

Abstract
Poly-ADP-ribose polymerase-1 (PARP-1)'s multiple roles in the cell span from maintaining life to inducing death. The processes PARP-1 is involved in include, but are not limited to DNA repair, DNA transcription, mitosis, and cell death. Of PARP-1's different cellular functions, its active role in cell death is of particular interest to designing therapies for diseases. Genetic deletion of PARP-1 revealed that PARP-1 over activation underlies cell death in experimental models of stroke, diabetes, inflammation and neurodegeneration. Since interfering with PARP-1 mediated cell death will be clinically beneficial, great effort has been invested into designing PARP-1 inhibitors and understanding mechanisms downstream of PARP-1 over activation. PARP-1 overactivation may kill by depleting cellular energy through nicotinamide adenine dinucleotide (NAD+) consumption, and by releasing the cell death effector apoptosis-inducing factor (AIF). Unexpectedly, recent evidence shows that poly-ADP ribose (PAR) polymer itself, and not the consumption of NAD+ is the source of cytotoxicity. Thus, PAR polymer acts as a cell death effector downstream of PARP-1-mediated cell death signaling. We coined the term parthanatos after Thanatos, the personification of death in Greek mythology, to refer to PAR-mediated cell death. In this review, we will summarize the proposed mechanisms by which PARP-1 overactivation kills. We will present evidence for parthanatos, and the questions raised by these recent findings. It is evident that further understanding of parthanatos opens up new avenues for therapy in ameliorating diseases related to PARP-1 over activation.
Keywords: PARP-1, PAR polymer, PARG, Parthanatos, Cell Death, AIF, Review
2. BACKGROUND
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme important for cellular homeostasis, and is involved in diseases such as stroke, myocardial infarction, and Parkinson disease (PD). Under physiological conditions, PARP-1 is important for DNA repair, genomic stability and transcription. In pathological conditions that cause severe genomic stress such as ischemia-reperfusion injury, inflammation, myocardial infarction, glutamate excitotoxicity, and PD, PARP-1 over-activation leads to cell death. Cell demise from PARP-1 over-activation has been attributed to depletion of cellular energy, release of the death effector apoptosis-inducing factor (AIF) from the mitochondria, and production of excess poly-ADP ribose (PAR) polymer, a novel death signal. We coined the term parthanatos from Thanatos, the personification of death in Greek mythology to describe cell death initiated by PAR polymer. In this review, we first summarize proposed mechanisms by which PARP-1 kills, including NAD depletion, AIF release and excess PAR. Then we present the evidence for and possible basis of parthanatos.
2.1. PARPs
Poly-ADP ribosylation is a post-translational modification catalyzed by poly-ADP ribose polymerases (PARPs). PARPs form homopolymers of PAR which can be linear or branched, and free or attached to proteins (1). Due to PAR's anionic charge, non-covalent or covalent binding of PAR to proteins alters the function of the target protein (2), which is possibly one mechanism by which PARPs modulate cellular processes such as DNA repair, DNA transcription, mitosis, and cell death.
PARP-1 is the founding member of the PARP superfamily. The crystal structure of the catalytic domain of chicken PARP-1 shows structural homology with the active site of bacterial ADP-ribosylating toxins (3). The conserved beta-alpha-loop-beta-alpha NAD+ fold in the catalytic domain of PARP-1 was used to extensively search the NCBI non-redundant protein database for other PARPs (4). With this approach, 17 PARP family members have been identified that can potentially carry out poly-ADP ribosylation activities (5). These PARPs vary extensively in cell localization, regulation and function, but can be broadly classified into 3 groups as discussed in Hassa et al (1). In this review, we will focus on PARP-1, which has been strongly implicated in several experimental models of stroke, diabetes, inflammation and neurodegeneration. For a more detailed discussion on the different members of the PARP family, please refer to excellent reviews by Schreiber et al (5) and by Hassa and Hottiger (6).
2.2. Synthesis of PAR
PARP-1 has several main features (Figure 1) consisting of a DNA binding domain of two zinc zippers that recognize DNA strand breaks, a bipartite nuclear localization signal containing a caspase-cleavage site, an automodification domain with a BRCT motif for protein-protein interactions, and a C-terminal catalytic site which contains the well-conserved NAD+-fold present in mono-ADP ribosylating toxins (4). PARP-1 synthesizes PAR, a polymer of ADP-ribose linked by glycosidic bonds. PAR synthesis was first described by Chambon et al. (7) as nicotinamide mononucleotide (NMD)-induced and involving DNA-dependent incorporation of ATP into a nuclear extract with poly-adenine as the possible product. Further analysis revealed that poly-ADP ribose, not poly-adenine, is the product of the reaction (8-11). This is because the observed molar ratio of the product is 1 adenine:2 ribose: 2 phosphate, and that the product is susceptible to phosphodiesterase but not to alkaline hydrolysis (10, 11). In addition, it was further discovered that the ADP-ribose moiety of NAD+, not ATP, is incorporated in the reaction which proceeds with the concomitant release of nicotinamide (10).
Figure 1.
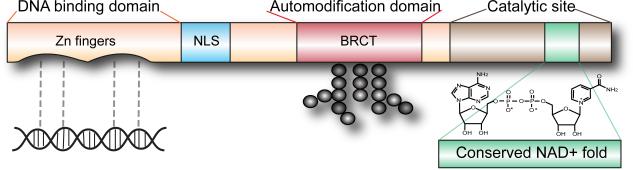
PARP domain. PARP-1 uses NAD+ to form polymers of ADP-ribose on various protein acceptors. The major domains of PARP-1 include: (1) two zinc fingers responsible for PARP-1's detection of DNA breaks, (2) nuclear localization signal (NLS) containing a caspase-cleavage site, (3) an automodification domain with a BRCT motif for protein-protein interactions, (4) a catalytic site which contains the PARP signature NAD+-fold.
