

Abstract
Chronic wasting disease (CWD) is an efficiently transmitted, fatal, and progressive prion disease of cervids with an as yet to be fully clarified host range. While outbred domestic cats (Felis catus) have recently been shown to be susceptible to experimental CWD infection, the neuropathologic features of the infection are lacking. Such information is vital to provide diagnostic power in the event of natural interspecies transmission and insights into host and strain interactions in interspecies prion infection. Using light microscopy and immunohistochemistry, we detail the topographic pattern of neural spongiosis (the “lesion profile”) and the distribution of misfolded prion protein in the primary and secondary passage of feline CWD (FelCWD). We also evaluated cellular and subcellular associations between misfolded prion protein (PrPD) and central nervous system neurons and glial cell populations. From these studies, we (1) describe the novel neuropathologic profile of FelCWD, which is distinct from either cervid CWD or feline spongiform encephalopathy (FSE), and (2) provide evidence of serial passage-associated interspecies prion adaptation. In addition, we demonstrate through confocal analysis the successful co-localization of PrPD with neurons, astrocytes, microglia, lysosomes, and synaptophysin, which, in part, implicates each of these in the neuropathology of FelCWD. In conclusion, this work illustrates the simultaneous role of both host and strain in the development of a unique FelCWD neuropathologic profile and that such a profile can be used to discriminate between FelCWD and FSE.
Keywords: prion, chronic wasting disease, immunohistochemistry, interspecies, cat, feline spongiform encephalopathy, transmissible spongiform encephalopathy, adaptation, species barrier
Chronic wasting disease (CWD) is a naturally occurring transmissible prion disease of cervids, including white-tailed deer, mule deer, elk, and moose.42 CWD, like other transmissible spongiform encephalopathies (TSEs), including scrapie, bovine spongiform encephalopathy (BSE), and human Creutzfeldt-Jakob disease (CJD), is caused by the coerced conversion of normal protease-sensitive prion protein (PrPC) to a misfolded protease-resistant conformation (PrPD), which progressively accumulates in the central nervous system (CNS) and lymphoid system.44,47,50 CWD is uniquely characterized by a high degree of horizontal transmissibility that is due, in part, to the efficient shedding of infectious prions in a number of cervid excreta, including urine, saliva, and feces.17,18,38 However, despite the facile nature by which CWD is horizontally spread, the breadth of CWD’s possible host range, including the risk of interspecies transmission to humans and other cohabitant species, including scavengers and predators, remains unknown.
The interspecies spread of prions is dependent on successful breaching of the prion species barrier.11 The major determinants of the species barrier are believed to be many, although amino acid sequence of the host prion protein (PrP) and conformational characteristics of the infecting strain are believed to be the most significant.1 While robust, the species barrier is not absolute, and there are multiple examples of its breaching, most notably the interspecies transmission of BSE-origin prions: (a) to humans as variant Creutzfeldt-Jakob disease (vCJD) and (b) to cats (both domestic and wild) as feline spongiform encephalopathy (FSE).23 It is noteworthy that the BSE origin of both vCJD and FSE was largely provided by the detailed neuropathologic characterization of the affected humans and animals.22 In CWD, evidence of the in vivo plasticity of the species barrier has been illustrated by successful interspecies infection of cattle, sheep, goats, ferret, mink, squirrel monkeys, transgenic mice, and recently domestic cats.19,39 However, despite these studies, the entirety of the natural CWD host range has yet to be fully clarified. Relevant to the work described here is the potential role that sympatric species, including large cats, might play in the spread of CWD, particularly in light of studies providing compelling evidence that mountain lions (Puma concolor) selectively prey on CWD-infected deer.31
TSE neuropathology consists of gray matter spongiosis, glial cell accumulation and activation, neuronal loss, and accumulation of the abnormal, misfolded isoform of the prion protein (denoted PrPD).25 It is well established that there can be substantial host- and strain-conferred variation in the morphology, severity, and distribution of this pathology, which is most commonly identified through evaluation of the lesion profile and the map of PrPD distribution.6,13,25,34 Such neuropathologic profiling has been used in natural and experimental TSE research, including in CWD and FSE, to (1) identify a number of distinct prion strains, (2) implicate BSE as the etiology of vCJD and FSE, and (3) discriminate between 2 clinically identical TSEs in sheep (scrapie and BSE).10,14,16,24,35,55–57 As the recognition of natural interspecies prion transmission can be difficult, experimentally deriving predictive lesion and PrPD profiles would greatly enhance the ability should such events occur.40
The goals of this study were to describe the neuropathologic features associated with breaching of the cervid-feline prion species barrier in primary and secondary passage feline CWD (FelCWD). To accomplish these aims, we (1) generated the lesion profile of primary and secondary passage FelCWD, (2) characterized the immunohistochemical distribution of PrPD in primary and secondary passage FelCWD, and (3) evaluated the cellular and subcellular patterns of PrPD deposition using multilabel immunofluorescence histochemistry and confocal microscopy. As a result, we demonstrate a sufficiently unique neuropathologic profile that provides novel insights into CWD neuropathogenesis, illustrates the phenomenon of serial passage–associated adaptation of CWD in a new host, and will be useful to identify future natural transspecies feline CWD infections.
