

Abstract
The multifunctional nature of coated pit receptors predicts that these proteins will contain multiple domains. To establish the genetic basis for these domains, we have determined the exon organization of the gene for the low-density lipoprotein (LDL) receptor. This gene is more than 45 kilobases in length and contains 18 exons, most of which correlate with functional domains previously defined at the protein level. Thirteen of the 18 exons encode protein sequences that are homologous to sequences in other proteins: five of these exons encode a sequence similar to one in the C9 component of complement; three exons encode a sequence similar to a repeat sequence in the precursor for epidermal growth factor (EGF) and in three proteins of the blood clotting system (factor IX, factor X, and protein C); and five other exons encode nonrepeated sequences that are shared only with the EGF precursor. The LDL receptor appears to be a mosaic protein built up of exons shared with different proteins, and it therefore belongs to several supergene families.
Cell surface receptors are multifunctional proteins with binding sites that face the external environment and effector sites that couple the binding to an intracellular event. Many receptors have an additional function: they transport bound ligands into cells (1). Such receptor-mediated endocytosis requires that the proteins have specific domains that allow them to cluster within clathrin-coated pits on the plasma membrane and in many cases to recycle to the cell surface after ligand delivery (2).
The multiple functions of coated pit receptors imply that they will have multiple domains, each with a single function. The structural features responsible for these functions are currently the subject of intense study. Recent insights have emerged from the complementary DNA (cDNA) cloning of the messenger RNA’s (mRNA) for several receptors and the subsequent determination of their amino acid sequences. These studies have revealed surprising homologies between the primary structures of receptors and other proteins. For example, the receptor for plasma low-density lipoprotein (LDL), a cholesterol transport protein, contains one region that is homologous to the precursor of a peptide hormone, epidermal growth factor (EGF) (3, 4), and another region that is homologous to complement component C9, the terminal component of the complement cascade (5). The cell surface receptor for immunoglobulin A/immunoglobulin M is homologous to the immunoglobulins themselves (6). Finally, the receptor for EGF is homologous to a viral and cellular gene, erb-B, that produces a protein with tyrosine kinase activity (7).
These findings suggest that coated pit receptors share functional domains with other proteins. One likely mechanism for such sharing is through the duplication and migration of exons during evolution (8). Although the cDNA’s for five coated pit receptors have been isolated and sequenced (4, 6, 9), the organizations of the genes encoding these proteins are not yet known. The elucidation of the gene structures of coated pit receptors should reveal the relationships between exons and protein domains and provide insight into the evolution of this important class of cell surface molecules.
Here, we report the exon organization of the gene for the human LDL receptor, a classic example of a cell-surface protein that mediates endocytosis through coated pits. A close correlation between functional domains in the LDL receptor protein and the exon-intron organization of the gene is revealed. In an accompanying report (10), we show that genomic sequences that are shared between the human LDL receptor and the human EGF precursor have a similar exon-intron organization, suggesting that coated pit receptor genes may have been assembled during evolution from itinerant exons encoding discrete protein domains.
Protein domains of LDL receptor
We recently isolated a full-length 5.3-kilobase (kb) cDNA for the human LDL receptor (4). The amino acid sequence as deduced from the nucleotide sequence revealed that the receptor is synthesized as a precursor of 860 amino acids. The first 21 amino acids constitute a typical hydrophobic signal sequence that is cleaved from the protein prior to its appearance on the cell surface, leaving an 839-amino-acid mature protein with five recognizable domains.
The first domain of the mature receptor contains the binding site for apoproteins B and E of LDL and of related lipoproteins. This domain consists of ~300 amino acid residues (4), which is assembled from multiple repeats of 40 residues each. Each repeat has six cysteine residues, all of which are involved in disulfide bonds (4). This repeated 40-amino-acid unit bears a strong homology to a single 40-amino-acid sequence that occurs within the cysteine-rich region of human complement component C9, a plasma protein of 537 amino acids (5).
The second domain of the human LDL receptor is a sequence of about 400 amino acids that was found to be homologous to the precursor for mouse EGF (3, 4). The EGF precursor is a protein of 1217 amino acids that may be synthesized as a membrane protein with a short cytoplasmic tail at the COOH-terminus (11–14). The EGF sequence of 53 amino acids lies immediately external to the hydrophobic membrane segment, from which it is presumably released by proteolysis. In earlier studies we found that the human LDL receptor was homologous to a large part of the external domain of the mouse EGF precursor, but not to EGF itself. Within a stretch of 400 amino acids, 33 percent of the residues are identical between the two proteins (3, 4).
The third domain of the LDL receptor is a sequence of 48 amino acids that contains 18 serines and threonines, many of which appear to serve as attachment sites for O-linked carbohydrate chains (3, 4). The fourth domain is a 22-amino-acid hydrophobic membrane-spanning region, and the fifth domain is a 50-amino-acid COOH-terminal cytoplasmic tail (3, 4).