To form PAR, PARP-1 first forms ADP-ribose by hydrolyzing NAD+ and releasing nicotinamide. PAR is then covalently bound to proteins through an ester bond between the first ADP-ribose and an amino acid (Glu, Asp) in the acceptor proteins. Polymerization of PAR is through catalysis of ribose-ribose 2’-1’ glycosidic bonds with branching occurring on average of one branch per 20-50 ADP-ribose units (1). Depending on the stimuli, PAR formed can vary in length and in the frequency of branching. This structural heterogeneity by PAR may be in part responsible in distinguishing among the life and death functions of PARP-1 (1). The half-life of PAR within the cell depends on the activity of PARG (poly-(ADP) ribose glycohydrolase) or AH3 lyase (human ADP-ribosylhydrolase 3). Upon genotoxic stress for instance, PAR is rapidly degraded by PARG or ARH3 lyase which breaks down PAR by cleaving the glycosidic bonds between ADP-ribose units (1). Thus, given the processes by which the levels and structure of PAR can be varied, in addition to other factors regulating PARP-1 activity, PARP-1 is capable of achieving varied responses within the cell depending on the stimuli.
2.3. PARP-1 in life and death
PARP-1 function ranges from supporting survival to inducing death. One of the ways by which PARP-1 regulates its function is through regulating the formation, structure and degradation of PAR (Figure 2). In the presence of mild DNA damage, the catalytic activity of PARP-1 is increased by more than 500-fold causing ADP-ribosylation of PARP-1 and its substrates (2). Its substrates include, but are not limited to histones, DNA helicases, high mobility group proteins, topoisomerases I and II, single-strand break repair factors, base-excision repair factors and several transcription factors (reviewed in (2)). However, with excess genotoxic stress, PARP-1 is overactivated, produces excess PAR, leading to cell death.
Figure 2.

PAR polymer metabolism. In the initiation step of PAR synthesis (1), PARP-1 converts NAD+ to ADP-ribose and nicotinamide. The ADP-ribose is then attached to a glutamic acid residue in the protein acceptor. Structural heterogeneity of the PAR polymer is achieved by elongation (2) and branching (3) at the 2’-OH and 2”-OH of the ribose moiety respectively. PAR breakdown (4) is catalyzed by the endoglycosidic and exoglycosidic activities of PARG. Removal of the final ADP-ribose in the protein acceptor is catalyzed by mono-ADP-ribosyl-protein lyase.
PARP-1 knockout (KO) mouse models were generated by three independent groups, and exhibited phenotypes highlighting the physiological and pathological roles of PARP-1 (12). PARP-1 KO mouse models were all found to be hypersensitive to alkylating agents and ionizing radiation, possibly due to defective DNA repair (12). In contrast, PARP-1 KO mice are protected from LPS-induced shock, ischemic injury and MPTP excitotoxicity. Thus, these genetic models highlight a dual role for PARP-1 in the cell depending on the stimuli: PARP-1 can support survival by facilitating DNA repair or can mediate pathological cascades as a result of inflammation or excitotoxicity (2). Since PARP-1's role in cell death has been found to underlie experimental models of disease, understanding how PARP-1's activity can lead to cell death in several pathological contexts will help in expanding therapeutic options for PARP-1 related diseases.
3. PARP-1 IS A REGULATOR OF CELL DEATH
3.1. PARP-1 in neuronal injury due to excitotoxicity
PARP-1 activation is downstream of overproduction of nitric oxide (NO). Neuronal damage following ischemia is known to be mediated by overactivation of N-methyl-D-aspartic acid (NMDA) glutamate receptors due to excess glutamate released by injured neurons (13). This phenomenon known as excitotoxicity underlies neuronal death and injury in experimental models of stroke, PD, and other neurodegenerative disorders (13, 14). The key steps underlying NMDA excitotoxicity have been described. Neuronal injury leads to over activation of NMDA glutamate receptors that in turn results in an excessive influx of calcium. Excess calcium then leads to over activation of neuronal nitric oxide synthase (nNOS). nNOS plays an important role in excitotoxicity since primary brain cultures obtained from nNOS KO mice or wild type cultures treated with NOS inhibitors are resistant to NMDA excitotoxicity (15-19). Mice lacking nNOS are also resistant against middle cerebral artery occlusion (MCAO)-induced neuronal injury or an intrastriatal injection of an excitotoxic dose of NMDA (18, 20).
NO induces ADP-(ribosyl)ation in isolated nuclei providing a mechanistic link between PARP-1 and NO (21). Excess NO by reacting with superoxide anion to produce the oxidant peroxynitrite causes DNA damage that activates PARP-1 (15, 22, 23). PARP-1 activation plays a prominent role in NMDA excitotoxicity as PARP inhibitors protect neurons from NMDA excitoxicity and NO-mediated toxicity (21, 24, 25). Furthermore, PARP-1 KO mice are profoundly resistant to stroke and NMDA excitotoxicity (24-26). Interestingly, PARP-1 KO mice or wild type mice treated with PARP inhibitors are similarly neuroprotected when treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (27-29) suggesting that inhibiting PARP-1 for neuroprotective purposes is not only relevant to acute neuronal injury such as stroke, but also to neurodegenerative disorders such as PD.
PARP-1 overactivation leads to neuronal cell death. Since PARP-1 consumes NAD+, Berger and colleagues postulated that PARP-1 overactivation leads to massive consumption of NAD+, followed by ATP depletion (30). However, in light of recent data, this hypothesis seems to be insufficient, as described in the next section. Hence, other mechanisms by which PARP-1 kills are proposed such as the release of the death effector, AIF (31, 32). Recent findings reveal that PAR polymer produced by PARP-1 acts as a cell death effector by inducing the release of AIF (33, 34). This, PAR polymer and not consumption of NAD+ causes cell death.
3.2. PARP-1 activation induces NAD depletion
According to the suicide hypothesis, PARP-1 kills primarily by NAD+ depletion (30). The suicide hypothesis stems from studies showing that PARP-1 overactivation causes energy depletion. Indeed, inhibiting PARP-1 through pharmacological inhibitors or genetic deletion restores NAD+ levels and viability (31). In neurons for example, different neuronal populations treated with a variety of toxic stimuli exhibit protection and concomitant energy preservation with PARP inhibitors (31, 35). Moreover, PARP-1 KO mice, which are resistant to transient cerebral ischemia-induced damage, exhibit reduced PAR and preserved NAD+ levels (31, 35).