Materials and Methods
Animals Used in This Study
The animal experiments were conducted under the guidelines developed by the Colorado State University Animal Care and Use Committee. Four-month-old, outbred domestic cats were provided by the Andrea D. Lauerman Specific Pathogen Free (SPF) colony, Department of Microbiology, Immunology and Pathology (DMIP), College of Veterinary Medicine and Biological Sciences (CVMBS), Colorado State University (CSU). All animals were handled in strict accordance with good animal practice as defined by relevant national and/or local animal welfare bodies, and all animal work was approved by Colorado State University Animal and Care Use Committee (ACUC approval numbers 02-151A, 08-175A, and 11-2622A).
Inoculum Preparation
The details of the inoculation protocol have been previously described and are summarized here.37
Primary passage deer-origin CWD (CerCWD)
For primary passage studies, CWD-positive brain inoculum from a free-ranging mule deer (989-09147) was provided by Dr Terry Spraker at the Colorado State Veterinary Diagnostic Laboratory (CSVDL). This brain inoculum was a homogenate prepared from multiple regions of the brain, and the infectivity of this material has been confirmed in previous bioassay studies.38 CWD-negative brain inoculum for sham controls was obtained from 2 white-tailed deer (UGA 1 and 2) born and raised in a CWD-free area provided by the Warnell School of Forestry and Natural Resources, University of Georgia, Athens (UGA).
Secondary passage feline-adapted CWD (FelCWD)
Inocula for feline-adapted secondary passage studies consisted of pooled brain homogenate from 2 primary passage cats (4152 and 4137) confirmed as CWD positive by Western blot and immunohistochemistry (IHC) or 1 primary passage CWD-negative sham-inoculated cat (4149).
Animal Inoculation, Evaluation, Euthanasia, and Necropsy
Primary and secondary passage
For the primary passage studies, cohorts (n = 5) of SPF domestic cats were inoculated with undiluted CerCWD brain intracranially (IC; 0.5 g into the left parietal cortex). For the secondary passage studies, FelCWD brain pool was IC inoculated into cohorts (n = 5) of SPF domestic cats (0.5 g into the left parietal cortex). For each of the primary and secondary passage studies, age-matched sham controls were inoculated in the same manner with brain from CWD-negative deer (primary passage) or a CWD-negative domestic cat (secondary passage). All IC inoculations were performed by hand under general anesthesia. For the studies described below, brain tissues were available for only 4 animals from each of the primary passage, secondary passage, and sham-inoculated groups. One of the brains from both the primary and secondary passage animal was frozen for alternative studies and inoculum preparation.
Monitoring and sample collection
Following inoculation, cats were monitored daily for behavioral changes and euthanized at or before onset of late-stage clinical signs. At study termination, tissues from each cat were harvested, fixed, and/or frozen for the detection of FelCWD.