Genomic cloning of LDL receptor
A series of bacteriophage λ and cosmid clones that span most of the LDL receptor gene were isolated (Fig. 1). These clones include the 18 exons that encode the LDL receptor protein and most of the 17 introns that separate them. Within two introns (located between exons 1 and 2 and between exons 6 and 7), there are gaps that are not covered by the genomic clones.
Fig. 1.

Map of the human LDL receptor gene. The gene is shown in the 5′ to 3′ orientation at the top of the diagram and is drawn to scale. Exons are denoted by filled-in areas, and introns by open areas. The regions encompassed by genomic DNA inserts in the seven bacteriophage λ and two cosmid clones are indicated at the bottom. Cleavage sites for 13 selected restriction endonucleases are shown. Asterisks denote sites that are present in the cDNA. The encircled Pvu II site is polymorphic in human populations (30). The diagonal line between exons 1 and 2 represents a gap of unknown size not present in any of the genomic clones. Additional cleavage sites for the restriction enzymes shown may be present in this gap and in intron 6 (Table 1, legend). The λ clones were isolated from 1.2 × 107 plaques of a human genomic bacteriophage λ library (31). Cos1 was isolated from 6 × 106 colonies of a human cosmid library (32). Cos26 was isolated from 0.9 × 106 colonies of a human cosmid library (33). The libraries were screened with 32P-labeled probes derived from the human LDL receptor cDNA, pLDLR-2 (4). Probes were isotopically labeled by nick translation (34) or hexanucleotide priming (35) and screening was carried out with standard procedures (34). Positive clones were plaque-purified or isolated as single colonies. Thirty fragments from the nine genomic clones were subcloned into pBR322 and characterized by restriction endonuclease digestion, Southern blotting (34), and DNA sequencing of exon-intron junctions (see Table 1). The restriction map was verified by comparing overlapping and independently isolated genomic clones and by Southern blotting analysis of genomic DNA isolated from normal individuals.
Exons within the cloned genomic DNA were identified in plasmid sub-clones by restriction mapping and by Southern blotting with cDNA probes (Fig. 1). The nucleotide sequences at the exon-intron boundaries (Table 1) were established by DNA sequence comparison of cDNA and genomic subclones. The 5′ donor and 3′ acceptor splice sites in each of the 17 introns conform to the GT … AG rule (G, guanine; T, thymine; A, adenine) and agree well with consensus sequences compiled for the exon-intron boundaries of other genes (15).
Table 1.
Exon-intron organization of the human LDL receptor gene. The nucleotide sequences of exon-intron junctions were determined by the enzymatic method (36) with the use of recombinant bacteriophage M13 clones containing genomic fragments as templates and the universal primer (37) together with a family of 25 LDL receptor–specific oligonucleotide primers. Exons 1 to 3 and 5 to 17 were sequenced in their entirety; intron 13 was sequenced in its entirety. Exon sequences are in capital letters; intron sequences are in lowercase letters. The number shown immediately below the DNA sequence denotes the nucleotide position at which the intron interrupts the LDL receptor mRNA (4). The numbers shown in parentheses in the extreme right-hand column denote the position of the indicated amino acid in the mature LDL receptor protein (4). The size for intron 1 is a minimal estimate based on concordance between data obtained from Southern blotting experiments with genomic DNA and data obtained from the restriction maps of the genomic clones. Approximately 2 kb of intron 6 was present in the genomic clones (Fig. 1); the amount of DNA not present in the genomic clones (0.7 kb) was estimated by Southern blotting of genomic DNA using probes derived from exons 6 and 7.
| Exon num- ber |
Exon size (bp) |
Sequence at exon-intron junction | Intron length (kb) |
Amino acid inter- rupted |
|
|---|---|---|---|---|---|
| 5′ Splice donor | 3′ Splice acceptor | ||||
| 1 | 145–160 | >10 | Val (2) | ||
| 2 | 123 | 2.5 | Leu (43) | ||
| 3 | 123 | 2.4 | Pro (84) | ||
| 4 | 381 | 0.95 | Ala (211) | ||
| 5 | 123 | 0.85 | Val (252) | ||
| 6 | 123 | 2.7 | Gly (293) | ||
| 7 | 120 | 0.45 | Asp (333) | ||
| 8 | 126 | 1.2 | Gly (375) | ||
| 9 | 172 | 0.9 | Ser (432) | ||
| 10 | 228 | 2.6 | Gly (508) | ||
| 11 | 119 | 0.6 | Asp (548) | ||
| 12 | 140 | 3.0 | Glu (594) | ||
| 13 | 142 | 0.134 | Gly (642) | ||
| 14 | 153 | 2.8 | Glu (693) | ||
| 15 | 171 | 5.5 | Ala (750) | ||
| 16 | 78 | 1.4 | Val (776) | ||
| 17 | 158 | 1.7 | Ser (828) | ||
| 18 | 2535 | ||||
Characterization of 5′ end of LDL receptor gene
Figure 2 shows the nucleotide sequence of the 5′ end of the human LDL receptor gene, with the A of the initiator methionine codon designated as position +1 and the nucleotide positions to the 5′ side of this region designated by negative numbers. To determine the point at which transcription initiates, we performed an S1 nuclease analysis with poly(A)+ RNA (polyadenylated) isolated from SV40-transformed human fibroblasts or adult human adrenal glands (Fig. 3). The probe was a single-stranded DNA labeled with 32P at the 5′ end and encompassing nucleotides −682 to +43 of the genomic sequence (Fig. 2). The sizes of the protected fragments were estimated by comparison with a dideoxynucleotide sequence established with an oligonucleotide primer and a recombinant bacteriophage M13 template (right-hand lanes in Fig. 3). We observed a cluster of three major S1 nuclease-protected fragments whose lengths corresponded to protection up to positions −79 to −89 of the genomic sequence. A small amount of an additional S1 nuclease-protected product was seen at a position corresponding to −57. Poly(A)+ RNA from fibroblasts and adrenal tissue gave the same S1 nuclease-protected fragments. All of the protected fragments were drastically reduced in amount when the poly(A)+ RNA was obtained from fibroblasts grown in the presence of sterols, conditions that reduce the amount of receptor mRNA (4).
