

Abstract
Post-Golgi trafficking of mature VLDL (very-low-density lipoprotein) is crucial in maintaining normal TAG (triacylglycerol) homoeostasis of hepatocytes; however, the mechanism that regulates the exit of mature VLDL from the TGN (trans-Golgi network) is not known. We developed an in vitro TGN-budding assay that allowed us to examine the formation of secretory vesicles from the TGN in primary rat hepatocytes. We isolated TAG-rich PG-VTVs (post-TGN VLDL transport vesicles) using a continuous sucrose density gradient. PG-VTVs were distributed in low-density fractions, whereas protein transport vesicles were present in relatively higher-density fractions of the same sucrose gradient. EM revealed large intact PG-VTVs ranging 300–350 nm in size. The biogenesis of PG-VTVs from the TGN required cytosol, ATP, GTP hydrolysis and incubation at 37 °C. PG-VTVs concentrated the VLDL proteins: apolipoproteins apoB100, apoAIV, apoAI and apoE, but did not contain either albumin or transferrin. Proteinase K treatment did not degrade VLDL core proteins, suggesting that PG-VTVs were sealed. PG-VTVs were able to fuse with and deliver VLDL to the PM (plasma membrane) in a vectorial manner. We conclude that we have identified a new TGN-derived vesicle, the PG-VTV, which specifically transports mature VLDL from the TGN to the PM.
Keywords: apolipoprotein B, endoplasmic reticulum, post-Golgi VLDL transport vesicle (PG-VTV), trans-Golgi network (TGN), triacylglycerol, very low-density lipoprotein (VLDL)
INTRODUCTION
Among its many functions, the liver is responsible for the conversion of potentially cytotoxic FFAs [non-esterified (‘free’) fatty acids] into more physiologically pragmatic TAGs (triacylglycerols) that are subsequently incorporated into VLDL (very-low-density lipoprotein) before secretion into the aqueous environment of the blood. This is imperative not only due to the incongruity between the non-polar hydrophobic chemical composition of TAGs and the aqueous environment of the blood, but it is also significant as it creates a process which can be regulated, fitting to the variable dietary needs of organisms. VLDLs are synthesized in the ER (endoplasmic reticulum) and transported to the Golgi apparatus via a unique VTV (VLDL transport vesicle) where they undergo maturation before their eventual secretion from the hepatocyte into the plasma [1].
VLDL biogenesis is a complex multi-step process beginning at the level of the ER where FFAs are incorporated into TAG and apoB100 (apolipoprotein B100) is co-translationally translocated into the ER lumen [2]. The newly synthesized apoB100 is lipidated and this process is facilitated by MTP (microsomal TAG transfer protein) [3,4]. Since apoB100 is regulated by its degradation, this lipidation also serves to inhibit the direction of the newly synthesized apoB100 to the ERAD (ER-associated protein degradation) pathway [5]. This newly synthesized VLDL particle is then targeted to the Golgi via the VTV, a novel COPII (coatamer protein II)-dependent vesicle characterized previously by our group [1]. Upon fusion of the VTV with the Golgi and delivery of the VTV cargo, nascent VLDL, into the Golgi lumen, the VLDL particle undergoes several important modifications including its acquisition of apoE (apolipoprotein E), apoAI (apolipoprotein AI), and its further lipidation as shown by its lighter buoyant density and larger size [6–8]. Moreover, its core protein apoB100 is phosphorylated and additionally glycosylated as determined by resistance to endoglycosidase H [6–8]. These modifications that take place in the Golgi lumen result in the production of mature VLDL particles, which are then transported to the PM (plasma membrane) and secreted into the circulatory system. The transport of mature VLDL from the TGN (trans-Golgi network) to the PM is required for their secretion; however, the molecular mechanisms that regulate the release of mature VLDL from the TGN and their subsequent export to the PM remains to be explored. Understanding the mechanism that controls the eventual secretion of VLDL particles is of high significance because abnormalities associated with this step result in dyslipidaemia, hepatic steatosis and atherosclerosis, among other metabolic disorders.
The Golgi apparatus, especially the TGN, is the site where various cargos are sorted and directed to their various intracellular destinations such as the PM and endocytic compartment. In their seminal work, Simons and colleagues demonstrated that multiple cargos destined to different subcellular organelles are sorted in the TGN before their packaging in distinct TGN-derived vesicles [9–11]. The process of cargo selection and its intracellular transport is crucial for maintaining cellular protein and lipid homoeostasis, and a plethora of information is available on how specific cargos are sorted in COPII-coated and COPI (coatamer protein I)-coated vesicles [12–14]. COPII-coated vesicles transport newly synthesized proteins from the ER to the Golgi, whereas COPI-coated vesicles are involved in retrograde transport from the Golgi to the ER. These vesicles have been studied extensively; however, it is surprising that not much work has been done on how different cargos are being exported from the TGN. Despite the fact that TGN-derived vesicles have been poorly studied at the molecular level, a number of regulatory factors involved in the fission process of these vesicles have been identified [15,16]. Data published in previous studies from our laboratory and Fisher’s group have established how nascent VLDL is transported from the ER to the Golgi; however, the process that mediates the post-TGN transport of mature VLDL remains to be elucidated [1,17–19].
To study the mechanism of VLDL exit from the hepatic TGN, we developed a cell-free TGN-budding assay that allowed us to isolate the TGN-derived vesicles, which contain mature VLDL particles. The present study describes the isolation and characterization of a novel transport vesicle of light buoyant density, the PG-VTV (post-TGN VTV), that vectorially transports mature VLDLs from the TGN to the PM in rat primary hepatocytes. Additionally, our results show that the PG-VTVs fuse with and deliver VLDLs to the PM for their secretion from the hepatocyte.
EXPERIMENTAL
Materials
[3H]Oleic acid (45.5 Ci/mM) was purchased from PerkinElmer Life Sciences. Protease inhibitor cocktail tablets (catalogue number 04693116001) were purchased from Roche Applied Science. Gel electrophoresis and immunoblotting reagents were from Bio-Rad Laboratories. ECL reagents were obtained from GE Healthcare Life Sciences. Other reagents used were of analytical grade and purchased from Fisher Scientific. Sprague–Dawley rats (150–200 g) were acquired from Harlan. All procedures involving animals were conducted according to the guidelines of the University of Central Florida’s Institutional Animal Care and Use Committee (IACUC) and strictly followed the IACUC-approved protocol.
Antibodies
Rabbit anti-apoB (apolipoprotein B) polyclonal antibodies were generated commercially (Protein Tech Group) using a synthetic peptide corresponding to amino acids 2055–2067 of rat apoB. Goat anti-apoAIV (apolipoprotein AIV), anti-apoE, anti-calnexin, anti-GOS28, anti-Rab11, anti-SNAP23 (23 kDa synaptosome-associated protein) and anti-syntaxin polyclonal antibodies, and mouse anti-Sec22b and anti-TGN38 monoclonal antibodies were purchased from Santa Cruz Biotechnology. Rabbit polyclonal antibodies against apoAI and mouse monoclonal antibodies against alpha 1 Sodium Potassium ATPase were purchased from Abcam. Rabbit anti-Sar1 and anti-VAMP7 (vesicle-associated membrane protein 7; amino acids 105–123) polyclonal antibodies were generated commercially and have been described previously [1,20]. Rabbit anti-rat albumin antibodies were purchased from Bethyl Laboratories. HRP (horseradish peroxidase)-conjugated goat anti-(mouse IgG), goat anti-(rabbit IgG) and rabbit anti-(goat IgG) antibodies were purchased from Sigma.