Based on what is known regarding bioenergetics, NAD+ depletion causes ATP depletion; and the resulting drop in cellular energy leads to cell demise. (2, 31). NAD+ is known to be a cofactor in several cellular metabolic processes needed for generating ATP such as glycolysis and the tricarboxylic acid cycle (2). In addition, NAD+ resynthesis requires at least 2-4 molecules of ATP while NAD+ depletion blocks glyceraldehyde 3-dehydrogenase activity leading to the cell investing ATP in glycolysis, but without the return in NAD+ (2, 36, 37). Thus, in support of the suicide hypothesis, PARP-1 activation leads to a block in the glycolysis. Indeed, replenishment of glycolytic and tricarboxylic acid cycle (TCA cycle) intermediates and substrates such as alpha-ketoglutarate or pyruvate, are neuroprotective (31, 38). Also, administering NAD+ to cells or overexpression of NAD+ biosynthetic genes seem to rescue PARP-1 dependent cell death, suggesting that indeed, the NAD+ decline associated with PARP-1 overactivation can cause cell demise (31). However, NAD+ replenishment probably prevents cell death through SIRT1 which is unrelated to energy levels (1, 39, 40).
Recent findings suggest that the compartmentalization of NAD+ within cells also must be considered when evaluating death induced by NAD+ depletion. The mitochondrial pool of NAD+ appears to be more relevant for cell death rather than the cytosolic and nuclear pools, as cell death is rescued upon preservation of NAD+ in the mitochondria by cyclosporin A, or replenishment of NAD+ by overepression of the biosynthetic nicotinic acid mononucleotide adenylyltransferase (Namnt) (41, 42). The drop in NAD+ levels following PARP-1 overactivation reflects whole cell NAD+. Thus, conclusions made from these studies need to be reexamined in light of the recent finding that the mitochondrial levels of NAD+ remain at physiological levels following genotoxic stress and can support viability even when nuclear and cytoplasmic pools of NAD+ are depleted (42). Whether PARP-1-dependent cell death exclusively depends on mitochondrial NAD+ depletion remains to be determined, since overexpression of Namnt to replenish mitochondrial stores of NAD+ leads to a partial rescue of cell death only (41). Reduction in cell death by cyclosporin A may also not be attributed solely to NAD+ preservation in the mitochondria, as it blocks mitochondrial permeability transition, which in itself is an important player in cell death signaling.
Several studies question whether PARP-1 overactivation kills primarily by NAD+ depletion. Paschen et al (43) showed that NAD+ decline does not always correlate with ATP decline in a model of ischemia reperfusion. Moreover, Goto et al (44) showed that PARP-1 KO mice have reduced infarct size after MCAO ischemia compared with wild type animals, but there is no difference between groups in the changes in the energy status as measured by the water diffusion coefficient. Also, Bax and calpain KO cells are protected from N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) to the same degree as wild-type controls treated with the PARP inhibitor DPQ (45). This is in spite of a massive drop in the NAD+ levels in Bax and calpain KO cells that is not seen in the DPQ-treated cells. These findings suggest that NAD+ depletion due to PARP-1 activation is not sufficient to account for cell demise.
3.3. PARP-1 activation induces AIF release
PARP-1 overactivation leading to AIF release from the mitochondria was first demonstrated by Yu et al (32). In this study, PARP-1 KO mouse embryonic fibroblasts and neurons fail to release AIF. Furthermore, PARP-1 dependent cell death is reduced when AIF-depleting antibodies are delivered into the cells. Most importantly, AIF translocation has been well-documented in several experimental models of PARP-1 mediated cell death.
AIF was purified from supernatants of mitochondria induced to undergo permeability transition (46). This ubiquitously-expressed mitochondrial protein was isolated for its ability to induce nuclear fragmentation, and was shown to translocate from the mitochondria to the nucleus to induce chromatin condensation and DNA fragmentation (47). The AIF gene is localized to the A6 region of mouse X chromosome, and is composed of 17 exons (48). The AIF gene expresses a 67 kDa protein that is believed to be converted to a 57 kDa protein upon cleavage of its putative mitochondrial localization sequence (49). However, recent studies indicate that it is processed to a 62-kDa form upon cleavage of its mitochondrial localization sequence, and it is only processed to the 57-kDa form after a cell death stimulus (50, 51). Analysis of its sequence and its crystal structure reveal a glutathione reductase-like fold with an FAD-binding domain and an NADH binding domain as well as a C-terminal domain composed of five antiparallel β-strands and two α-helices (52). The structure of the AIF protein reveals an overall positive electrostatic potential and homology to Bph4, a ferredoxin reductase in a dioxygenase from bacteria (52). Analysis of recombinant AIF reveals NADH-oxidase activity that can catalyze formation of superoxide anions (53) further suggesting a role for AIF in redox processes within the mitochondria.
The exact function of AIF in the mitochondria has been further clarified through the use of genetic knockdowns and conditional deletion of AIF. Gaining insights to AIF's in vivo function was initially precluded by AIF's essential role in embryonic development (54). Thus, models such as the Harlequin (Hq) mutant mouse have been useful. Hq mice contain a proviral insertion in the AIF gene that results in an 80% reduction of AIF protein levels (55). Neonatal Hq mice exhibited 18% less respiratory chain complex I and 30% less catalase compared with WT mice (56). These mice develop a late-onset degeneration in the cerebellar granule cells and retinal ganglion cells (57). In addition, there is marked oxidative stress in the brain and retina (55), suggesting that AIF acts as a free radical scavenger in the mitochondria (55). Later studies point that this increased levels of reactive oxygen radicals in Hq mice may be due to a role for AIF in the complex I activity of the electron transport chain (53, 58). This notion was confirmed by conditional deletion of AIF specifically in cardiac and skeletal muscle of mice, which resulted in impaired activity and decreased protein expression of mitochondrial complex I (57). In addition to a role for AIF in redox processes, conditionally deleting AIF in embryonic telencephalon produced neurons with fragmented mitochondria, abnormal cristae structure and decreased survival (59).