Fig. 2.

Nucleotide sequence of the 5′ end of the human LDL receptor gene. Nucleotide position +1 is assigned to the A of the ATG codon specifying the initiator methionine; negative numbers refer to 5′ flanking sequences. Amino acids encoded by the first exon and the position of intron 1 are indicated on the bottom line. Vertical arrows above the sequence indicate sites of transcription initiation as determined by S1 nuclease mapping (Fig. 3). Vertical arrows below the sequence indicate sites of transcription initiation as determined by primer extension (Fig. 4). Asterisks denote an apparent S1 nuclease hypersensitive site (above sequence) or a strong stop point for reverse transcriptase (below sequence). Two AT-rich regions that are located 20 to 30 nucleotides upstream of the mRNA start points are boxed. Solid horizontal arrows denote three imperfect direct repeats of 16 nucleotides each. Dashed arrows denote two imperfect inverted repeats of 14 nucleotides each. The DNA sequence was determined by a combination of the chemical (36) and enzymatic (37) methods. Two M13 subclones derived from the bacteriophage genomic clone λ34 were used as templates together with the universal primer (38) and five LDL receptor-specific oligonucleotide primers to establish the sequence by the enzymatic method. Selected regions were then sequenced again by the chemical method; 90 percent of the sequence was determined on both strands of the DNA.
Fig. 3.

Sites of transcription initiation in the human LDL receptor gene as determined by S1 nuclease analysis. The indicated amount of yeast tRNA (lanes 1 and 2), poly(A)+ RNA from SV40-transformed human fibroblasts (lanes 3 to 6), or poly(A)+ RNA from adult adrenal glands (lane 7) was annealed to a 5′ end-labeled with 32P, single-stranded DNA probe corresponding to nucleotides −682 to +43 of Fig. 2. The fibroblasts were grown in the presence or absence of sterols as indicated. The RNA-DNA hybrids were digested with S1 nuclease, and the resistant products were subjected to electrophoresis through a 10 percent polyacrylamide-7M urea gel and detected by autoradiography for 72 hours at −20°C. SV40-transformed fibroblasts were set up in roller bottles (3 × 106 cells per bottle) and grown under standard conditions with fetal calf serum (10 percent) for 48 hours (39). On day 2, one-half of the roller bottles were switched to medium containing 10 percent calf lipoprotein-deficient serum and 10 µM compactin in the absence of sterols. The other half of the roller bottles were switched to medium containing 10 percent newborn calf serum in the presence of 25-hydroxycholesterol (3 µg/ml) plus cholesterol (12 µg/ml). The cells were incubated for 24 hours and harvested for the preparation of poly(A)+ RNA (4). Adult adrenal glands (obtained from human cadavers at the time of removal of kidneys for transplantation) were frozen at −70°C until preparation of poly(A)+ RNA. The 5′ end-labeled, single-stranded 35P probe of 725 nucleotides was prepared by priming an M13 clone containing a fragment of the LDL receptor gene corresponding to nucleotides −686 to +66 (Fig. 2) with a 32P-labeled synthetic oligonucleotide complementary to nucleotides +19 to +43 (Fig. 2). The synthetic oligonueleotide was labeled at the 5′ end with [γ-32P]ATP (7000 Ci/mmol) and polynucleotide kinase (34) to a specific radioactivity of ~5 × 106 cpm/pmol. After primer extension with the Klenow fragment of DNA polymerase I in the presence of each of the four deoxynucleoside triphosphates (15 µM), the resulting double-stranded DNA was cleaved with Bam HI, and the radioactive probe fragment was purified by electrophoresis on a denaturing polyacrylamide gel and subsequent electroelution (34). A portion of the probe (105 cpm) was coprecipitated with the indicated amount of tRNA or poly(A)+ RNA in ethanol at −70°C. The precipitated material was resuspended in 20 µl of 80 percent formamide, 0.4M NaCl, 40 mM 1,4-piperazinediethane-sulfonic acid (pH 6.4), and 1 mM EDTA and hybridized at 65°C for 36 hours. The samples were diluted with 9 volumes of 0.25M NaCl, 30 mM potassium acetate (pH 4.5), 1 mM ZnSO4, and 5 percent glycerol; treated with 200 units of S1 nuclease (Bethesda Research Laboratories) at room temperature for 60 minutes (40); precipitated with ethanol; and analyzed on a sequencing gel. The protected fragments were compared with the adjacent dideoxy nucleotide–derived sequencing ladder obtained with the same primer and M13 template used to generate the probe. Numbers on the left denote the estimated nucleotide position corresponding to the 5’ end of the protected fragments according to the numbering scheme of Fig. 2. The sequence in the −78 to −97 region is shown on the right.