Preparation of radiolabelled hepatic ER and cis- and trans-Golgi
Radiolabelled hepatic subcellular organelles were prepared using the same methods as described previously [1,18]. In brief, rat primary hepatocytes were collected by the perfusion of rat liver with Ca2+ -free Kreb’s buffer [119 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4 and 25 mM NaHCO3 (pH 7.4)] followed by perfusion with collagenase (11 000 units in 150 ml of Kreb’s buffer per liver). Rat primary hepatocytes in buffer B [136 mM NaCl, 11.6 mM KH2PO4, 7.5 mM KCl, 0.5 mM DTT and 8 mM Na2HPO4 (pH 7.2)] were incubated with [3H]oleate (100 μCi) complexed with 10 % BSA in PBS for 35 min at 37 °C, followed by washing with 2 % BSA in PBS twice to remove excess [3H]oleate. Cells were then resuspended in buffer C [0.25 M sucrose in 10 mM Hepes (pH 7.2), 5 mM EDTA and protease inhibitor cocktail] and homogenized using a Parr bomb at 1100 psi (1 psi = 6.9 kPa) for 40 min. A sucrose step gradient was used to separate the ER and cis- and trans-Golgi fractions as detailed previously [1,18,21].
Preparation of hepatic cytosol
Hepatic cytosol was prepared from freshly isolated rat primary hepatocytes utilizing the same procedure as discussed previously [1,18,21]. Freshly isolated rat primary hepatocytes were washed with cytosol buffer [25 mM Hepes, 125 mM KCl, 2.5 mM MgCl2, 0.5 mM DTT and protease inhibitor cocktail (pH 7.2)] and then homogenized using a Parr bomb at 1100 psi for 40 min. The resultant sample was pelleted by ultracentrifugation at 40 000 rev./min (100 000 g) for 95 min (Beckman 70.1 Ti rotor) and the supernatant (cytosol) was collected. The cytosol was dialysed overnight against cold cytosol buffer at 4 °C. The cytosol was then concentrated to a final concentration of protein ~ 20 mg/ml using a centricon filter (Amicon) and ultra-filtration membrane (Millipore) with a 10 kDa cut-off.
Preparation of hepatic PM
The PM fraction was prepared using the same methodology as reported by several groups [22–24]. Briefly, freshly harvested rat liver was cut into small pieces (3 mm × 5 mm × 0.5 mm; 4–5 g), washed with and homogenized in buffer C [0.25 M sucrose in 10 mM Hepes (pH 7.2), 5 mM EDTA and protease inhibitor cocktail] using a Parr bomb. Homogenate was then centrifuged at 600 g for 10 min. The PNS (post-nuclear supernatant) was collected and centrifuged at 38 400 g for 10 min (Fiberlite™ F21S-8 × 50y rotor). The pellet was resuspended in 1.67 M sucrose solution and adjusted to 1.37 M sucrose using a refractometer. The sample was then laid under a discontinuous sucrose step gradient of 1.09 M sucrose and 0.25 M sucrose, then ultracentrifuged at 106 600 g (Beckman SW 32 Ti) for 16 h at 4 °C. The sample was collected at the interface of the 0.25 M and 1.09 M sucrose layer, its density was adjusted to 0.43 M sucrose and then overlaid on top of a sucrose step gradient (0.69 M, 0.9 M and 1.12 M). The gradient was ultracentrifuged at 124 800 g (Beckman SW 41 Ti rotor) for 2 h at 4 °C and the purified PM was collected at the interface of the 1.12 M and 0.9 M sucrose layer by aspiration. Purified PM fractions were flipped inside out by repeated freeze–thaw cycles as established previously [24].
Preparation of hepatic whole cell lysate
Rat hepatic whole cell lysate was prepared by lysing primary rat hepatocytes with RIPA buffer (Thermo Scientific) supplemented with protease inhibitor and centrifuged at 13 000 g for 15 min as reported recently [18]. The resultant supernatant was then collected [18].
In vitro TGN-budding assay
A cell-free in vitro TGN-budding assay was utilized to generate PG-VTVs. Rat hepatic TGN membranes (200 μg) containing either [3H]TAG or [14C]TAG and [3H]protein were incubated with hepatic cytosol (500 μg), an ATP-generating system (1 mM ATP, 5 mM phosphocreatine and 5 units of creatine phosphokinase), 1 mM GTP, 1 mM E600, 5 mM Mg2+, 5 mM DTT and 5 mM Ca2+. The reaction mixture volume was adjusted to 500 μl by the addition of transport buffer [30 mM Hepes, 0.25 M sucrose, 2.5 mM magnesium acetate and 30 mM KCl (pH 7.2)] for 30 min at 37 °C in the absence of acceptor PM. The reaction was terminated by placing tubes on ice and the density of the reaction mixture was adjusted to 0.1 M sucrose using ice-cold 10 mM Hepes. The reaction mixture was then laid over a continuous sucrose gradient (0.1–0.86 M sucrose in 10 mM Hepes), then ultracentrifuged at 115 000 g (Beckman SW 41 Ti rotor) for 2 h at 4 °C. Fractions of 500 μl were collected by aspiration, TAG was extracted from each fraction and the associated d.p.m. values were determined using a liquid scintillation counter [1,18].
In vitro PG-VTV–PM fusion assay
PG-VTVs (150 μg) containing [3H]TAG were incubated with rat hepatic PM (150 μg), cytosol (500 μg), an ATP-regenerating system, Mg2+, Ca2+, DTT, E600, transport buffer [2.5 mM Ca2+, 5mM Mg2+, 2mM DTT, 30 mM KCl and 30 mM Hepes (pH 7.2)] and incubated at 37 °C for 35 min. The reaction was terminated by placing the tubes on ice. The density of the reaction mixture was adjusted to 0.43 M sucrose with 0.69 M sucrose solution in 10 mM Hepes, overlaid on a sucrose step gradient (0.69 M, 0.9 M and 1.12 M sucrose in 10 mM Hepes) and ultracentrifuged at 124 800 g (Beckman SW 41 Ti rotor) for 2 h at 4 °C. PM fractions (500 μl) were collected at the interface of the 1.12 M and 0.9 M sucrose layer by aspiration. [3H]TAG was extracted from the PM and the associated d.p.m. value was measured using a liquid scintillation counter.
Extraction of radiolabelled TAG
Radiolabelled TAG was extracted from the PG-VTVs or the PM using the same method as described by Kumar and Mansbach [25]. Briefly, 1.5 ml of reagent 1 [isopropanol/heptane/water at 80:20:2 (by vol.)] was added to the PG-VTVs or the PM suspension (200 μl) and the reagent was vortex-mixed. After 5 min incubation at room temperature (23 °C), 1 ml of n-heptane and 0.5 ml of water was added. The top heptane layer was collected and washed twice with 2 ml of reagent 2 [ethanol/water/0.1 M sodium hydroxide at 50:50:10 (by vol.)]. The heptane layer (0.5 ml) was placed in a scintillation vial and dried before measuring the radioactivity.
Measurement of TAG and protein radioactivity
Radioactivity associated with [3H]TAG was quantified in terms of d.p.m. using the single-isotope mode on the liquid scintillation analyser (TriCarb, Model 2910, PerkinElmer Life and Analytical Sciences). Proteins were precipitated with TCA (trichloroacetic acid) and radioactivity was measured as described previously [1,21,26]. For doubly labelled TGN-budding assays, we used the dual-isotope mode on the same liquid scintillation analyser.
Proteinase K treatment
Vesicles were treated with proteinase K at a final concentration of 0.1 mg/ml for 30 min at 4 °C in the absence or presence of 2 % (v/v) Triton X-100. After incubation, proteinase K was neutralized with an excess of PMSF as described previously [1,21,26].