Similar to cytochrome c, AIF assumes a deadly role once released from the mitochondria. Whereas cytochrome c is known to kill by the canonical apoptotic pathway of apoptosome formation (60), the mechanism by which AIF kills is not clear. Translocation of AIF to the nucleus induces chromatin condensation and DNA fragmentation possibly through cyclophilin A, even in the presence of caspase inhibitors (48, 49, 54, 61). This nuclear apoptosis can be blocked by microinjecting neutralizing antibodies against AIF into the cytosol (32, 49), inhibition of cysteine proteases (62) and overexpression of hsp70 (63, 64). Translocation of AIF into the nucleus, and not its loss from the mitochondria, kills cells (59). AIF translocation to the nucleus has been observed in different cell types induced to die, including a myriad of cases of neuronal cell death (65), experimental models of Parkinson's disease (66), excitotoxicity (67), perinatal hypoxia-ischemia (68, 69), brain trauma (70), and ischemic stroke (65, 71-73). AIF is a key in neuronal cell death paradigms, since mice with reduced levels of AIF through siRNA or through the Hq mutation have reduced levels of AIF have decreased infarct size after ischemic injury (51, 56, 65).
4. PARTHANATOS, A MESSENGER OF DEATH
4.1 PAR polymer is toxic to cells
Neurons subjected to NMDA excitotoxicity and Hela cells subjected to genotoxic stress using MNNG display a surge of PAR polymer (33, 34). When accumulation of PAR polymer is antagonized by immunodepletion using antiserum to PAR or when PAR polymer is degraded through PARG overexpression, cell death is decreased in both NMDA treated neurons and MNNG treated Hela cells (33, 34). These results suggest that PAR polymer is a key contributor to cell death. Similarly in vivo, PAR polymer contributes to neuronal death observed in a mouse model of stroke, since transgenic mice overexpressing PARG subjected to transient MCAO, experience smaller infarcts compared with wild-type mice (33). Conversely, mutant mice with only one copy of PARG show increased infarct volume compared with wild-type mice (33).
How does excess PAR mediate cell death? PAR is known to mediate cell death in part by inducing AIF release (34). PAR releases AIF from the mitochondria in vitro when PAR generated from MNNG-treated nuclei is incubated with isolated mitochondria (34). Moreover, delivery of PAR into the cell induces AIF translocation and cell death (33, 34). Cell death is mediated specifically by PAR because AIF release is abolished when PAR is catabolized by PARG or phosphodiesterase I (PDI) which degrade PAR polymers. In addition, overexpression of PARG prevents NMDA-induced AIF translocation (33, 34).
4.2 Properties of parthanatos
Parthanatos shares cytological and morphological features of apoptosis and ncecrosis, but is the result of a distinct molecular mechanism. Parthanatos is caspase-independent (33, 34). Downstream of PARP-1 overactivation and complex PAR accumulation, PAR migrates from the nuclei to the cytosol, leading to AIF translocation from the mitochondria to the nucleus (33, 34, 45).
PAR toxicity is dependent on the length and complexity of the PAR polymer (33, 34). Through direct delivery of PAR polymer in neurons and Hela cells, it was found that PAR polymers of size greater than 60 ADP-ribose units are more toxic than less complex polymers (33). Delivery of >60 ADP-ribose polymer kills cells in a dose-dependent manner with toxicity observed at 20 nm PAR and near complete killing at 80 nm PAR (33). Similar levels of PAR polymer are achieved with NMDA excitotoxicity and MNNG-induced cell death (33). Delivery of equivalent concentrations of poly-adenine which has a similar negative charge to PAR polymer is not toxic to cells indicating that the toxicity is not due to the negative charge of PAR polymer (33). Since PAR complexity seems to be closely tied to cell death, it would be interesting to further study how PAR complexity is regulated, and how its production varies across the heterogeneous PARP family.
4.3 Insights from PARG (poly-(ADP) ribose glycohydrolase) deletion
The toxicity of PAR polymers is highlighted in studies of PARG KOs. Though there are several PARPs that have been characterized, there are only two proteins so far that have been identified to catabolize PAR. PARG and the recently identified ARH3 degrade PAR polymers (1, 74). The PARG gene is known to encode 4 isoforms, the nuclear 110- kDa isoform, the cytoplasmic 102-kDa and 99-kDa isoforms (detected only in humans) and the 59-kDa isoform (60-kDa in mice) (1). Mice lacking exons 2 and 3 which only leaves the PARG 59 kDa intact are viable and normal, but show an increase in PAR levels and infarct volumes when subjected to distal MCAO (75). However, a 65-kDa version of PARG accumulates in the mitochondria of these mutant mice due to an alternative translational start site (5). Since these mice do not completely lack PARG, they cannot be used as a tool to assess the effects of accumulation of PAR through PARG deletion. On the other hand, deletion exon 4 creating a null mutation in all PARG isoforms leads to embryonic lethality at day 3.5 (76), suggesting that PARG is essential for survival (5). Trophoblasts isolated from early PARG KO embryos survive only in the presence of the PARP inhibitor, benzamide (76). This survival stems from inhibiting the accumulation of PAR, thereby highlighting the toxicity of PAR. In addition, these cells were hypersensitive to MNNG and menadione, chemicals that are known to induce PARP-1 overactivation and PAR accumulation (76). Similarly, in Drosophila, a loss-of-function mutation of PARG results in larval lethality, and this effect can be rescued by raising the temperature from 25 to 29°C (77). Adult PARG-deficient flies show neurodegeneration, reduced locomotor activity and a short lifespan (77), further highlighting how PAR accumulation is toxic.
4.4 Structural species of PAR and its functional relevance
Unlike most other posttranslational modifications, poly-ADP ribosylation is structurally heterogeneous in that different sizes of PAR polymer can bind to proteins either through covalent or non-covalent binding (1). The ADP-ribose units are linked by glycosidic ribose-ribose 1’-2’ bonds and can stretch from 200-400 units in vitro and in vivo (1). PAR polymers may be bound or free, linear or branched, short or long. How this structural heterogeneity is controlled by the different PARPs is not known (1). Given that there are several PARPs, but only 2 PARGs identified so far, it is conceivable that the polymers produced by the PARP family members are structurally distinct and this plays a role in the diverse functions of the different PARPs. Structural heterogeneity in the PAR produced by different PARPs may account for the overlapping and non-redundant functions of PARP-1 and PARP-2. Mice lacking PARP-1 or PARP-2 are viable, but mice lacking both PARPs are embryonic lethal (78). Interestingly, knocking out PARP-1 alone is sufficient to protect completely from endotoxic and excitotoxic stimuli (12), whereas knocking out PARP-2 protects from focal but not global ischemia (79). Thus, perhaps PARP-2 is able to produce PAR polymers that are necessary for viability, but not the PAR that is cytotoxic in specific paradigms.