Neuropathology
Brains from CWD-infected (primary and second passage) and sham-inoculated (n = 4 per group) domestic cats (Felis catus) were collected at necropsy and sagitally sectioned. One half brain was frozen while the remainder was fixed in 10% neutral buffered formalin (primary passage studies) or paraformaldehyde-lysineperiodate (PLP, second passage studies) for 2 to 5 days followed by storage in 60% ethanol. The fixed half brain was coronally sectioned at 4- to 5-mm intervals, and tissue sections were placed into plastic tissue cassettes. Prior to routine processing and embedding, so as to abrogate preexistent PrPC immunoreactivity, cassetted tissues were immersed in 88% formic acid (FA) for 1 hour. For neuropathologic examination, 6-μm-thick, paraffin-embedded tissue sections were routinely stained with hematoxylin and eosin (HE).
Lesion profiling
Spongiosis scoring was performed and lesion profiles were generated using a modified variation of previously published criteria.13 For each lesion profile, the particular neuroanatomic regions were selected based on (1) the ease by which a particular section could be reproducibly identified and (2) correlation with previously published CWD or FSE lesion profile studies.49,55 The sites evaluated were the (1) caudate nucleus, (2) ventromedial nucleus of the thalamus, (3) medial geniculate nucleus, (4) frontal cortex, (5) pons, (6) parasympathetic nucleus of the vagus, (7) cerebellar vermis, and (8) hippocampus. For each site, the severity of vacuolar lesions was graded 0 (no lesions) to 4 (extensive vacuolization) using a representative composite of photomicrographs. All 12 areas were graded by 3 observers, one of whom is a board-certified veterinary pathologist (D.M.S.). To generate lesion profiles, mean scores for each area were calculated using data from all 3 observers and plotted ± SEM. Last, to compare our data with previously published FSE studies, we extracted spongiosis scoring data from a publication by Wells et al.55
Immunohistochemistry (IHC)
For all IHC experiments, 6-μm-thick, paraffin-embedded tissue sections were mounted on positively charged glass slides; warmed in a 64°C oven; deparaffinized in xylene; and rehydrated through graded ethanol. Following rehydration, to further abrogate normal protein immunoreactivity and enhance antigen retrieval, slides were immersed in 88% FA for 90 minutes. Antigen retrieval was accomplished by heat-induced epitope retrieval (HIER) using an automated antigen-retrieval system (The Retriever Electron Microscopy Sciences, Hatfield, PA) and a citrate buffer solution (10 mM sodium citrate, 0.05% Tween 20, pH 6.0). Of note, in this report, we follow the lead of others and use PrPC to denote the normal, α helical–rich form and the term PrPD (“disease-associated PrP”) to describe the abnormal, β sheet–rich form.25
To examine the effect of proteinase K (PK) on antigen unmasking and PrPD detection sensitivity, sequential brain sections from representative brain regions from both the primary and second passage CWD-infected cats were equilibrated in Tris-buffered saline (TBS) and immersed in 1 of 3 concentrations of PK (2, 8, and 20 μg/mL in TBS) for 15 minutes at 37°C prior to HIER. Digestion was stopped by serial washes in TBS. These PK-digested sections were compared with matched sections treated with a 30-minute immersion in FA.
For the detection of PrPD, a 2-step immunostaining procedure with tyramide signal amplification (TSA) was used. Following slide rehydration and HIER, endogenous peroxidase (EP) activity was quenched with 3% hydrogen peroxide (H2O2) in methanol and sections were blocked with a proprietary protein block (TNB; Perkin-Elmer, Waltham, MA) for 30 minutes each. Slides were sequentially incubated with a 1:300 dilution of the anti–prion protein antibody, L42 (R-Biopharm, Darmstadt, Germany), which is a mouse monoclonal prion protein antibody raised against amino acids 145 to 163 of the ovine prion protein20 and a horseradish peroxidase (HRP)–conjugated, anti–mouse secondary antibody (EnVision+; DakoCytomation, Carpinteria, CA). Between all incubation steps, slides were washed 3 times (5 minutes each) in TNT wash buffer (0.1M Tris-HCl [pH 7.5], 0.15M NaCl, and 0.05% Tween-20). Slides were then sequentially incubated with the remaining TSA reagents (Perkin-Elmer). Antibody deposition was visualized using the chromagen 3-amino-9-ethylcarbazole (AEC; DakoCytomation), and slides were counterstained with hematoxylin, incubated with a bluing reagent (0.1% sodium bicarbonate), and coverslipped with an aqueous mounting media. All steps were performed at room temperature.