EM analysis
The negative staining technique of EM was used to examine the morphology of PG-VTVs using exactly the same methodology as we reported previously [1,26,27]. A formvar-carbon-coated nickel grid was placed on a drop of concentrated PG-VTV fraction for 2–3 min, and rinsed with PBS and water. The grid was then stained with 0.5 %aqueous uranyl acetate, air-dried and examined at a magnification of ×10 000 using an FEI Morgagni 268(D) transmission electron microscope.
To examine the morphology of the post-fusion PM by EM, we followed the same procedure as described previously [26,27]. Briefly, post-fusion PM was pelleted by centrifugation, fixed first in 2.5 % glutaraldehyde and then in 1 % osmium tetroxide buffered with 0.2 M imidazole (pH 7.2). Post-fixation, the PM samples were embedded in Spur medium. The thin sections were stained with uranyl acetate and lead citrate and examined using a FEI Morgagni 268(D) transmission electron microscope at a magnification of ×6000.
Gel electrophoresis and immunoblot analysis
Samples (30 μg) were solubilized in two loads of Laemmli buffer and resolved by SDS/PAGE [18,27]. The proteins were then transblotted on to nitrocellulose membranes (Bio-Rad Laboratories) overnight at 4 °C. The membrane was then blocked with 10 % (w/v) non-fat dried skimmed milk powder in PBS-T (PBS with 0.05% Tween 20), incubated with specific primary antibodies and then the appropriate HRP-conjugated secondary antibodies. The protein was detected using ECL reagents and exposing to film (MIDSCI).
Statistical analysis
Comparisons between means were carried out using a statistical package supplied by GraphPad Software (GraphPad Prism 5 Software for Mac OS X version) using either a two-tailed Student’s t test or ANOVA.
RESULTS
Purity of subcellular organelles
In order to generate PG-VTVs in vitro with confidence, the purity of isolated subcellular organelles needed to be assessed using specific marker proteins. The cis-Golgi fraction contained GOS28, a marker protein for cis-Golgi, but not calnexin or Rab11, ruling out ER and endosomal/lysosomal contamination (Figure 1). The trans-Golgi fraction contained most of the trans-Golgi marker, TGN38, whereas the cis-Golgi fraction contained a relatively small amount of this marker protein (Figure 1). The ER fraction contained calnexin, but not the Golgi-specific proteins GOS28 and TGN38, neither did it contain Rab11, an endosomal/lysosomal protein (Figure 1). These results strongly indicate the purity of the Golgi fractions to be used in the in vitro budding assays.
Figure 1. Western blot analysis to determine the purity of subcellular organelles prepared from primary hepatocytes.

Protein samples (30 μg each) containing hepatic whole cell lysate (WCL), ER and cis-and trans-Golgi separated by SDS/PAGE (12 % gel), transblotted and probed with indicated antibodies. The results are representative of four independent experiments.
In vitro biogenesis of PG-VTV
In an attempt to investigate how mature VLDLs exit the TGN, we first established an in vitro TGN-budding assay that allows us to monitor the formation of TGN-derived vesicles. We achieved this by adopting the same methodology as we used in developing an in vitro ER-budding assay to study the transport of nascent VLDLs from the ER to the Golgi [1]. Hepatic TGN membranes containing [3H]TAG as a marker for VLDL were incubated for 30 min at 37 °C with cytosol, GTP and an ATP-regenerating system. After 30 min incubation, the reaction mixture was resolved on a continuous sucrose density gradient (0.1–0.86 M) by ultracentrifugation. Under these conditions, it was expected that the VLDL-containing vesicles would appear in the lightest density fractions and the unreacted TGN membranes be pelleted. The entire sucrose gradient was fractionated into 0.5 ml fractions by aspiration, [3H]TAG was extracted from each fraction and the associated d.p.m. value was determined. As shown in Figure 2(A), the low-density fractions contained the highest [3H]TAG d.p.m. indicating the presence of putative TAG-rich mature VLDL carrying TGN-derived vesicles. In the presence of cytosol, 22–25 % of the nascent [3H]TAG was shifted from the TGN to the first four fractions containing putative PG-VTVs. For the sake of clarity, we will refer to these vesicles as PG-VTVs.
Figure 2. PG-VTV biogenesis and their distribution in continuous sucrose density gradient.

(A) Hepatic TGN containing [3H]TAG isolated from primary hepatocytes was incubated at 37 °C with or without rat hepatic cytosol in the presence of GTP and an ATP-regenerating system. The reaction mixture was resolved on a continuous sucrose gradient (0.1–0.86 M) and fractions of 0.5 ml were collected. The [3H]TAG was extracted and d.p.m. was measured from each fraction (0.5 ml). Data shown are the means ± S.E.M. (n = 4). (B) A budding assay similar to (A) was performed using doubly labelled TGN membranes that contain [14C]TAG and [3H]protein. Radiolabelled TAG was extracted and the protein was precipitated with TCA from each fraction and the [14C]TAG d.p.m. (left y-axis) and [3H]protein d.p.m. (right y-axis) were measured. Data shown are the means ± S.E.M. (n = 4). (C) Distribution of apoB100, apoAI, apoAIV, apoE, albumin, transferrin and GOS28 across the continuous sucrose density (0.1–0.86 M) gradient. A similar experiment to (A) was performed and the reaction mixture was resolved on a continuous sucrose gradient. An equal volume (100 μl) of each fraction and 30 μg of protein of whole cell lysate (WCL) were separated by SDS/PAGE (4–20 % gels) and probed with the indicated antibodies. The results are representative of four independent experiments. Cyto, cytosol.
We have shown previously that nascent VLDL particles and secretory proteins are transported from the ER to the Golgi in two different types of vesicles [1]. This raises the possibility that mature VLDL and secretory proteins exit the TGN in two different vesicles. Therefore we decided to examine the distribution of both PG-VTVs and TGN-derived PTVs (protein transport vesicles) in the same continuous sucrose density gradient. To distinguish the two populations of vesicles, we used doubly labelled ([14C]TAG and [3H]protein) TGN membranes in the TGN-budding assay. The data presented in Figure 2(B) revealed that the maximal [14C]TAG d.p.m. was present in fractions 1–4, whereas most of the [3H]protein d.p.m. appeared in fractions 6–8, suggesting the presence of PG-VTVs and PTVs in these fractions respectively. Of the total [14C]TAG and [3H]protein d.p.m. present in the TGN, ~ 25 % of [14C]TAG was released to the first four fractions, whereas 25–27 %of [3H]protein d.p.m. was released to fractions 6–8 in the presence of cytosol GTP and an ATP-regenerating system. We observed that a small amount of [3H]protein d.p.m. was present in fractions 1–4 (Figure 2B), indicating the presence of nascent proteins in the PG-VTVs. These results provided the first evidence that mature VLDLs and secretory proteins exit the TGN in distinct vesicles.
To provide additional evidence that putative PG-VTVs appear in the lightest density fractions of the gradient, we probed the sucrose gradient fractions by Western blotting for the presence of apoB100, a core protein of VLDL [28], as well as for apoAIV, apoAI and apoE, additional marker proteins of mature VLDL (Figure 2C). Figure 2(C) shows that apoB100, apoAIV, apoAI and apoE are present in the light density fractions of the sucrose gradient. Since these proteins appeared in the same fractions that contained highest [3H]TAG d.p.m., we deduced that PG-VTVs are present in these fractions. To verify the specificity of these results, we examined the fractions for the presence of GOS28, a cis-Golgi-resident protein [29], and albumin and transferrin, marker proteins for secretory protein-containing vesicles [1,17,30]. Our results show that albumin and transferrin are present primarily in higher sucrose-density fractions of the gradient, which is the expected place for TGN-derived PTVs (Figure 2C) and is consistent with our previous observations [1]. GOS28 appeared only in the whole cell lysate (Figure 2C). These data suggest that mature VLDL particles exit the hepatic TGN in distinct vesicles that do not contain albumin or transferrin. Moreover, our results indicate that PG-VTVs have lighter buoyant density compared with PTVs.