Though it is not known how excess PAR leads to release of AIF from the mitochondria, it is conceivable that it is through covalent and non-covalent binding of excess PAR to substrates, which ultimately results in the disruption or promotion of certain protein-protein and DNA-protein interactions. Non-covalent binding of PAR to proteins is stable, in vitro PAR binding is resistant to strong acids, chaotropes, detergents, and high salt concentrations (80, 81). PAR binding is selective depending on the PAR chain length and the presence of binding sites on the substrates (1, 81-83). For instance, PAR binds to histones with the following relative affinity: branched polymers > long, linear polymers > short, linear polymers (80). Binding affinity of PAR to different histones isoforms has this hierarchy: H1 > H2A > H2B = H3 > H4 (80). Recent studies indicate that PAR binds to a variety of proteins (1, 83). It is likely that identification of PAR substrates will be key in understanding parthanatos.
4.5 PAR and mitochondria
Downstream of PARP-1 overactivation is AIF release from the mitochondria (32), indicating that there is nuclear-mitochondrial crosstalk that occurs in PARP-1 mediated cell death (Figure 3). Upon PARP-1 overactivation, excess PAR, free or bound, shuttles outside of the nucleus and binds to specific cytosolic or mitochondrial proteins. PAR binding to these proteins ultimately leads to AIF release from the mitochondria. Targets modified by PAR possibly include proteins with roles in AIF release, mitochondrial membrane permeabilization or mitochondrial function.
Figure 3.

PARP-1 overactivation leads to cell death. In the presence of death stimuli such as excessive DNA damage (1), PARP-1 overactivation (2) leads to the release of the death effector AIF from the mitochondria (3). The biochemical events mediating this nuclear-mitochondrial crosstalk are not completely known. Excess free or protein-bound complex PAR polymer may move from the nuclei to the cytosol where it disrupts protein-protein interactions. Since loss of mitochondrial membrane potential was observed in PARP-1 mediated cell death, PAR possibly binds cytosolic or mitochondrial proteins with roles in AIF release, mitochondrial membrane permeabilization or mitochondrial function. Other events downstream of PARP-1 overactivation include calpain activation, and Bax translocation to the mitochondria. These events appear to be important for AIF release: calpain is hypothesized to cleave AIF which is then released from mitochondria through pores formed by Bax.
Mitochondria maintain physiological integrity of cells through their important role in energy generation. However, upon cellular stress, mitochondria become permeabilized, and participate in cell death signaling by releasing death effectors such as cytochrome c, AIF, second mitochondria-derived activator of caspase/ direct inhibitor of apoptosis binding protein with a low pI (Smac/DIABLO), and Omi stress-regulated endoprotease/ high temperature requirement protein A2 (Omi/HtrA2) (84). Release of these proapoptotic factors is thought to occur through outer membrane permeabilization (OMP) or opening of the permeability transition pore (PTP) (84).
Mitochondrial permeability transition (PT) and loss of mitochondrial membrane potential are early events in PARP-1 dependent cell death (32). PT follows the formation of mitochondrial permeability transition pore (PTP). Although there are conflicting reports about the composition of the PTP, it is thought that the PTP is a proteinacious pore that is composed of adenine nucleotide translocase (ANT) from the inner mitochondrial membrane, voltage dependent anionic channel (VDAC) and cyclophilin D from the mitochondrial matrix (84). PTP can be inhibited by the immunophilin cyclosporin A (85) or bongkrekic acid (86). Cyclosporin A inhibits PARP-1 dependent cell death in astrocytes following MNNG treatment (87). In addition, blocking PTP by cyclosporin A can preserve mitochondrial NAD+ pools (87). Moreover, mice lacking cyclophilin D are protected against ischemia-reperfusion injury (88-90). These results taken together suggest that PT may underlie PARP-1 mediated cell death. However, additional studies are needed to determine whether inhibiting PT is sufficient to prevent AIF release and cell death.
OMP involves the Bcl-2 family of proteins with members classified into pro-apoptotic or anti-apoptotic. One of the proapoptotic members, Bax, has been implicated in AIF release (45). Upon death stimuli, Bax translocates from the cytosol to the mitochondria, where it form pores on the outer mitochondrial membrane through oligomers of itself or through association with other Bcl-2 members (84). Fibroblasts lacking Bax show compromised AIF release from the mitochondria, and are protected against PARP-1 mediated cell death (45). Thus, Bax activation is downstream of PARP-1 overactivation and induces OMP needed for AIF release (45). However, it is possible that Bax deficiency merely delayed the onset of cell death. This was not clear from this study, since it did not investigate events beyond 12 h. Previous data from our laboratory, indicates that Bcl-2 overexpression does not completely protect cells against PARP-1 dependent cell death, but only delays the onset of cell death (32). A deficiency in Bax may produce a similar effect by preserving mitochondrial integrity following PARP-1 activation. Thus, further studies are needed to explore in detail the role of Bax and other Bcl-2 proteins in PARP-1 dependent cell death.
In addition to OMP, evidence shows that cleavage of AIF by calpains is needed for AIF to be completely released from the mitochondria (45, 91). Calpains are calcium-dependent proteases that are believed to cleave AIF causing its release from the inner mitochondrial membrane (45). Mutation of AIF on its calpain-cleavage site protects against oxygen-glucose deprivation induced neuronal death (51). Moreover, overexpressing calpastatin, a calpain inhibitor, protected neurons against cell death from transient global ischemia (51). In MNNG-treated fibroblasts, calpain activation was found to be downstream of PARP-1 overactivation and upstream of Bax activation (45). These findings hint at an active role for calpains downstream of PARP-1 overactivation, but it is not clear how these events are interconnected. Specifically, what is the direct relationship among excess PAR, calpain activation and Bax translocation (45)? In addition, it remains to be identified whether calpain inhibition or genetic knockdown is indeed a factor for cell survival in PARP-1 dependent cell death, or whether calpain inhibition simply delays the onset of cell death and AIF-translocation.