To evaluate the topographic distribution of PrPD in FelCWD, a total of 33 areas were examined: (1) cerebral cortex, (2) cerebral white matter, (3) septal nucleus, (4) caudate nucleus, (5) putamen, (6) claustrum, (7) pars supracommissuralis of the hippocampus, (8) thalamic nuclei, (9) hypothalamic nuclei, (10) internal capsule, (11) corpus callosum, (12) hippocampus, (13) rostral colliculus, (14) caudal colliculus, (15) substantia nigra, (16) cuneiform nucleus, (17) rostral cerebellar peduncle, (18) tegmental field, (19) periaqueductal gray matter, (20) nucleus coeruleus, (21) pontine nuclei, (22) pyramidal tract, (23) trapezoid body, (24) raphe nucleus, (25) cochlear nucleus, (26) cerebellar nucleus, (27) vestibular nucleus, (28) nucleus of the solitary tract, (29) parasympathetic nucleus of the vagus, (30) cerebellar white matter, (31) cerebellar molecular layer, (32) cerebellar granular layer, and (33) cerebellar Purkinje layer.
Similar to previous CWD and FSE studies, the severity of the PrPD deposition was semiquantitatively graded: 0 (no PrPD seen), + (mild PrPD deposition), ++ (moderate PrPD deposition), or +++ (marked PrPD deposition).21,51 For the morphologic classification of PrPD, a modified version of a previously published protocol was used.53 Using this protocol, PrPD deposits were classified into 1 of 7 categories: (1) intraneuronal (IN, PrPD within the neuronal perikaryon), (2) perineuronal (PN, PrPD along the periphery of the neuronal perikaryon), (3) linear (Li, PrPD deposited along neuronal protrusions, principally axons), (4) stellate (St, star-shaped PrPD deposits, presumed to be associated with glial cells), (5) finely granular (FG, small punctate PrPD deposits), (6) coarsely granular (CG, cohesive aggregates of PrPD that are larger than FG), and (7) clumped (Cl, the largest aggregates of PrPD).
Co-localization Studies
For dual-label studies, 6-μm-thick tissue sections were mounted on positively charged glass slides, deparaffinized, rehydrated, subjected to HIER, quenched of EP activity, and blocked as described above. Slides were then sequentially incubated with the anti–prion protein antibody L42 (diluted 1:300) for 2 hours; an Alexa Fluor 568 (Invitrogen, Carlsbad, CA) conjugated secondary antibody; a second primary antibody (summarized in Table 1) for 2 hours; an anti–rabbit, HRP-conjugated labeled polymer (EnVision+; DakoCytomation); and the components of a fluorescent TSA kit (Alexa Fluor 488 TSA+; Invitrogen). At the completion of immunostaining, slides were washed; nuclei were labeled with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Invitrogen); and slides were mounted with fluorescent mounting medium (ProLong Gold Antifade Reagent; Invitrogen). Between all incubation steps, slides were washed 3 times (5 minutes each) in TNT wash buffer. All steps were performed at room temperature. To evaluate for the co-localization of PrPD with cell phenotype antibodies, dual-labeled sections were evaluated with a confocal laser-scanning microscope (Zeiss Laser Scanning Axiovert Confocal Microscope, LSM510 meta Carl Zeiss Microscopy, Thornwood, NY).
Table 1.
Summary of Antibodies Used in Studies.
| Antibody | Source | Dilution | Clonality | Labeled Structures |
|---|---|---|---|---|
| L42 | R-Biopharm | 1:300 | Mouse IgG1 | Prion protein (PrPD) |
| GFAP | Abcam | 1:1000 | Rabbit (polyclonal) | Glial fibrillar acid protein (astrocytes) |
| LAMP1 | Abcam | 1:500 | Rabbit (polyclonal) | Lysosome-associated membrane protein 1 (lysosomes) |
| Iba1 | Abcam | 1:200 | Rabbit (polyclonal) | Ionized calcium binding adaptor molecule 1 (microglia) |
| NFM | Abcam | 1:500 | Rabbit (polyclonal) | 160-kD neurofilament medium (neurons) |
| Synaptophysin | Abcam | 1:50 | Rabbit (polyclonal) | Synaptophysin |
NFM = Neurofilament M.