Next, we sought to determine the factors that are required for the formation of the PG-VTVs. As shown in Figure 3(A), the removal of cytosol from the reaction resulted in complete cessation of PG-VTV genesis, indicating the requirement of cytosol. Of the total [3H]TAG d.p.m. in the TGN, ~ 25 %was shifted to the PG-VTV fraction in the presence of cytosol; however, only 4–5 % of [3H]TAG was released without cytosol ( − Cyto). To ascertain that the cytosolic factor(s) required for the biogenesis of the PG-VTVs is protein, we either boiled the cytosol or treated it with proteinase K before use in the TGN-budding assay. The results presented in Figure 3(A) clearly indicate that both boiled cytosol and proteinase-K-treated cytosol did not support the formation of the PG-VTV because only ~ 4 % [3H]TAG was transferred from the TGN to the PG-VTV fraction under these conditions. These data suggest that cytosolic protein(s) are needed for the biogenesis of the PG-VTV. When the TGN-budding assay was carried out at 4 °C, we observed a sharp decrease in PG-VTV formation (Figure 3A), only ~ 7 % of the total [3H]TAG d.p.m. was released to the PG-VTV fraction, suggesting that the PG-VTV formation occurs at 37 °C.
Figure 3. Determinants of PG-VTV biogenesis.

(A) An in vitro TGN-budding assay similar to Figure 2(A) was performed in which hepatic TGN containing [3H]TAG was incubated with rat hepatic cytosol ( + Cyto), without cytosol ( − Cyto), with boiled rat hepatic cytosol (Boiled Cyto) or with rat hepatic cytosol treated with proteinase K (Cyto + Prot K) in the presence of GTP and an ATP-regenerating system at 37 °C. The TGN-budding assay carried out at 4 °C with rat hepatic cytosol in the presence of GTP and an ATP-regenerating system (4 °C). Data shown are the means ± S.D. (n = 4). Bars labelled with different symbols are significantly different at P < 0.002 as determined using one-way ANOVA. (B) Similar to (A), an in vitro TGN-budding assay was performed with rat hepatic cytosol at 37 °C in the presence of GTP and an ATP-regenerating system ( + ATP), in the absence of an ATP-regenerating system ( − ATP), or ATP replaced with: 20 units/ml apyrase, ATPγS (10 μM) or ATPγS (25 μM). Data are shown as the means ± S.D. (n = 4). Bars labelled with different symbols are significantly different at P < 0.005 as determined using one-way ANOVA. (C) Time course of ATP-dependent PG-VTV biogenesis. A budding assay similar to Figure 2(A) was performed for the indicated time points and the [3H]TAG from the first four fractions was extracted and [3H]TAG d.p.m. was measured. The budding efficiencies were calculated as relative to the highest [3H]TAG d.p.m. of the first four fractions. Data are shown as the means ± S.E.M. (n = 4). (D) An in vitro TGN-budding assay was carried out with rat hepatic cytosol at 37 °C in the presence of GTP and an ATP-regenerating system (control) or GTP replaced with GTPγS (10 μM), GTPγS (20 μM) or GTPγS (50 μM). Data shown as the means ± S.D. (n = 4). Bars labelled with different symbols are significantly different at P < 0.002 as determined using one-way ANOVA.
To ascertain that the formation of PG-VTVs from the hepatic TGN requires ATP, we either replaced the ATP-regenerating system with 5 mM glucose and 25 units/ml hexokinase (Figure 3B) or used 20 units/ml apyrase (Figure 3B) instead of ATP. Under either condition, the PG-VTV budding activity was significantly reduced (Figure 3B). We estimated that, of the total [3H]TAG d.p.m. in the TGN, 24–25 %( + ATP) and ~ 6 %( − ATP and apyrase) shifted to the PG-VTV fractions, suggesting that the PG-VTV requires ATP for its formation. To find out whether ATP hydrolysis is needed for the biogenesis of the PG-VTV, we probed the effect of ATPγ S, a non-hydrolysable analogue of ATP, on the PG-VTV budding activity. As shown in Figure 3(B), ATP replacement with 10 μM or 25 μM ATPγS did not inhibit the budding activity (25–26 %of total [3H]TAG d.p.m. in the TGN was released to the PG-VTV fractions), indicating that ATP hydrolysis is not required for the formation of the PG-VTV. The data presented in Figure 3(C) demonstrate the time course of ATP-dependent biogenesis of the PG-VTV. As shown in Figure 3(C), ~ 50 % of budding activity was achieved in 5 min; however, the maximal budding activity was observed at the 30 min time point. The extent of PG-VTV budding did not increase after 30 min when the incubation was continued for 120 min (Figure 3C). In an attempt to determine whether GTP hydrolysis is required for the formation of the PG-VTV from hepatic TGN membranes, we investigated the effect of GTPγS, a non-hydrolysable analogue of GTP, on the PG-VTV budding activity. In the presence of GTP (control), ~ 24 % of the total [3H]TAG d.p.m. shifted from the TGN to the PG-VTV fraction, whereas when GTP was replaced with different concentrations of GTPγS, only 3–5 % [3H]TAG was measured in the PG-VTV fractions (Figure 3D). We determined that the PG-VTV budding activity was reduced by ~ 80 % when GTP was replaced with GTPγS (10 μM) and the maximal reduction (~ 90 %) in budding activity was observed with 20 μM GTPγS (Figure 3D). These data suggest that the biogenesis of the PG-VTV from the TGN is GTP hydrolysis dependent.
Characterization of PG-VTVs
To characterize the TGN-derived PG-VTVs, we first needed to assess the morphology of the vesicles present in the light density fractions. To visualize the putative budded vesicles, peak [3H]TAG fractions 1–4 (Figure 2A) were concentrated and examined by EM. Figure 4(A) shows intact vesicles ranging in diameter from 300 to 350 nm. Some of these vesicles have bright circular areas, which may be an artefact of the staining process. Since intact vesicles (PG-VTVs) were adsorbed on EM grids, we did not expect to visualize internal content of the PG-VTVs, VLDL particles, using the negative staining technique. Next, we examined 104 vesicles from different fields and measured their diameters. The data presented in Figure 4(B) show the size distribution of PG-VTVs. The average diameter of PG-VTVs was 320 ± 12 nm. These observations suggest that the hepatic TGN-derived PG-VTVs are larger in size than the PTVs budded from the TGN, which average between 78 and 100 nm in size [11].
Figure 4. Morphology of the PG-VTV.

(A) PG-VTVs were generated from the hepatic TGN as in Figure 2(A). PG-VTVs (fractions 1–4) were examined by the EM using the negative-staining technique (see the Experimental section for details). Scale bars, 100 nm. (B) The diameters of 104 PG-VTVs from different fields were measured.
One of the characteristics of vesicle biogenesis is that budded vesicles should be sealed. To answer whether PG-VTVs were sealed or not, we incubated the vesicular fractions with proteinase K at a final concentration of 0.1 mg/ml for 30 min at 4°C in the absence or presence of Triton X-100. Post-incubation, PMSF was used in excess to neutralize proteinase K. As shown in Figure 5(A), the treatment of PG-VTVs with proteinase K did not decrease the intensity of protein bands for apoB100, apoAIV and apoAI as compared with mock-treated PG-VTVs. However, when the PG-VTVs were treated with proteinase K in the presence of 2 %(v/v) Triton X-100, which disrupts the vesicular membranes, we did not observe any protein band for apoB100, apoAIV and apoAI (Figure 5A), indicating that PG-VTVs have acquired an intact membrane during budding from hepatic TGN membranes. These data strongly suggest that VLDL proteins are enclosed by a lipid layer and that PG-VTVs are sealed.