5. CONCLUSION
Parthanatos underlies PARP-1 mediated cell death through the actions of PAR polymer inducing AIF translocation from the mitochondria to the nucleus. The identification of PAR polymer as a novel death signal opens up new avenues for therapy in ameliorating diseases related to PARP-1 overactivation. How PAR polymer relates to other events implicated in AIF release such as mitochondrial membrane permeabilization, calpain activation, and Bax translocation requires further study. Since PAR mediates binding, the next steps involve the identification of specific PAR targets and to determine how binding by PAR leads to cytotoxicity.
ACKNOWLEDGEMENTS
This work is supported by Grant Number R36AG031572 from the National Institute on Aging. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Abbreviations
- PARP
poly-(ADP) ribose polymerase
- PAR
poly-(ADP) ribose
- PARG
poly-(ADP) ribose glycohydrolase
- AIF
apoptosis-inducing factor
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- NMDA
N-methyl-D-aspartic acid
- NAD
nicotinamide adenine dinucleotide
- KO
knock-out
- Namnt
nicotinic acid mononucleotide adenylyltransferase
- PTP
permeability transition pore
REFERENCES
- 1.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. doi:10.1128/MMBR.00040-05 http://dx.doi.org/10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–68. doi:10.1042/0264-6021:3420249 http://dx.doi.org/10.1042/0264-6021:3420249. [PMC free article] [PubMed] [Google Scholar]
- 3.Ruf A, Mennissier de Murcia J, de Murcia G, Schulz GE. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc Natl Acad Sci U S A. 1996;93:7481–5. doi: 10.1073/pnas.93.15.7481. doi:10.1073/pnas.93.15.7481 http://dx.doi.org/10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–93. doi: 10.1002/bies.20085. doi:10.1002/bies.20085 http://dx.doi.org/10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28. doi: 10.1038/nrm1963. doi:10.1038/nrm1963 http://dx.doi.org/10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 6.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADPribose polymerases. Front Biosci. 2008;13:3046–82. doi: 10.2741/2909. doi:10.2741/2909 http://dx.doi.org/10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 7.Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. doi:10.1016/0006-291X(63)90024-X http://dx.doi.org/10.1016/0006-291X(63)90024-X. [DOI] [PubMed] [Google Scholar]
- 8.Chambon P, Doly JD, Strosser MT, Mandel P. On the Formation of a Novel Adenylic Compound by Enzymatic Extracts of Liver Nuclei. Biochem Biophys Res Commun. 1966;25:638–643. doi:10.1016/0006-291X(66)90502-X http://dx.doi.org/10.1016/0006-291X(66)90502-X. [Google Scholar]
- 9.Fujimura S, Hasegawa S, Shimizu Y, Sugimura T. Polymerization of the adenosine 5'-diphosphate-ribose moiety of nicotinamide-adenine dinucleotide by nuclear enzyme. I. Enzymatic reactions. Biochim Biophys Acta. 1967;145:247–59. doi: 10.1016/0005-2787(67)90043-3. No doi match found. [DOI] [PubMed] [Google Scholar]
- 10.Nishizuka Y, Ueda K, Nakazawa K, Hayaishi O. Studies on the polymer of adenosine diphosphate ribose. I. Enzymic formation from nicotinamide adenine dinuclotide in mammalian nuclei. J Biol Chem. 1967;242:3164–71. No doi match found. [PubMed] [Google Scholar]
- 11.Reeder RH, Ueda K, Honjo T, Nishizuka Y, Hayaishi O. Studies on the polymer of adenosine diphosphate ribose. II. Characterization of the polymer. J Biol Chem. 1967;242:3172–9. No doi match found. [PubMed] [Google Scholar]
- 12.Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. No doi match found. [DOI] [PubMed] [Google Scholar]
- 13.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–22. doi: 10.1056/NEJM199403033300907. doi:10.1056/NEJM199403033300907 http://dx.doi.org/10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 14.Olney JW. Neurotoxicity of excitatory amino acids. In: McGeer EG, Olney JW, McGeer PL, editors. Kainic acid as a tool in neurobiology. Raven Press; New York: 1978. Reference not parsed. [Google Scholar]
- 15.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–71. doi: 10.1073/pnas.88.14.6368. doi:10.1073/pnas.88.14.6368 http://dx.doi.org/10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–61. doi: 10.1523/JNEUROSCI.13-06-02651.1993. No doi match found. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson TM, Dawson VL, Snyder SH. Nitric oxide as a mediator of neurotoxicity. NIDA Res Monogr. 1993;136:258–71. discussion 271-3. Reference not parsed. [PubMed] [Google Scholar]
- 18.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–5. doi: 10.1126/science.7522345. doi:10.1126/science.7522345 http://dx.doi.org/10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 19.Schulz JB, Matthews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, et al. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419–29. doi: 10.1523/JNEUROSCI.15-12-08419.1995. No doi match found. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–17. doi: 10.1523/JNEUROSCI.17-18-06908.1997. No doi match found. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–9. doi: 10.1126/science.8080500. doi:10.1126/science.8080500 http://dx.doi.org/10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 22.Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans. 1993;21:330–4. doi: 10.1042/bst0210330. No doi match found. [DOI] [PubMed] [Google Scholar]
- 23.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–4. doi: 10.1073/pnas.93.13.6770. doi:10.1073/pnas.93.13.6770 http://dx.doi.org/10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95. doi: 10.1038/nm1097-1089. doi:10.1038/nm1097-1089 http://dx.doi.org/10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 25.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–51. doi: 10.1097/00004647-199711000-00002. doi:10.1097/00004647-199711000-00002 http://dx.doi.org/10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, Dawson VL. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–11. doi: 10.1523/JNEUROSCI.20-21-08005.2000. No doi match found. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosi C, Marien M. Decreases in mouse brain NAD+ and ATP induced by 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP): prevention by the poly(ADP-ribose) polymerase inhibitor, benzamide. Brain Res. 1998;809:58–67. doi: 10.1016/s0006-8993(98)00829-4. doi:10.1016/S0006-8993(98)00829-4 http://dx.doi.org/10.1016/S0006-8993(98)00829-4. [DOI] [PubMed] [Google Scholar]