To determine which S1 nuclease-protected fragments reflected transcription initiation sites, we performed a primer extension experiment (Fig. 4). We used poly(A)+ RNA from cultured human A-431 carcinoma cells and SV40-transformed human fibroblasts grown in the absence or presence of sterols. To obtain a primer that would give the required sensitivity, we isolated a single-stranded, uniformly 32P-labeled fragment of 93 nucleotides that was complementary to the 5′ end of the protein coding region of the mRNA. When this primer was extended on the poly(A)+ mRNA template, three major products were observed whose length corresponded to transcription initiation sites between positions −84 to −93. In addition, we observed a shorter extension fragment corresponding to a 5′ end at position −29. When the A-431 cells or fibroblasts were grown in the presence of sterols, none of these primer-extended products was seen, confirming that they were derived from the receptor mRNA.
Fig. 4.

Sites of transcription initiation in the human LDL receptor gene as determined by primer extension analysis. Poly(A)+ RNA was obtained from human A-431 epidermoid carcinoma cells (lanes 1 and 2) or SV40-transformed human fibroblasts (lanes 3 and 4) that had been grown in the absence or presence of sterols. The RNA was annealed to a uniformly labeled, single-stranded 32P probe [corresponding to nucleotides +9 to +101, Fig. 2 and (4)] that served as a primer for extension by reverse transcriptase. The primer-extended products were subjected to electrophoresis through a 10 percent polyaerylamide-7M urea gel and detected after autoradiography for 60 hours at −20°C. The poly(A)+ RNA from A-431 cells and SV40-transformed fibroblasts cultured in the absence or presence of sterols was prepared as described in the legend of Fig. 3. A single-stranded, uniformly 32P-labeled primer complementary to nucleotides +9 to +101 of the human LDL receptor mRNA was derived from an M13 cDNA clone (38) as described (41). A portion (approximately 2 × 106 cpm) of the 93-nucleotide 32P-primer was precipitated with ethanol together with 10 µg of the indicated poly(A)+ RNA; resuspended in 5 µl of a buffer containing 50 mM tris-chloride (pH 8.0), 50 mM KCl, 5 mM MgCl2, and 20 mM dithiothreitol; and sealed in a glass capillary. The reaction mixture was denatured by boiling for 2 minutes, and primer-template complexes were allowed to form at 65°C for 4 hours. After this annealing period, the entire solution was transferred to a plastic microfuge tube containing 2.5 µl of a 1 mM solution of the four deoxynucleoside triphosphates and 6 units of avian myeloblastosis virus reverse transcriptase (Molecular Genetic Resources). Primer extension was allowed to occur for 50 minutes at 37°C and was stopped by the addition of 6 µl of a formamide-dye mix. The sample was boiled for 5 minutes, quickly chilled on ice, and subjected to electrophoresis on a sequencing gel. Size standards, shown on the right, were generated by dideoxy nucleotide sequencing (37) of a known M13 recombinant clone. Numbers on the left denote the calculated position corresponding to the limits of primer extension according to the numbering scheme of Fig. 2. The intense band at the bottom of lanes 1 to 4 represents the 32P-labeled primer used in the experiment.
The three sites of transcription initiation as determined by the S1 nuclease technique and the three sites determined by the primer extension are indicated in Fig. 2. In general, these sites are well correlated with each other, except that the S1 nuclease technique systematically yields fragments that are 4 to 5 base pairs shorter than those obtained by primer extension. This may represent some “nibbling” by the S1 nuclease at the ends of the protected fragments. In contrast, the single shorter fragments seen in the S1 nuclease and primer extension experiments did not correspond to each other (see asterisks in Fig. 2). It is possible that the shorter S1 nuclease-generated fragment represents an S1 nuclease hypersensitive site and that the shorter primer extended product arises as a consequence of a strong stop sequence for reverse transcriptase in the mRNA template.