Figure 5. Effect of proteinase K treatment on cargo proteins of the PG-VTV and the protein composition of the PG-VTV.

(A) PG-VTVs were incubated with proteinase K (0.1 mg/ml) in the presence or the absence of 2 % (v/v) Triton X-100 for 30 min at 4 °C. Protein samples (30 μg) from treated or untreated PG-VTVs were assessed by immunoblotting with the indicated antibodies. (B) Immunoblot analysis of PG-VTV proteins. Proteins samples (30 μg each) of the TGN and the PG-VTVs were immunoblotted for the indicated proteins. The results are representative of four experiments.
Another quality of transport carriers is that they concentrate their cargo proteins and specific SNARE (soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor) proteins which direct them to their destination. Also, these transport carriers exclude non-cargo proteins from their parent membrane. To investigate whether PG-VTVs meet these criteria, we probed the PG-VTVs for a number of VLDL, SNARE and TGN-resident proteins using Western blotting. We used equal amounts of the PG-VTV and TGN proteins for comparative analysis. As we expected, cargo proteins for PG-VTVs, such as apoB100, apoAI, apoE and apoAIV, were concentrated in PG-VTVs as compared with the TGN (Figure 5B). By contrast, the TGN-resident protein TGN38 was not present in the PG-VTVs, but gave a robust signal in the TGN (Figure 5B). As shown in Figure 5(B), VAMP7, a post-Golgi v-SNARE protein, was found to be concentrated in the PG-VTVs, whereas Sec22b, Sar1 (proteins typical for ER-to-Golgi vesicles), and albumin were completely absent in PG-VTVs, but were found in the TGN. SNAP23, a SNARE protein involved in the TGN-to-PM transport event, was present in the PG-VTVs; however, no increase in SNAP23 signal was observed compared with the TGN (Figure 5B). Since ER-derived VTVs and the TGN membranes contain MTPs, it was of interest to investigate whether MTPs were present in TGN-derived PG-VTVs. Our results show that MTP was present in the PG-VTVs; however, we did not see any significant difference in MTP levels in the PG-VTVs when compared with the TGN (Figure 5B). Taken together, these results strongly suggest that PG-VTVs accommodate their specific cargo and exclude other non-specific proteins. Moreover, these observations enabled us to conclude that PG-VTVs are the TGN-to-PM transport vesicles and are not the fragmented TGN membranes.
ApoAI acquisition occurs in cis-Golgi
Previous studies from our group have shown that nascent VLDLs from the ER do not contain apoAI [1]; however, the data presented in the present study (Figure 2C) suggest that mature VLDL particles exiting the TGN contain apoAI. It is not clear whether acquisition of apoAI on VLDL occurred in the cis-Golgi or trans-Golgi. To resolve this issue, we isolated VLDL particles from the purified fractions of the ER, VTV, cis-Golgi and trans-Golgi and probed for the presence of apoAI. Our results show that VLDL particles isolated from both the cis- and trans-Golgi contain apoAI (Figure 6), whereas VLDL isolated from the VTV and the ER membranes were devoid of apoAI, which is consistent with our previous report [27]. These results suggest that the acquisition of apoAI on VLDL occurs in the cis-Golgi.
Figure 6. ApoAI-recruitment to the VLDL particles occurs in the cis-Golgi.

Hepatic ER, ER-derived VTVs, cis-Golgi and trans-Golgi were incubated with 100 mM carbonate buffer (pH 11). The VLDL particles released from the: ER (ER-VLDL), VTV (VTV-VLDL), cis-Golgi (CG-VLDL) and trans-Golgi (TGN-VLDL) were floated by centrifugation and collected. De-lipidated VLDL proteins were immunoblotted with specific antibodies against apoAI and apoAI associated with the VLDL was quantitated by densitometry using ImageJ (NIH). The data are representative of four independent experiments. Bars labelled with different symbols have P < 0.001 using one-way ANOVA.
PG-VTVs mediate the targeted delivery of mature VLDL to the PM
One of the most important features of the functional transport carriers is that they should be able fuse with their target membranes and deliver their cargos to their destination. In this case, PG-VTVs should be able to fuse with the PM for VLDL delivery and eventual secretion. To determine whether PG-VTVs were fusogenic and able to deliver mature VLDL particles to the PM, we developed an in vitro fusion assay that allowed us to monitor the fusion between PG-VTVs and the PM. Before the fusion assay, we assessed the purity of our PM fractions. As shown in Figure 7(A), Na+/K+ -ATPase, a marker for the PM, was concentrated in the PM fraction, whereas a relatively small amount of Na+/K+ -ATPase was detected in the TGN fraction. Additionally, it was necessary to flip the cytosolic side of the isolated PM outward so that the cytosolic side can be exposed and PG-VTVs can fuse. We used a freeze–thaw method to flip the cytosolic side of the PM as reported by others [24].
Figure 7. PG-VTV fuses with the hepatic PM.
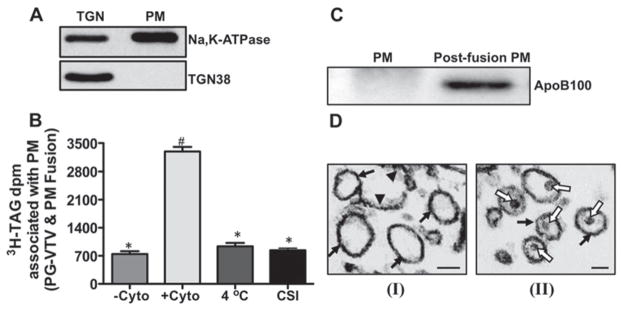
(A) Isolation and characterization of hepatic PM. The PM was isolated and its purity was determined by immunoblot analysis. Samples of the TGN and the PM (PM) (each containing 30 μg) were probed with specific antibodies against Na +/K + -ATPase and TGN38. The results are representative of four independent experiments. (B) PG-VTVs (150 μg) containing [3H]TAG were incubated with non-radiolabelled PM with the cytosolic side outward (300 μg) at 37 °C in the absence ( − Cyto) or presence ( + Cyto) of hepatic cytosol (500 μg) or at 4 °C in the presence of cytosol (500 μg) (4 °C). PG-VTVs (150 μg) containing [3H]TAG were incubated with non-radiolabelled CSI PM (300 μg) at 37 °C in the presence of hepatic cytosol (500 μg). The PM was separated and the associated [3H]TAG d.p.m. was measured. The data are representative of four independent experiments. Bars labelled with different symbols are significantly different at P < 0.002 as determined using one-way ANOVA. (C) The presence of apoB100 in the PM fraction after fusion reaction. Western blot analysis of the naive PM (PM) and the PM isolated after fusion reaction (post-fusion PM) in the presence of cytosol at 37 °C. The results are representative of four independent experiments. (D) Similar to (A), PG-VTVs containing [3H]TAG were incubated with non-radiolabelled PM with cytosolic side outward in the absence (I) or presence (II) of hepatic cytosol. Post-fusion, the PM was separated from the reaction mixture and examined by EM. The electron micrographs shown are representative of 25 different fields that were examined for each fusion reaction (without or with cytosol). Black arrows indicate vesicular PM, black arrowheads indicate broken/linear PM and white open arrows indicate VLDL particles inside the PM vesicles. Scale bars, 200 nm.
