of TCR and non-TCR LMO2 translocations. (F) FISH with RP11-646J21 (green) and RP11-278N12 (red) probes and whole chromosome painting probe for chromosome 2 revealed a translocation between LMO2 with the short arm of chromosome 2 in case 15 with 47,XY,t(1;1)(p33;q41),t(2;11)(p15;p15),i(7q),+12[10]. (G) FISH analysis revealed simultaneous involvement of TCRB and TCRA-TCRD in case 17. Dual color FISH experiments with probes RP11-114L10 (TCRB, red) and RP11-646J21 (LMO2, green) confirmed the rearrangement between 7q34 and 11p13 (left). Meanwhile, FISH with probes RP11-440N18 (MYC, red) and RP11-256C2 (TCRA-D, green) confirmed the rearrangement between 8q24 and 14q11 (right).
Furthermore, to explore undetectable cytogenetic abnormalities, we performed integrative genomic and transcriptional analyses on these 24 T-ALL patients with LMO2 rearrangements. FISH with RP11-646J21/278N12 probes revealed del(11p13p13) in 6 patients including 2 with del(11p12) according to routine karyotyping (Table 1). Array-CGH analysis confirmed respective 95 Kbp and 475 Kbp deletions including the upstream LMO2 region at 11p13 in 2 T-ALL samples (Figure 1B).
In addition, we performed whole genome sequencing in 2 LMO2 rearranged T-ALL patients (cases 6 and 10) without t(11;14)(p13;q11) or del(11)(p12p13). We performed sequencing to a mean coverage of 50× in each sample. In case 6 (normal karyotype), we identified fusion between 11p13 (33,856,828 bp) and 7q34 (142,494,025 bp). Two segments, respectively 7-bp and 16-bp, of unknown origin were inserted at 11p13 and 7q34 breakpoints. PCR and bidirectional Sanger sequencing confirmed the presence of chimeric product. The 11p13 breakpoint lay ~23 Kbp downstream of LMO2, while the 7q34 breakpoint lay within the TCRB gene. Dual color FISH experiments confirmed a balanced translocation between 7q34 and 11p13 (Figure 1C). Thus, we identified a rather rare translocation, t(7;11)(q35;p13), in case 6, which has been reported only in a very few T-ALL patients hitherto.10
In case 10, we identified a fusion between 11p13 (33,957,035 bp) and 14q32 (98,842,615 bp). PCR and bidirectional Sanger sequencing confirmed the presence of chimeric product. The 11p13 breakpoint lay ~43 Kbp upstream of LMO2. The 14q32 breakpoint was located between C14orf64 and C14orf177 genes, ~793 kbp downstream of BCL11B. We recently described a cluster of powerful T-cell enhancers in 3′-BCL11B which can activate homeobox oncogenes NKX2-5 and HOX11L2 by juxtaposition in cytogenetically identical t(5;14)(q35;q32.2).11–13 The corresponding region in mice has been shown to control specificity of T-cell expression therein.14 It is interesting to note that this patient had the highest LMO2 transcription level as shown by qRT-PCR in the 49 T-ALL patients mentioned above. We, therefore, propose that the LMO2 gene is deregulated by juxtaposition with 3´-BCL11B via a novel t(11;14)(p13;q32.2) rearrangement (Figure 1D). In T-ALL, cytogenetic alterations juxtaposing LMO2 with strong promoters and enhancers of T-cell receptor loci are recognized as the main activating mechanism. Placement of patient breakpoints often provides clues to the underlying leukemogenic mechanisms involved. The respective breakpoint regions at 11p13/LMO2 and 14q32.2/BCL11B are shown in Figure 1E. While the breakpoint at 14q32 lay amid the far distal downstream cluster which we reported previously where NK-family homeobox genes are activated,13 that at 11p13 lay upstream of those involved in TCR- LMO2 rearrangements where it clustered together with MBNL1 and STAG2 additional non- TCR-LMO2 partners which we described recently.7 FISH analysis with BAC clones in 11p13 and whole chromosome painting confirmed the translocation between LMO2 with the short arm of chromosome 2 in another patient with 47,XY,t(1;1)(p33;q41),t(2;11)(p15;p13),i(7q),+12[10] (case 15) (Figure 1F). Chromosome 2p15 has yet to be assigned a recurrent oncogene target in T-ALL to serve as candidate LMO2 partner in this case. Taken together, these findings imply that the imputed activation of LMO2 by non-TCR loci is mechanistically distinct from canonical translocation disease, a conclusion of potential therapeutic relevance.
Interestingly, we identified simultaneous involvement of TCRB and TCRA-TCRD in case 17 by FISH screening. Further FISH characterization indicated that LMO2 is activated via formation of t(7;11)(q35;p13), and MYC via t(8;14)(q24;q11) in this patient (Figure 1G).