Considered together, the S1 nuclease and primer extension experiments indicate that there is no intron in the 5′ untranslated region of the LDL receptor gene and that transcription initiates at three closely spaced sites located between positions −79 and −93. This conclusion is supported by Northern blotting results showing that synthetic oligonucleotide or restriction fragment probes derived from DNA sequences upstream of position −102 do not hybridize to the LDL receptor mRNA, whereas probes derived from sequences downstream of position −64 do hybridize to the mRNA (data not shown). Approximately 20 to 30 base pairs to the 5′ side of the mRNA initiation sites at −79 to −93 are two AT-rich sequences (TTGAAAT and TGTAAAT) that might serve as TATA boxes (16). The existence of two such closely spaced sites might explain the multiple closely spaced transcription initiation sites. To the 5′ side of these AT-rich sequences, we did not find the sequence CCAAT (C, cytosine), which is conserved in some but not all eukaryotic genes (16).
The findings derived from the experimental data shown in Figs. 3 and 4 confirm previous results that the amount of mRNA for the LDL receptor is reduced when cells are incubated with sterols (4, 17). If the control of receptor mRNA expression is similar to that of other genes, then the amount of mRNA is likely to be determined by the rate of transcription, and DNA sequences in the 5′ end of the gene are likely to be responsible for this regulation. In the 5′ end of the gene, there are three imperfect direct repeat sequences of 16 nucleotides each that are located just upstream of the clustered sites of transcription initiation (Fig. 2, solid arrows). This same region contains an imperfect inverted repeat sequence of 14 nucleotides (Fig. 2, dashed arrows). Repeat sequences of 12 nucleotides in the 5′ end of the mouse metallothionien gene have recently been shown to mediate heavy metal inducibility (18). Thus, the direct repeats noted above in the 5′ end of the LDL receptor gene may play a role as sterol regulatory elements. Linking the LDL receptor sequences to heterologous marker genes may provide insight into the regulation of mRNA expression by sterols.
Exon organization and protein domains
The arrows in Fig. 5 show the position of each intron in the LDL receptor gene in relation to the domains of the protein that were identified earlier on the basis of protein sequence and proteolysis studies (3, 4). The introns interrupt the protein coding sequence in such a way that many of the protein segments are revealed as products of individual exons. Exon 1 encodes the short 5′ untranslated region plus the signal sequence of the protein. The first intron interrupts the coding sequence two amino acids distal to the end of the signal sequence; thus, the signal sequence is contained in a discrete exon (Fig. 5).
Fig. 5.

Exon organization and protein domains in the human LDL receptor. The six domains of the protein are delimited by thick black lines and are labeled in the lower portion. The seven cysteine-rich, 40-amino acid repeats in the LDL binding domain (Fig. 6) are assigned the roman numerals I to VII. Repeats IV and V are separated by eight amino acids. The three cysteine-rich repeats in the EGF precursor homology domain (Fig. 8) are lettered A to C. The positions at which introns interrupt the coding region are indicated by arrowheads. Exon numbers are shown between the arrowheads.
An informative set of introns occurs in the cysteine-rich repeat region that contains the LDL binding domain. The initial analysis of the protein sequence derived from cDNA clones suggested a total of eight repeats in this region, of which the first seven were strongly homologous (4). The eighth repeat showed weaker homology but retained a cysteine-rich character. Analysis of the exon structure now reveals that the first seven of these sequences belong to one repeat sequence family (Figs. 5 and 6). Each repeat in this family is about 40 residues long, and each contains six cysteine residues spaced at similar intervals. In 19 of the 40 amino acid positions, a single amino acid occurs in more than 50 percent of the repeats, allowing a consensus sequence to be written (Fig. 6A). This consensus shows a striking preponderance of acidic residues; all of the charged residues that are conserved bear a negative charge (Fig. 6B). This clustering of negative charges is particularly strong in the sequence Asp-Cys-X-Asp-Gly-Ser-Asp-Glu, which occurs near the COOH-terminal end of each repeat. These clustered negative charges may account for the ability of the LDL receptor to bind to closely spaced positively charged residues on apoprotein E, a high affinity ligand for the LDL receptor (4, 19).
Fig. 6.

Location of introns in the cysteine-rich repeat region of the binding domain of the LDL receptor. The amino acids constituting each of the seven repeat units are numbered in the left column according to the translated sequence of the receptor cDNA (4). (A) Optimal alignment was made by the computer programs ALIGN and RELATE (4) with modifications based on the location of introns. Amino acids that are present at a given position in more than 50 percent of the repeats are boxed and shown as a consensus on the bottom line. Cysteine residues (C) in the consensus sequence are underlined. The positions at which introns interrupt the coding sequence of the gene are denoted by the encircled amino acids. The single letter amino acid code translates to the three letter code as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; Y, Tyr. (B) The net charge of each of the amino acids in (A) is shown. All of the conserved amino acids that are charged bear a negative charge; none are positively charged.
As originally noted by Stanley et al. (5), each of the seven 40–amino acid repeats in the LDL receptor is strongly homologous to a single 40-residue unit in complement factor C9. Of the 19 conserved amino acids in the LDL receptor repeats, 14 are found in the C9 sequence, including the highly conserved negatively charged cluster (Fig. 7).
Fig. 7.