PG-VTVs containing [3H]TAG were incubated with non-radiolabelled acceptor, hepatic PM, in the presence of hepatic cytosol, GTP and an ATP-regenerating system at 37 °C for 30 min. Post-incubation, the PM was isolated using a sucrose density gradient and the associated [3H]TAG was extracted and its d.p.m. measured. We found that a small amount, ~ 7 % of the total [3H]TAG d.p.m. in PG-VTV, was transferred to the PM in the absence of cytosol (Figure 7B). Also, when PG-VTVs and the PM were incubated at 4 °C, no significant increase in [3H]TAG d.p.m. was observed in the PM compared with when fusion assay was performed in the absence of cytosol (Figure 7B). However, when PG-VTVs were incubated with the PM in the presence of cytosol at 37 °C, a significant increase in [3H]TAG d.p.m. associated with the PM was observed (Figure 7B). Interestingly, as was expected, when PG-VTVs were incubated with PM that was not flipped inside-out [CSI (cytosolic side inward)], no fusion activity was noticed (Figure 7B, CSI). Under positive conditions ( + Cyto), ~ 35 % of the total [3H]TAG present in the PG-VTV was transferred to the PM, whereas under non-fusogenic conditions, as indicated in the parentheses, ~ 9 % (4 °C) and ~ 8 % (CSI) [3H]TAG was measured in the PM (Figure 7B). To support the observations that the PG-VTV fuses with and delivers mature VLDLs to the PM, we sought to probe the presence of apoB100 in the PM after the fusion. We performed the fusion assay in the presence of cytosol, GTP and an ATP-regenerating system (exactly the same as in + Cyto; Figure 7B) and isolated the PM (post-fusion PM). The data shown in Figure 7(C) show that apoB100 was not present in naive PM, which was not subjected to the fusion reaction; however, we observed a strong apoB100 signal in the post-fusion PM. These results suggest that PG-VTV was able to deliver VLDL-apoB100 to the PM.
However, these data did not distinguish between the two possibilities: first, the PG-VTV actually fused with the PM and delivered the mature VLDL, and secondly, the PG-VTV was bound to the PM, but did not fuse. To address this issue, we adopted a morphological approach, in which we carried out detailed EM analyses of the post-fusion PM under conditions that: (i) do not support the fusion (no cytosol or ATP) and (ii) support the fusion (with cytosol and ATP). Post-fusion assay, the PM was isolated from the reaction mixture and examined by EM. Figure 7(D) shows the morphology of the PM under non-fusion (i) and fusion (ii) conditions. Under both conditions, the PM appeared mostly as vesicular structures (Figure 7D, I and II, black arrows), which is consistent with observations reported previously by Hubbard et al. [31]. Interestingly, we identified particles resembling VLDLs in the PM vesicles (Figure 7D, II, white open arrows) when the fusion assay was performed under fusogenic conditions. No VLDL-like structure was seen in the PM when the fusion assay was carried out under non-fusogenic conditions (Figure 7D, I). These data strongly suggest that PG-VTVs were, in fact, able to fuse with and deliver their cargo to the PM.
PG-VTV delivers mature VLDL to the PM vectorially
The data presented in the present study show that the PG-VTV originates from the TGN and fuses with the PM. We questioned whether the transport of mature VLDL particles facilitated by PG-VTVs is unidirectional or bi-directional. To answer this question, we incubated PG-VTVs containing [3H]TAG with non-labelled TGN membranes in the presence of cytosol, GTP and an ATP-regenerating system at 37 °C for 30 min. After 30 min incubation, TGN membranes were isolated using a sucrose density gradient (see the Experimental section), TAG was extracted and [3H]TAG d.p.m. was determined. As shown in Figure 8, there was no significant shift in [3H]TAG d.p.m. from the PG-VTV to the TGN; however, when PG-VTVs were incubated with the PM, a significant amount of [3H]TAG d.p.m. was transferred to the PM (Figure 8). Of the total amount of [3H]TAG present in the PG-VTVs, ~ 7 % ( − Cyto), ~ 35 % (PGVTV + PM) and ~ 6 % (PGVTV + TGN) was transferred to the PM or the TGN under conditions indicated in parentheses. These results strongly suggest that the PG-VTV facilitates the transport of mature VLDL in an anterograde manner and does not support the retrograde transport.
Figure 8. PG-VTV fuses with hepatic PM vectorially.

PG-VTVs containing [3H]TAG were incubated at 37 °C with non-radiolabelled PM in the presence of cytosol (PGVTV + PM) or the absence of cytosol ( − Cyto) or in the presence of cytosol with hepatic TGN (PGVTV + TGN). Post-fusion, the PM and the TGN were separated and the associated [3H]TAG extracted and d.p.m. was determined. The data are representative of four independent experiments. Bars labelled with different symbols are significantly different at P < 0.005 as determined using one-way ANOVA.
DISCUSSION
Upon completion of their genesis in hepatic ER, nascent VLDL particles are transported to the Golgi using a dedicated vesicular system. Previous work from our group has shown that an ER-derived vesicle, the VTV, delivers the nascent VLDL particle into the Golgi lumen where inevitable modifications occur yielding a mature VLDL particle ready to be transported to the PM for its ultimate secretion into the blood stream [1]. The movement of mature VLDL particles from the TGN to the PM is required for their secretion; however, this physiologically important event has not been studied so far. To investigate how mature VLDL particles exit the TGN and how they are being transported to the PM in primary rat hepatocytes, we developed an in vitro system. The data of the present study describe a new post-Golgi vesicular system that is dedicated to the TGN-to-PM transport of mature VLDLs.
Multiple lines of evidence demonstrate that we were successful in developing an in vitro TGN-budding assay to monitor the formation of PG-VTVs from the hepatic TGN and that the budded vesicles were bona fide TGN-derived vesicles that contained mature VLDL particles: (i) EM revealed large intact vesicles ranging from ~ 300 to 350 nm in diameter. These vesicles were larger than post-TGN PTVs and were large enough to accommodate two to three mature VLDL particles, which average ~ 80 nm in their diameter [17,19]; (ii) the biogenesis of PG-VTVs was dependent on cytosol and an ATP-regenerating system; (iii) the formation of PG-VTV occurred when the budding assay was carried out at 37 °C, whereas no PG-VTV biogenesis was observed at 4 °C; (iv) PG-VTVs were impervious since proteinase K treatment did not degrade the VLDL core proteins apoB100, apoAI and apoAIV; (v) PG-VTVs were able to concentrate a cargo protein apoB100; (vi) PG-VTVs concentrated the post-Golgi v-SNARE protein VAMP7 and excluded the Golgi-resident proteins GOS28 and TGN38 and secretory cargo proteins that are transported by a different vesicle system namely albumin and transferrin; and (vii) PG-VTVs were able to fuse with the PM. All of these morphological, biochemical and functional features of PG-VTVs qualify them as typical post-Golgi transport vesicles and rule out the possibility of their artefactual genesis.
Vesicle budding and fusion are complex, but inevitable events for the transport of specific cargos between subcellular organelles and eventual secretion out of cells [32]. Regulation of vesicular transport at the level of the TGN is physiologically vital to the generation of basolateral and apical membranes in polarized cells, and for cargo sorting for transport from the Golgi [33–36]. The significance of the Golgi regulation has been shown with studies on protein-containing vesicles where Golgi lipids such as phosphoinositides have been shown to play a role in the recruitment of trafficking components such as clathrin coats [37,38], whereas glycosphingolipids and cholesterol levels have been hypothesized to play a role in segregating components into lipid rafts [39]. These studies have emphasized the importance of the continuous membrane flow, a characteristic of the Golgi, in directing various cargos for secretion [15], and have proposed that the Golgi-derived vesicles are initially formed by a tubule-like extension of the TGN membranes and their subsequent membrane fission resulting in vesicles [40,41]. Although much is known about the clathrin-coated vesicles that mediate PM-to-TGN vesicular transport, COPI-coated vesicles that facilitate retrograde Golgi-to-ER transport and more recently endosomes-to-TGN transport of secretory proteins, and COPII-coated vesicles that mediate anterograde ER-to-Golgi protein transport, the vesicles that mediate the TGN-to-PM transport are not yet fully characterized. This novel vesicle, the PG-VTV, is a new TGN-to-PM cargo carrier, unique in size, buoyant density, and cargo and membrane proteins responsible for the secretion of fully matured VLDL from hepatocytes.