- 28.Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6- tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A. 1999;96:5774–9. doi: 10.1073/pnas.96.10.5774. doi:10.1073/pnas.96.10.5774 http://dx.doi.org/10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwashita A, Yamazaki S, Mihara K, Hattori K, Yamamoto H, Ishida J, Matsuoka N, Mutoh S. Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and in mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Pharmacol Exp Ther. 2004;309:1067–78. doi: 10.1124/jpet.103.064642. doi:10.1124/jpet.103.064642 http://dx.doi.org/10.1124/jpet.103.064642. [DOI] [PubMed] [Google Scholar]
- 30.Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. doi:10.2307/3576299 http://dx.doi.org/10.2307/3576299. [PubMed] [Google Scholar]
- 31.Chiarugi A. Intrinsic mechanisms of poly(ADP-ribose) neurotoxicity: three hypotheses. Neurotoxicology. 2005;26:847–55. doi: 10.1016/j.neuro.2005.01.012. doi:10.1016/j.neuro.2005.01.012 http://dx.doi.org/10.1016/j.neuro.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–63. doi: 10.1126/science.1072221. doi:10.1126/science.1072221 http://dx.doi.org/10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 33.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–13. doi: 10.1073/pnas.0606526103. doi:10.1073/pnas.0606526103 http://dx.doi.org/10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–9. doi: 10.1073/pnas.0606528103. doi:10.1073/pnas.0606528103 http://dx.doi.org/10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–17. doi: 10.1016/j.nbd.2003.08.008. doi:10.1016/j.nbd.2003.08.008 http://dx.doi.org/10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–98. doi: 10.1016/s0165-6147(98)01193-6. doi:10.1016/S0165-6147(98)01193-6 http://dx.doi.org/10.1016/S0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 37.Sheline CT, Wei L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience. 2006;140:235–46. doi: 10.1016/j.neuroscience.2006.02.019. doi:10.1016/j.neuroscience.2006.02.019 http://dx.doi.org/10.1016/j.neuroscience.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 38.Ying W, Chen Y, Alano CC, Swanson RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab. 2002;22:774–9. doi: 10.1097/00004647-200207000-00002. doi:10.1097/00004647-200207000-00002 http://dx.doi.org/10.1097/00004647-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–30. doi: 10.1074/jbc.M506162200. doi:10.1074/jbc.M506162200 http://dx.doi.org/10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 40.Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291:H1545–53. doi: 10.1152/ajpheart.01124.2005. doi:10.1152/ajpheart.01124.2005 http://dx.doi.org/10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- 41.Alano CC, Tran A, Tao R, Ying W, Karliner JS, Swanson RA. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res. 2007;85:3378–85. doi: 10.1002/jnr.21479. doi:10.1002/jnr.21479 http://dx.doi.org/10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107. doi: 10.1016/j.cell.2007.07.035. doi:10.1016/j.cell.2007.07.035 http://dx.doi.org/10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paschen W, Olah L, Mies G. Effect of transient focal ischemia of mouse brain on energy state and NAD levels: no evidence that NAD depletion plays a major role in secondary disturbances of energy metabolism. J Neurochem. 2000;75:1675–80. doi: 10.1046/j.1471-4159.2000.0751675.x. doi:10.1046/j.1471-4159.2000.0751675.x http://dx.doi.org/10.1046/j.1471-4159.2000.0751675.x. [DOI] [PubMed] [Google Scholar]
- 44.Goto S, Xue R, Sugo N, Sawada M, Blizzard KK, Poitras MF, Johns DC, Dawson TM, Dawson VL, Crain BJ, Traystman RJ, Mori S, Hurn PD. Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke. 2002;33:1101–6. doi: 10.1161/01.str.0000014203.65693.1e. doi:10.1161/01.STR.0000014203.65693.1E http://dx.doi.org/10.1161/01.STR.0000014203.65693.1E. [DOI] [PubMed] [Google Scholar]
- 45.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol;2007;27:4844–62. doi: 10.1128/MCB.02141-06. doi:10.1128/MCB.02141-06 http://dx.doi.org/10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–41. doi: 10.1084/jem.184.4.1331. doi:10.1084/jem.184.4.1331 http://dx.doi.org/10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–23. doi: 10.1016/s0014-5793(00)01731-2. doi:10.1016/S0014-5793(00)01731-2 http://dx.doi.org/10.1016/S0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- 48.Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, Brothers G, Penninger JM, Kroemer G. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. Faseb J. 2001;15:758–67. doi: 10.1096/fj.00-0388com. doi:10.1096/fj.00-0388com http://dx.doi.org/10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 49.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. doi:10.1038/17135 http://dx.doi.org/10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 50.Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. Embo J. 2005;24:1375–86. doi: 10.1038/sj.emboj.7600614. doi:10.1038/sj.emboj.7600614 http://dx.doi.org/10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–93. doi: 10.1523/JNEUROSCI.2826-07.2007. doi:10.1523/JNEUROSCI.2826-07.2007 http://dx.doi.org/10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mate MJ, Ortiz-Lombardia M, Boitel B, Haouz A, Tello D, Susin SA, Penninger J, Kroemer G, Alzari PM. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9:442–6. doi: 10.1038/nsb793. doi:10.1038/nsb793 http://dx.doi.org/10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- 53.Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. Embo J. 2004;23:4679–89. doi: 10.1038/sj.emboj.7600461. doi:10.1038/sj.emboj.7600461 http://dx.doi.org/10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–54. doi: 10.1038/35069004. doi:10.1038/35069004 http://dx.doi.org/10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 55.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–74. doi: 10.1038/nature01034. doi:10.1038/nature01034 http://dx.doi.org/10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 56.Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, Nilsson M, Eriksson PS, Hagberg H, Culmsee C, Plesnila N, Kroemer G, Blomgren K. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007;14:775–84. doi: 10.1038/sj.cdd.4402053. doi:10.1038/sj.cdd.4402053 http://dx.doi.org/10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]
- 57.Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–72. doi: 10.1128/MCB.25.23.10261-10272.2005. doi:10.1128/MCB.25.23.10261-10272.2005 http://dx.doi.org/10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–8. doi: 10.1074/jbc.M010498200. doi:10.1074/jbc.M010498200 http://dx.doi.org/10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 59.Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, McBride HM, Penninger JM, Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006;25:4061–73. doi: 10.1038/sj.emboj.7601276. doi:10.1038/sj.emboj.7601276 http://dx.doi.org/10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–42. doi: 10.1038/sj.cdd.4401088. doi:10.1038/sj.cdd.4401088 http://dx.doi.org/10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–92. doi: 10.1126/science.1076194. doi:10.1126/science.1076194 http://dx.doi.org/10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- 62.Yuste VJ, Moubarak RS, Delettre C, Bras M, Sancho P, Robert N, d'Alayer J, Susin SA. Cysteine protease inhibition prevents mitochondrial apoptosis-inducing factor (AIF) release. Cell Death Differ. 2005;12:1445–8. doi: 10.1038/sj.cdd.4401687. doi:10.1038/sj.cdd.4401687 http://dx.doi.org/10.1038/sj.cdd.4401687. [DOI] [PubMed] [Google Scholar]