To determine the association of LMO2 rearrangements with other recurrent gene mutations in T-ALL, we investigated gene mutations by PCR and direct Sanger sequencing in a cohort of 88 T-ALL patients for whom genomic DNA was available, including 13 with LMO2 rearrangements. After excluding known polymorphisms and silent mutations, mutations of FBXW7, IL7R, NOTCH1, PHF6 and WT1 were respectively detected in 8 (9.1%), 4 (4.5%), 40 (45.5%), 12 (13.7%), and 4 (4.5%) of these 88 patients (Online Supplementary Table S1). There were no significant differences in the incidence of FBXW7 (P=0.168), IL7R (P=0.252), NOTCH1 (P=0.956), PHF6 (P=0.265), and WT1 mutated cases (P=0.252) with and without LMO2 rearrangements. We also sequenced the entire coding region of LMO2 in 117 T-ALL patients and found no somatic mutation. Taken together, LMO2 rearrangements were identified in 9.1% (24 of 264) of T-ALL patients of which 50% (12 of 24) were deemed cryptic. The LMO2 transcripts were significantly higher in cases with LMO2 rearrangements than without. Moreover, we detected non-TCR chromosome translocations activating LMO2 in 2 T-ALL patients, suggesting that non-TCR chromosome translocations activating LMO2 are recurrent in T-ALL at significant levels. It is worthy of note that we identified a novel t(11;14)(p13;q32.2) translocation which activates LMO2 by juxtaposition with remote leukemic enhancers of 3′-BCL11B using whole genome sequencing. Our results indicate that LMO2 is a novel partner gene of BCL11B in T-ALL besides TLX3 and NKX2-5.
Footnotes
Funding: this work was supported by grants from National Key Scientific Projects of China (2011CB933501), the Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Province’s Key Provincial Talents Program, the National Natural Science Foundation of China (81100372, 81200370), National Public Health Grand Research Foundation (No.201202017), and Foundation of Jiangsu Province Health Department (H200915).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dik WA, Nadel B, Przybylski GK, et al. Different chromosomal breakpoints impact the level of LMO2 expression in T-ALL. Blood. 2007;110(1):388–392. [DOI] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. [DOI] [PubMed] [Google Scholar]
- 3.McCormack MP, Rabbitts TH. Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2004;350(9):913–922. [DOI] [PubMed] [Google Scholar]
- 4.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional muta-genes combined with acquired somatic mutations causes leukemo-genesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Vlierberghe P, van Grotel M, Beverloo HB, et al. The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood. 2006;108(10):3520–3529. [DOI] [PubMed] [Google Scholar]
- 6.Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(1):75–87. [DOI] [PubMed] [Google Scholar]
- 7.Chen S, Nagel S, Schneider B, et al. Novel non-TCR chromosome translocations t(3;11)(q25;p13) and t(X;11)(q25;p13) activating LMO2 by juxtaposition with MBNL1 and STAG2. Leukemia. 2011;25(10):1632–1635. [DOI] [PubMed] [Google Scholar]
- 8.Shaffer LG, Slovak ML, Campbell LJ, eds. ISCN 2009: an international system for human cytogenetic nomenclature. Basel: Karger; 2009. [Google Scholar]
- 9.Cubedo E, Gentles AJ, Huang C, et al. Identification of LMO2 transcriptome and interactome in diffuse large B-cell lymphoma. Blood. 2012;119(23):5478–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitzgerald TJ, Neale GA, Raimondi SC, Goorha RM. Rhom-2 expression does not always correlate with abnormalities on chromosome 11 at band p13 in T-cell acute lymphoblastic leukemia. Blood. 1992;80(12):3189–3197. [PubMed] [Google Scholar]
- 11.MacLeod RA, Nagel S, Kaufmann M, Janssen JW, Drexler HG. Activation of HOX11L2 by juxtaposition with 3′-BCL11B in an acute lymphoblastic leukemia cell line (HPB-ALL) with t(5;14)(q35;q32.2). Genes Chromosomes Cancer. 2003; 37(1):84–91. [DOI] [PubMed] [Google Scholar]
- 12.Nagel S, Kaufmann M, Drexler HG, MacLeod RA. The cardiac homeobox gene NKX2–5 is deregulated by juxtaposition with BCL11B in pediatric T-ALL cell lines via a novel t(5;14)(q35.1;q32.2). Cancer Res. 2003;63(17):5329–5334. [PubMed] [Google Scholar]
- 13.Nagel S, Scherr M, Kel A, et al. Activation of TLX3 and NKX2–5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3′-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res. 2007;67(4):1461–1471. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Zhang JA, Dose M, et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood. 2013;122(6):902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]