Our data clearly suggest that PG-VTVs are biochemically and morphologically different from other TGN-derived vesicles. PG-VTVs do not carry hepatic secretory proteins albumin and transferrin and their size is significantly larger than other TGN-derived vesicles, which average 78–100 nm in diameter [11]. This raises an obvious question: why does the liver use a different vesicular system to export mature VLDLs from the TGN to the PM? One possibility is that the VLDL synthesis and secretion are regulated by many factors and depend on the availability of FFAs, whereas protein synthesis and transport are constant in nature [42,43]. Another possibility is that the size of the mature VLDL particle is large and therefore, it cannot be accommodated in relatively smaller TGN-derived PTVs; however, Polishchuk et al. [44] have shown that outsized cargo particles such as procollagen-I are transported from the TGN to the PM in large tubular TGN-derived carriers that also contain the secretory protein marker VSV-G. Interestingly, these large TGN-to-PM carriers excluded other cargo proteins from the TGN [44]. Transport of different cargos from the TGN in distinct vesicles has been very well established [15,45]. Not only do specific vesicles deliver specific cargos to the PM, but it has also been demonstrated that discrete TGN-derived vesicles are involved in cargo transport to the apical and basolateral areas of the PM [9,11]. These observations support our findings that the mature VLDL particle exits the TGN in a distinct vesicle, the PG-VTV.
The delivery of specific cargos to their destinations is a primary task of the secretory vesicles. Consistent with this characteristic, data discussed in the present study demonstrate that the PG-VTV fuses with the PM to deliver mature VLDL particles for their eventual secretion (Figure 9). PG-VTVs, however, did not fuse with hepatic TGN membranes, indicating that PG-VTVs are involved in anterograde TGN-to-PM trafficking of mature VLDL particles. Additionally, the presence of apoAI in TGN-derived PG-VTVs, but not in the ER-derived VTVs, indicates that VLDL particles acquire apoAI in the Golgi lumen. These data are consistent with previous reports demonstrating Golgi-mediated apoAI acquisition on the surface of lipoproteins of small intestinal origin, the chylomicrons [46,47].
Figure 9. Proposed model of post-Golgi VLDL trafficking in primary hepatocytes.

Mature VLDL particles exit the TGN in a specialized larger vesicle, the PG-VTV. PG-VTV is targeted to and fuses with hepatic PM in order to secrete mature VLDL particles into the circulatory system. Mature VLDL particles are not sorted into the TGN-derived smaller PTV that carries secretory proteins such as albumin and transferrin to the PM.
In the present study, we have identified and characterized a new member of the TGN-derived vesicle family, the PG-VTVs that specifically exports mature VLDLs to the PM in primary hepatocytes. EM data revealed that the size of the PG-VTV is larger (~ 300–350 nm) than other TGN-derived vesicles. The distribution of PG-VTVs in the lighter density fractions of the sucrose gradient indicates the presence of TAG-loaded vesicles because of their light buoyant density compared with other TGN-derived vesicles. Interestingly, these vesicles do not contain secretory proteins, albumin and transferrin and unidirectionally traverse to the cell surface to fuse with the PM. Our data show that mature VLDL particles exit the TGN in a specialized vesicle that delivers them to the PM (Figure 9). The data shown in the present study suggest that specific cargos can trigger the formation of distinct vesicles from the TGN.
Acknowledgments
We thank Dr Deborah Altomare (University of Central Florida, Orlando, FL, U.S.A.) for proofreading and editing the paper before submission.
FUNDING
This work was supported by the NIDDK (National Institute of Diabetes and Digestive and Kidney Diseases) of the NIH (National Institutes of Health) [grant number RO1 DK-81413 (to S.A.S.)]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDDK or NIH.
Abbreviations
- apoAI
apolipoprotein AI
- apoAIV
apolipoprotein AIV
- apoB
apolipoprotein B
- apoB100
apolipoprotein B100
- apoE
apolipoprotein E
- COPI
coatamer protein I
- COPII
coatamer protein II
- CSI
cytosolic side inward
- ER
endoplasmic reticulum
- FFA
non-esterified (‘free’) fatty acid
- HRP
horseradish peroxidase
- MTP
microsomal triacylglycerol transfer protein
- PG-VTV
post-TGN VLDL transport vesicle
- PM
plasma membrane
- PTV
protein transport vesicle
- SNAP23
23 kDa synaptosome-associated protein
- SNARE
soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor
- TAG
triacylglycerol
- TCA
trichloroacetic acid
- TGN
trans-Golgi network
- VAMP7
vesicle-associated membrane protein 7
- VLDL
very-low-density lipoprotein
- VTV
VLDL transport vesicle
Footnotes
AUTHOR CONTRIBUTION
Tanvir Hossain, Aladdin Riad and Shaila Siddiqi carried out most of the experiments and analysed the results. Shadab Siddiqi conceived, designed and performed the experiments. Aladdin Riad, Sampath Parthasarathy and Shadab Siddiqi analysed the results and wrote the paper.
References
- 1.Siddiqi SA. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochem J. 2008;413:333–342. doi: 10.1042/BJ20071469. [DOI] [PubMed] [Google Scholar]
- 2.Shelness GS, Ingram MF, Huang XF, DeLozier JA. Apolipoprotein B in the rough endoplasmic reticulum: translation, translocation and the initiation of lipoprotein assembly. J Nutr. 1999;129:456S–462S. doi: 10.1093/jn/129.2.456S. [DOI] [PubMed] [Google Scholar]
- 3.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 4.Hussain MM, Rava P, Pan X, Dai K, Dougan SK, Iqbal J, Lazare F, Khatun I. Microsomal triglyceride transfer protein in plasma and cellular lipid metabolism. Curr Opin Lipidol. 2008;19:277–284. doi: 10.1097/MOL.0b013e3282feea85. [DOI] [PubMed] [Google Scholar]
- 5.Olofsson SO, Boren J. Apolipoprotein B secretory regulation by degradation. Arterioscler Thromb Vasc Biol. 2012;32:1334–1338. doi: 10.1161/ATVBAHA.112.251116. [DOI] [PubMed] [Google Scholar]
- 6.Higgins JA. Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the Golgi complex. FEBS Lett. 1988;232:405–408. doi: 10.1016/0014-5793(88)80780-4. [DOI] [PubMed] [Google Scholar]
- 7.Gusarova V, Seo J, Sullivan ML, Watkins SC, Brodsky JL, Fisher EA. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J Biol Chem. 2007;282:19453–19462. doi: 10.1074/jbc.M700475200. [DOI] [PubMed] [Google Scholar]
- 8.Swift LL, Farkas MH, Major AS, Valyi-Nagy K, Linton MF, Fazio S. A recycling pathway for resecretion of internalized apolipoprotein E in liver cells. J Biol Chem. 2001;276:22965–22970. doi: 10.1074/jbc.M100172200. [DOI] [PubMed] [Google Scholar]
- 9.Keller P, Simons K. Post-Golgi biosynthetic trafficking. J Cell Sci. 1997;110:3001–3009. doi: 10.1242/jcs.110.24.3001. [DOI] [PubMed] [Google Scholar]