- 63.Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. doi:10.1038/ncb0901-839 http://dx.doi.org/10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 64.Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR, Liu J. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. doi:10.1038/sj.jcbfm.9600080 http://dx.doi.org/10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- 65.Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-Ribose) polymerase and bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–72. doi: 10.1523/JNEUROSCI.2818-05.2005. doi:10.1523/JNEUROSCI.2818-05.2005 http://dx.doi.org/10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci. 2003;991:132–9. doi: 10.1111/j.1749-6632.2003.tb07471.x. No doi match found. [DOI] [PubMed] [Google Scholar]
- 67.zCheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAXindependent mechanisms. J Neurosci. 2005;25:1324–34. doi: 10.1523/JNEUROSCI.4261-04.2005. doi:10.1523/JNEUROSCI.4261-04.2005 http://dx.doi.org/10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu C, Qiu L, Wang X, Hallin U, Cande C, Kroemer G, Hagberg H, Blomgren K. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86:306–17. doi: 10.1046/j.1471-4159.2003.01832.x. doi:10.1046/j.1471-4159.2003.01832.x http://dx.doi.org/10.1046/j.1471-4159.2003.01832.x. [DOI] [PubMed] [Google Scholar]
- 69.Zhu C, Wang X, Qiu L, Peeters-Scholte C, Hagberg H, Blomgren K. Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor in neonatal rat cerebral hypoxia-ischaemia. J Neurochem. 2004;90:462–71. doi: 10.1111/j.1471-4159.2004.02500.x. doi:10.1111/j.1471-4159.2004.02500.x http://dx.doi.org/10.1111/j.1471-4159.2004.02500.x. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–91. doi: 10.1046/j.1471-4159.2002.00975.x. doi:10.1046/j.1471-4159.2002.00975.x http://dx.doi.org/10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- 71.Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–50. doi: 10.1097/01.WCB.0000087090.01171.E7. doi:10.1097/01.WCB.0000087090.01171.E7 http://dx.doi.org/10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- 72.Komjati K, Mabley JG, Virag L, Southan GJ, Salzman AL, Szabo C. Poly(ADP-ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis-inducing factor in stroke. Int J Mol Med. 2004;13:373–82. No doi match found. [PubMed] [Google Scholar]
- 73.Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:458–66. doi: 10.1097/00004647-200404000-00011. doi:10.1097/00004647-200404000-00011 http://dx.doi.org/10.1097/00004647-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 74.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281:705–13. doi: 10.1074/jbc.M510290200. doi:10.1074/jbc.M510290200 http://dx.doi.org/10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 75.Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W, Cortes U, Wang ZQ, Moroni F, Chiarugi A. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J Cereb Blood Flow Metab. 2006;26:684–95. doi: 10.1038/sj.jcbfm.9600222. doi:10.1038/sj.jcbfm.9600222 http://dx.doi.org/10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- 76.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101:17699–704. doi: 10.1073/pnas.0406182101. doi:10.1073/pnas.0406182101 http://dx.doi.org/10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2004;101:82–6. doi: 10.1073/pnas.2237114100. doi:10.1073/pnas.2237114100 http://dx.doi.org/10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–63. doi: 10.1093/emboj/cdg206. doi:10.1093/emboj/cdg206 http://dx.doi.org/10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kofler J, Otsuka T, Zhang Z, Noppens R, Grafe MR, Koh DW, Dawson VL, de Murcia JM, Hurn PD, Traystman RJ. Differential effect of PARP-2 deletion on brain injury after focal and global cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:135–41. doi: 10.1038/sj.jcbfm.9600173. doi:10.1038/sj.jcbfm.9600173 http://dx.doi.org/10.1038/sj.jcbfm.9600173. [DOI] [PubMed] [Google Scholar]
- 80.Panzeter PL, Realini CA, Althaus FR. Noncovalent interactions of poly(adenosine diphosphate ribose) with histones. Biochemistry. 1992;31:1379–85. doi: 10.1021/bi00120a014. doi:10.1021/bi00120a014 http://dx.doi.org/10.1021/bi00120a014. [DOI] [PubMed] [Google Scholar]
- 81.Fahrer J, Kranaster R, Altmeyer M, Marx A, Burkle A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007;35:e143. doi: 10.1093/nar/gkm944. doi:10.1093/nar/gkm944 http://dx.doi.org/10.1093/nar/gkm944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275:40974–80. doi: 10.1074/jbc.M006520200. doi:10.1074/jbc.M006520200 http://dx.doi.org/10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 83.Gagne JP, Hunter JM, Labrecque B, Chabot B, Poirier GG. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem J. 2003;371:331–40. doi: 10.1042/BJ20021675. doi:10.1042/BJ20021675 http://dx.doi.org/10.1042/BJ20021675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. doi:10.1152/physrev.00013.2006 http://dx.doi.org/10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 85.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–72. doi:10.1023/A:1006879618176 http://dx.doi.org/10.1023/A:1006879618176. [PubMed] [Google Scholar]
- 86.Halestrap AP, Brennerb C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–25. doi: 10.2174/0929867033457278. doi:10.2174/0929867033457278 http://dx.doi.org/10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 87.Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–902. doi: 10.1074/jbc.M313329200. doi:10.1074/jbc.M313329200 http://dx.doi.org/10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- 88.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. doi:10.1038/nature03317 http://dx.doi.org/10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 89.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. doi:10.1038/nature03434 http://dx.doi.org/10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 90.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–10. doi: 10.1073/pnas.0505294102. doi:10.1073/pnas.0505294102 http://dx.doi.org/10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–54. doi: 10.1074/jbc.M413269200. doi:10.1074/jbc.M413269200 http://dx.doi.org/10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]