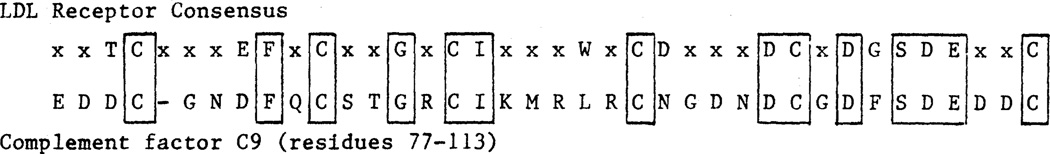
Comparison of consensus sequence in the binding domain of the LDL receptor (Fig. 6A) with the homologous sequence from complement factor C9 (5).
Support for the seven-repeat model of the binding domain for the LDL receptor comes from analysis of intron placement (Fig. 6A, circled residues). Introns are located immediately before the first repeat (two residues after the signal sequence cleavage point) and precisely after repeats I, II, V, VI, and VII (Fig. 6). Thus, four of the repeats are each encoded by individual exons. The other three repeats (III, IV, and V) are all contained in a single exon. The introns between repeats III, IV, and V must have been lost after the primordial exon had undergone duplication to produce the seven repeats. Loss of introns during evolution has been postulated to occur (20). Interestingly, all of the introns in this region interrupt codons at the same location, that is, after the first base of the codon (Table 1). Thus, any or all of the exons in the binding domain could possibly be spliced in or out of the mRNA without disturbing the reading frame. Such alternative splicing could produce receptors with different affinities or specificities for apoproteins B or E, the two ligands for the LDL receptor. Differential in-frame splicing of exons in protein coding regions has been reported for several other genes (21).
In previous studies we found that the sequence of amino acid residues 290 to 690 of the human LDL receptor was homologous to the sequence of the mouse EGF precursor (4). This region contains three repetitive sequences of about 40 amino acids, that are designated A, B, and C in Fig. 5 and displayed in detail in Fig. 8. Each of these repeats contains six cysteine residues spaced at similar intervals. The A, B, and C sequences are homologous to repeat sequences designated 1 to 4 in the EGF precursor (11). A similar sequence occurs in three proteins of the blood clotting system: factor X, factor IX, and protein C (Fig. 8). Doolittle et al. (13) originally discovered that the cysteine-rich sequences shown in Fig. 8 were shared between the EGF precursor and the three proteins of the blood clotting system. A striking feature of repeats A, B, and C of the LDL receptor is that each of these structures is contained within a single exon (Fig. 5). A similar exon-intron organization for repeats 2, 3, and 4 occurs in the human EGF precursor gene (10). Moreover, recent studies by Anson et al. (22) indicate that the repeat found in human factor IX is also encoded by a discrete exon.
Fig. 8.

Amino acid alignment of segments A, B, and C from the LDL receptor with homologous regions from the EGF precursor and several proteins of the blood clotting system. The number of amino acids comprising the variable region in the middle of each sequence is shown in parentheses. The standard one-letter amino acid abbreviations are used (Fig. 6). Amino acids that are present at a given position in more than 50 percent of the sequences are boxed and shown as a consensus at the bottom line. Cysteine residues (C) are underlined in the consensus sequence. Sequence data for the LDL receptor was taken from (4); sequence data for the other proteins were taken from the original references cited in (13, 23).
The repeat sequence shown in Fig. 8 is not the same as the recently described “growth factor-like” repeat found in EGF, α-transforming growth factor, tissue plasminogen activator, and the 19-kD vaccinia virus protein (13, 23). The proteins of the blood clotting system—factor IX, factor X, and protein C—appear to contain one copy of the growth factor-like sequence (13, 23) and one copy of the LDL receptor-like sequence shown in Fig. 8, whereas the EGF precursor contains four copies of the growth factor-like sequence and four copies of the LDL receptor-like sequence (13).
The LDL receptor undergoes several posttranslational carbohydrate processing events that cause a shift in its apparent molecular weight on sodium dodecyl sulfate–polyacrylamide gels from 120,000 to 160,000 during synthesis and transport to the cell surface (24). Most of this apparent molecular weight increase is due to the addition of complex carbohydrate chains in O-linkage to serine and threonine residues (24). The majority of these O-linked sugars have been localized by protease and lectin blotting studies to a region of the receptor that contains 18 clustered serine and threonine residues (3). This domain is located just above the transmembrane sequence of the protein. In the receptor gene, exon 15 encodes 58 amino acids that encompass all 18 of the clustered serine and threonine residues (Fig. 5).
A hydrophobic sequence of 22 amino acids flanked by arginine and lysine residues forms the transmembrane domain of the human LDL receptor (4). Deletion of this region in a naturally occurring mutation results in the synthesis of a receptor that is secreted from the cell (25). The transmembrane domain is encoded by exons 16 and 17 (Fig. 5). The intron that separates these exons interrupts the codon specifying the ninth residue of this 22–amino acid hydrophobic domain.