- 10.Klemm RW, Ejsing CS, Surma MA, Kaiser HJ, Gerl MJ, Sampaio JL, de Robillard Q, Ferguson C, Proszynski TJ, Shevchenko A, Simons K. Segregation of sphingolipids and sterols during formation of secretory vesicles at the trans-Golgi network. J Cell Biol. 2009;185:601–612. doi: 10.1083/jcb.200901145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wandinger-Ness A, Bennett MK, Antony C, Simons K. Distinct transport vesicles mediate the delivery of plasma membrane proteins to the apical and basolateral domains of MDCK cells. J Cell Biol. 1990;111:987–1000. doi: 10.1083/jcb.111.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes H, Stephens DJ. Assembly, organization, and function of the COPII coat. Histochem Cell Biol. 2008;129:129–151. doi: 10.1007/s00418-007-0363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen D, Schekman R. COPII-mediated vesicle formation at a glance. J Cell Sci. 2011;124:1–4. doi: 10.1242/jcs.069773. [DOI] [PubMed] [Google Scholar]
- 14.Kuehn MJ, Herrmann JM, Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- 15.Bard F, Malhotra V. The formation of TGN-to-plasma-membrane transport carriers. Annu Rev Cell Dev Biol. 2006;22:439–455. doi: 10.1146/annurev.cellbio.21.012704.133126. [DOI] [PubMed] [Google Scholar]
- 16.Yeaman C, Ayala MI, Wright JR, Bard F, Bossard C, Ang A, Maeda Y, Seufferlein T, Mellman I, Nelson WJ, Malhotra V. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat Cell Biol. 2004;6:106–112. doi: 10.1038/ncb1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusarova V, Brodsky JL, Fisher EA. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem. 2003;278:48051–48058. doi: 10.1074/jbc.M306898200. [DOI] [PubMed] [Google Scholar]
- 18.Tiwari S, Siddiqi S, Siddiqi SA. CideB protein is required for the biogenesis of very low density lipoprotein (VLDL) transport vesicle. J Biol Chem. 2013;288:5157–5165. doi: 10.1074/jbc.M112.434258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiwari S, Siddiqi SA. Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol. 2012;32:1079–1086. doi: 10.1161/ATVBAHA.111.241471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddiqi SA, Mahan J, Siddiqi S, Gorelick FS, Mansbach CM., 2nd Vesicle-associated membrane protein 7 is expressed in intestinal ER. J Cell Sci. 2006;119:943–950. doi: 10.1242/jcs.02803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siddiqi S, Mani AM, Siddiqi SA. The identification of the SNARE complex required for the fusion of VLDL-transport vesicle with hepatic cis-Golgi. Biochem J. 2010;429:391–401. doi: 10.1042/BJ20100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodson S, Brenchley G. Similarities of the Golgi apparatus membrane and the plasma membrane in rat liver cells. J Cell Sci. 1976;20:167–182. doi: 10.1242/jcs.20.1.167. [DOI] [PubMed] [Google Scholar]
- 23.Touster O, Aronson NN, Jr, Dulaney JT, Hendrickson H. Isolation of rat liver plasma membranes. Use of nucleotide pyrophosphatase and phosphodiesterase I as marker enzymes. J Cell Biol. 1970;47:604–618. doi: 10.1083/jcb.47.3.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmgren MG, Askerlund P, Fredrikson K, Widell S, Sommarin M, Larsson C. Sealed inside-out and right-side-out plasma membrane vesicles: optimal conditions for formation and separation. Plant Physiol. 1990;92:871–880. doi: 10.1104/pp.92.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar NS, Mansbach CM. Determinants of triacylglycerol transport from the endoplasmic reticulum to the Golgi in intestine. Am J Physiol. 1997;273:G18–G30. doi: 10.1152/ajpgi.1997.273.1.G18. [DOI] [PubMed] [Google Scholar]
- 26.Siddiqi SA, Gorelick FS, Mahan JT, Mansbach CM., 2nd COPII proteins are required for Golgi fusion but not for endoplasmic reticulum budding of the pre-chylomicron transport vesicle. J Cell Sci. 2003;116:415–427. doi: 10.1242/jcs.00215. [DOI] [PubMed] [Google Scholar]
- 27.Rahim A, Nafi-Valencia E, Siddiqi S, Basha R, Runyon CC, Siddiqi SA. Proteomic analysis of the very low density lipoprotein (VLDL) transport vesicles. J Proteomics. 2012;75:2225–2235. doi: 10.1016/j.jprot.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chahil TJ, Ginsberg HN. Diabetic dyslipidemia. Endocrinol Metab Clin North Am. 2006;35:491–510. doi: 10.1016/j.ecl.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Subramaniam VN, Peter F, Philp R, Wong SH, Hong W. GS28, a 28-kilodalton Golgi SNARE that participates in ER–Golgi transport. Science. 1996;272:1161–1163. doi: 10.1126/science.272.5265.1161. [DOI] [PubMed] [Google Scholar]
- 30.Sztul ES, Howell KE, Palade GE. Intracellular and transcellular transport of secretory component and albumin in rat hepatocytes. J Cell Biol. 1983;97:1582–1591. doi: 10.1083/jcb.97.5.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard AL, Wall DA, Ma A. Isolation of rat hepatocyte plasma membranes. I Presence of the three major domains. J Cell Biol. 1983;96:217–229. doi: 10.1083/jcb.96.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 33.Muth TR, Caplan MJ. Transport protein trafficking in polarized cells. Annu Rev Cell Dev Biol. 2003;19:333–366. doi: 10.1146/annurev.cellbio.19.110701.161425. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Boulan E, Kreitzer G, Musch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005;6:233–247. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Boulan E, Musch A. Protein sorting in the Golgi complex: shifting paradigms. Biochim Biophys Acta. 2005;1744:455–464. doi: 10.1016/j.bbamcr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Graham TR, Burd CG. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011;21:113–121. doi: 10.1016/j.tcb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 2003;114:299–310. doi: 10.1016/s0092-8674(03)00603-2. [DOI] [PubMed] [Google Scholar]
- 38.De Matteis M, Godi A, Corda D. Phosphoinositides and the Golgi complex. Curr Opin Cell Biol. 2002;14:434–447. doi: 10.1016/s0955-0674(02)00357-5. [DOI] [PubMed] [Google Scholar]
- 39.Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- 40.Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 41.Baron CL, Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science. 2002;295:325–328. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- 42.Mason TM. The role of factors that regulate the synthesis and secretion of very-low-density lipoprotein by hepatocytes. Crit Rev Clin Lab Sci. 1998;35:461–487. doi: 10.1080/10408369891234246. [DOI] [PubMed] [Google Scholar]
- 43.Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) 2010;7:35. doi: 10.1186/1743-7075-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polishchuk EV, Di Pentima A, Luini A, Polishchuk RS. Mechanism of constitutive export from the Golgi: bulk flow via the formation, protrusion, and en bloc cleavage of large trans-Golgi network tubular domains. Mol Biol Cell. 2003;14:4470–4485. doi: 10.1091/mbc.E03-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wakana Y, van Galen J, Meissner F, Scarpa M, Polishchuk RS, Mann M, Malhotra V. A new class of carriers that transport selective cargo from the trans-Golgi network to the cell surface. EMBO J. 2012;31:3976–3990. doi: 10.1038/emboj.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siddiqi SA, Siddiqi S, Mahan J, Peggs K, Gorelick FS, Mansbach CM., 2nd The identification of a novel endoplasmic reticulum to Golgi SNARE complex used by the prechylomicron transport vesicle. J Biol Chem. 2006;281:20974–20982. doi: 10.1074/jbc.M601401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mansbach CM, Siddiqi SA. The biogenesis of chylomicrons. Annu Rev Physiol. 2010;72:315–333. doi: 10.1146/annurev-physiol-021909-135801. [DOI] [PMC free article] [PubMed] [Google Scholar]