Results
Susceptibility of Domestic Cats to Infection With CWD Prions
These findings have been previously reported and are only briefly summarized here.37 Subtle clinical signs associated with TSE infection developed in 2 of 5 primary passage cats at 40 to 42 months postinoculation (PI) and included stilted gait, weight loss, anorexia, polydipsia, patterned motor behaviors, head and tail tremors, and ataxia. These cats were euthanized at 45 and 47 months PI. The remaining cats were euthanized at 25 and 86 months PI, for planned collection and study termination reasons, respectively. All 5 of the IC-inoculated second passage cats developed progressive signs associated with TSE infection at 20 to 24 months PI and were euthanized 23 to 27 months PI.
Serial Passage of CWD in Domestic Cats Is Associated With a Unique and Increasingly Severe Lesion Profile
To generate FelCWD lesion profiles, examiners (D.M.S., M.F., and V.F.) used an internally generated scoring to minimize the intergrader variation inherent in the subjective task of vacuolar lesion scoring.35 In primary passage cats, there was minimal to mild spongiosis through all of the examined brain regions, with the most severe pathology seen in the parasympathetic nucleus of the vagus (DMNV) (Fig. 1, Table 2). In secondary passage cats, there was moderate to marked spongiosis across nearly all examined areas, with most brain regions demonstrating more severe spongiosis than primary passage counterparts. Similar to the primary passage cats, the most severe spongiosis in the secondary passage cats was observed in the DMNV. In both primary and secondary passage FelCWD, the spongiosis was largely found in the neuropil with lesser amounts observed within neurons (primarily in the nuclei of the medulla oblongata, including the DMNV). In the neuropil, the vacuoles were largely small, discrete, and round. Within the neuronal perikarya, the vacuoles were single to multiple, large, and also well defined.
Figure 1.
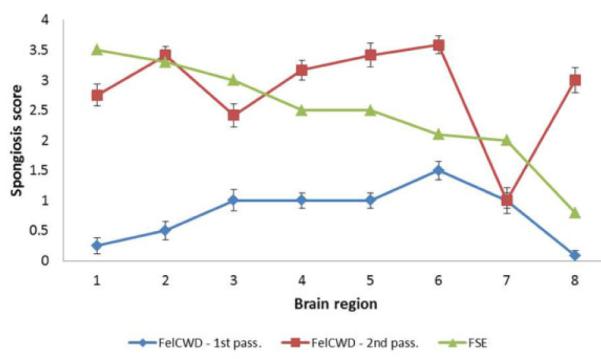
Comparison of the lesion profiles from primary and secondary passage feline chronic wasting disease (CWD) and feline spongiform encephalopathy (FSE). Note the more severe spongiosis in the secondary passage FelCWD (red line) compared with the primary passage FelCWD (blue line). Moreover, note the uniqueness of both primary and secondary passage FelCWD compared with FSE (blue and red lines vs green line). The specific brain regions evaluated were the (1) caudate nucleus, (2) ventromedial nucleus of the thalamus, (3) medial geniculate nucleus, (4) frontal cortex, (5) pons, (6) parasympathetic nucleus of the vagus, (7) cerebellar vermis, and (8) hippocampus. To generate lesion profiles, mean scores for each area were calculated using data from all 3 observers and plotted ± SEM.
Table 2.