The last protein domain in the LDL receptor consists of 50 amino acids located on the cytoplasmic side of the plasma membrane (3, 4). The amino acid sequence of this region is highly conserved between LDL receptors of different species (26). This domain serves to target the LDL receptor to coated pits on the cell surface (27). The cytoplasmic domain is encoded by exons 17 and 18. Exon 17 encodes 13 amino acids of the transmembrane domain and the first 39 amino acids of the cytoplasmic domain. Exon 18, the largest exon in the gene (Table 1), encodes the remaining 11 amino acids at the COOH-terminus of the protein and the 2.5 kb of DNA sequence that corresponds to the 3′ untranslated region of the mRNA, including three copies of the Alu family of middle repetitive DNA sequences (4, 25).
LDL receptor as a member of two supergene families
On the basis of studies of immunoglobulins and related proteins, Hood et al. (28) have defined the concept of a supergene family as “a set of … genes related by sequence (implying common ancestry), but not necessarily related in function. ” By this criterion the LDL receptor is a member of at least two supergene families. The LDL binding domain belongs to a supergene family whose only other member now known is complement component C9. The three repeated sequences in the domain of EGF precursor homology belong to a supergene family that includes the three proteins of the blood clotting system (factor IX, factor X, and protein C) as well as the EGF precursor. In the LDL receptor each of these regions is contained on one or more exons whose intron boundaries imply that they were free to move within the genome and to join other genes, thus creating a supergene family.
On the basis of these findings, it may become necessary to expand the concept of supergene families to include regions of proteins and to consider that a given protein may contain discrete regions derived from the exons of different supergene families. As originally proposed by Gilbert (8), the existence of introns permits functional domains encoded by discrete exons to shuffle between different proteins, thus allowing proteins to evolve as new combinations of preexisting functional units. The LDL receptor is a vivid example of such a mosaic protein.
Implications for genetics of familial hypercholesterolemia
The elucidation of the structure of the LDL receptor gene should be useful in further studies of mutations in this gene that underlie familial hypercholesterolemia (FH), a common cause of atherosclerosis and heart attacks (29). In the one mutation so far described at the molecular level, the defect involves a 5-kb deletion that removes several exons near the 3′ end of the gene (25). This deletion resulted from a recombination between two middle repetitive sequences of the Alu family. The finding of repeated exons in the LDL binding domain and in the EGF precursor homology region raises the possibility that some FH mutations may have arisen from deletions or duplications resulting from unequal crossing-over and recombination between these homologous segments. The availability of a detailed gene map should now permit the characterization of other mutations through Southern blotting and cloning of genomic DNA isolated from cells of FH patients.
Acknowledgments
We thank Daphne Davis, James Cali, and Gloria Brunschede for technical assistance, M. Lehrman and H. Hobbs for helpful discussions, and T. Maniatis, F. Grosveld, and Y. W. Kan for providing the λ and cosmid libraries. Supported by NIH research grants HL 20948 and HL 31346, a fellowship from the Deutsche Forschungsgemeinschaft (T.C.S.), and a Research Career Development Award from the NIH (HL 01287) (D.W.R.).
References and Notes
- 1.Goldstein JL, Anderson RGW, Brown MS. Nature (London) 1979;279:679. doi: 10.1038/279679a0. [DOI] [PubMed] [Google Scholar]
- 2.Brown MS, Anderson RGW, Goldstein JL. Cell. 1983;32:663. doi: 10.1016/0092-8674(83)90052-1. [DOI] [PubMed] [Google Scholar]
- 3.Russell DW, et al. ibid. 1984;37:577. [Google Scholar]
- 4.Yamamoto T, et al. ibid. 1984;39:27. [Google Scholar]
- 5.Stanley KK, et al. EMBO J. 1985;4:375. doi: 10.1002/j.1460-2075.1985.tb03639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; DiScipio RG, et al. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7298. [Google Scholar]
- 6.Mostov KE, Friedlander M, Blobel G. Nature (London) 1984;308:37. doi: 10.1038/308037a0. [DOI] [PubMed] [Google Scholar]
- 7.Downward J, et al. ibid. 1984;307:421. [Google Scholar]
- 8.Gilbert W. ibid. 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 9.Ullrich A, et al. ibid. 1984;309:418. [Google Scholar]; Holland EC, Leung JO, Drickamer K. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7338. doi: 10.1073/pnas.81.23.7338. [DOI] [PMC free article] [PubMed] [Google Scholar]; McClelland A, Kuhn LC, Ruddle FH. Cell. 1984;39:267. doi: 10.1016/0092-8674(84)90004-7. [DOI] [PubMed] [Google Scholar]; Schneider C, et al. Nature (London) 1984;311:675. doi: 10.1038/311675b0. [DOI] [PubMed] [Google Scholar]
- 10.Südhof TC, Russell DW, Goldstein JL, Brown MS, Sanchez-Pescador R, Bell GI. Science. 1985;228:893. doi: 10.1126/science.3873704. [DOI] [PubMed] [Google Scholar]
- 11.Scott J, et al. ibid. 1983;221:236. [Google Scholar]
- 12.Gray A, Dull TJ, Ullrich A. Nature (London) 1983;303:722. doi: 10.1038/303722a0. [DOI] [PubMed] [Google Scholar]
- 13. Doolittle RF, Feng D-F, Johnson MS. ibid. 1984;307:558. doi: 10.1038/307558a0. Doolittle RF. Trends in Biochem. Sci. in press. Doolittle et al. described ten repeat units in the mouse EGF precursor that fall into two classes A and B. The four class A repeats, designated g–j, correspond to the “growth factor-like” repeats found in α-transforming growth factor, tissue plasminogen activator, and the 19-kD vaccinia virus (23). Four of the six class B repeats, which Doolittle, Feng, and Johnson designated c to f, correspond, respectively, to repeats 1 to 4 of Scott et al. (11). These four repeats show the greatest homology with repeats A to C in the LDL receptor (Fig. 8). The three proteins of the blood clotting system (factor IX, factor X, and protein C) contain one copy each of the class A and B repeats (see figure 2 in Doolittle et al.).
- 14.Rall LB, et al. Nature (London) 1985;313:228. doi: 10.1038/313228a0. [DOI] [PubMed] [Google Scholar]
- 15.Mount SM. Nucleic Acids Res. 1982;10:459. doi: 10.1093/nar/10.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shenk T. Curr. Topics Microbiol. Immunol. 1981;93:25. doi: 10.1007/978-3-642-68123-3_3. [DOI] [PubMed] [Google Scholar]
- 17.Russell DW, et al. Proc. Natl. Acad. Sci. U.S.A. 1983;80:7501. doi: 10.1073/pnas.80.24.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart GW, et al. ibid. 1984;81:7318. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]; Carter AD, et al. ibid. :7392. [Google Scholar]
- 19.Innerarity TL, et al. J. Biol Chem. 1984;259:7261. [PubMed] [Google Scholar]
- 20.Doolittle WF. Nature (London) 1978;272:581. [Google Scholar]
- 21.King CR, Piatigorsky J. Cell. 1983;32:707. doi: 10.1016/0092-8674(83)90056-9. [DOI] [PubMed] [Google Scholar]; Medford RM, et al. ibid. 1984;38:409. doi: 10.1016/0092-8674(84)90496-3. [DOI] [PubMed] [Google Scholar]; Nawa H, Kotani H, Nakanishi S. Nature (London) 1984;312:729. doi: 10.1038/312729a0. [DOI] [PubMed] [Google Scholar]
- 22.Anson DS, et al. EMBO J. 1984;3:1053. doi: 10.1002/j.1460-2075.1984.tb01926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blomquist MC, Hunt LT, Barker WC. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7363. doi: 10.1073/pnas.81.23.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tolleshaug H, et al. Cell. 1982;30:715. doi: 10.1016/0092-8674(82)90276-8. [DOI] [PubMed] [Google Scholar]; Cummings RD, et al. J. Biol. Chem. 1983;258:15261. [PubMed] [Google Scholar]
- 25.Lehrman MA, et al. Science. 1985;227:140. doi: 10.1126/science.3155573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto T, et al. in preparation. [Google Scholar]
- 27.Lehrman MA, et al. Cell. in press. [Google Scholar]
- 28.Hood L, et al. ibid. 1985;40:225. [Google Scholar]
- 29.Goldstein JL, Brown MS. In: The Metabolic Basis of Inherited Disease. Stanbury JB, et al., editors. Vol. 5. New York: McGraw-Hill; 1983. pp. 672–712. [Google Scholar]
- 30.Hobbs H, et al. in preparation. [Google Scholar]
- 31.Lawn RM, et al. Cell. 1978;15:1157. doi: 10.1016/0092-8674(78)90043-0. [DOI] [PubMed] [Google Scholar]
- 32.Lau Y-F, Kan YW. Proc. Natl Acad. Sci. U.S.A. 1983;80:5225. doi: 10.1073/pnas.80.17.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grosveld FG, et al. Nucleic Acids Res. 1982;10:6715. doi: 10.1093/nar/10.21.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory; 1982. pp. 1–545. [Google Scholar]
- 35.Feinberg AP, Vogelstein B. Anal. Biochem. 1983;132:6. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 36.Maxam AM, Gilbert W. Methods Enzymol. 1980;5:499. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 37.Sanger F, Nicklen S, Coulson AR. Proc. Natl Acad. Sci. U.S.A. 1977;74:5463. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messing J. Methods Enzymol. 1983;101:20. doi: 10.1016/0076-6879(83)01005-8. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein JL, Basu SK, Brown MS. ibid. 1983;98:241. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 40.Berk A, Sharp P. Cell. 1977;12:721. doi: 10.1016/0092-8674(77)90272-0. [DOI] [PubMed] [Google Scholar]
- 41.Church GM, Gilbert W. Proc. Natl. Acad. Sci. U.S.A. 1984;81:1991. [Google Scholar]