Comparison of Lesion Scores by Brain Region in Cervid Chronic Wasting Disease–Infected Cats (FelCWD—First Passage), Cats Infected With FelCWD (FelCWD—Second Passage), and Cats With Feline Spongiform Encephalopathy (FSE).
| FelCWD—First | FelCWD—Second | ||
|---|---|---|---|
| Brain Region | Passage | Passage | FSE |
| 1. Caudate nucleus | 0 | 2.8 | 3.5 |
| 2. Ventromedial nucleus of the thalamus |
0.5 | 3.4 | 3.3 |
| 3. Medial geniculate nucleus | 1 | 2.4 | 3.0 |
| 4. Cerebral cortex—frontal lobe |
1 | 3.2 | 2.5 |
| 5. Pons | 1 | 3.4 | 2.5 |
| 6. Parasympathetic nucleus of the vagus |
1.5 | 3.6 | 2.1 |
| 7. Cerebellar vermis | 1 | 1 | 2.0 |
| 8. Hippocampus | 0 | 3 | 0.8 |
Topography and Morphology of PrPD in Primary Passage FelCWD
Preliminary work evaluating the antigen retrieval effects of varying concentrations of PK determined that immersion in 20 μg/ml PK prior to HIER resulted in optimal enhancement of PrPD IHC. Based on this, all subsequent PrPD mapping studies used 20 μg/ml PK. PrPD was detected in all 4 examined primary passage cats, and the deposits were mild to moderate in severity, with aggregates found in 8 of the 33 (24%) examined sites (Fig. 2a). Affected sites were the cortical gray matter (2/4 cats), the basal nuclei (caudate nucleus and putamen) (1/4), the septal nuclei (2/4), the pars supracommissuralis of the hippocampus (2/4), the hippocampus (2/4), the nucleus of the solitary tract (4/4), and the parasympathetic nucleus of the vagus (4/4). Morphologically, 6 of the 7 patterns of PrPD deposition were identified (all except for stellate) (Table 3, Figure 3). The most common patterns were finely granular and coarsely granular, both of which were found in 7 of 8 (88%) affected areas, followed by interstitial (6/8, 75%) and clumped (3/8, 38%). The linear pattern of PrPD deposition was the least common identified pattern (1/8, 13%).
Figure 2.

Brain; cat, primary and secondary passage FelCWD. In primary passage cats, misfolded prion protein (PrPD) was identified in the cerebral cortex (CC), pars supracommissuralis of the hippocampus (PS), basal nuclei (BN), septal nuclei (SN), hippocampus (HI), nucleus of the solitary tract (NST), and parasympathetic nucleus of the vagus (PaNV) (a). Note the increased scope of PrPD deposition in the secondary passage cats with additional deposits of PrPD found in the internal capsule (IC), thalamus (Th), hypothalamus (Hy), rostral colliculus (RC), periaqueductal gray matter (PAG), substantia nigra (SNi), caudal colliculus (CCo), nucleus coereleus (NuC), cuneiform nucleus (CN), raphe nuclei (RaN), rostral cerebellar peduncle (RCP), tegmental field (TF), pontine nucleus (PN), and pyramidal tract (PY), cerebellum (Cbm), cerebellar nuclei (CeN), vestibular nucleus (VN), cochlear nucleus (CoN), and the trapezoid body (TB) (b).
Table 3.
Distribution and Morphology of Misfolded Prion Protein (PrPD) in the Brains of Chronic Wasting Disease–Infected Cats.
| Neuroanatomic Location | Primary Passage (n = 4) | Secondary Passage (n = 4) |
|---|---|---|
| Telencephalon | ||
| Cerebral cortex | 0/+ – CG, IN | ++/+++ – FG, CG, IN, PN |
| Cerebral white matter | 0 | + – FG, CG, IN, PN, St |
| Septal nucleus | + – FG, CG, Cl, IN | +/++ – FG, CG, IN, PN, Li |
| Basal nuclei | ||
| Caudate nucleus | 0/+ – FG, CG, Cl, IN | ++ – FG, CG, IN, PN, Li |
| Putamen | 0/+ – FG, CG, Cl, IN | ++ – FG, CG, IN, PN, Li |
| Claustrum | 0 | +/++ – FG, CG, IN, PN, Li |
| Pars supracommissuralis | ++ – FG, PN, Li | +++ – FG, CG, IN, PN, Li |
| Diencephalon | ||
| Corpus callosum | 0 | 0 |
| Thalamic nucleia | 0 | ++/+++ – CG, Cl, Li, St, PN |
| Hypothalamic nucleib | 0 | +/++ – CG, Cl, Li, IN, PN |
| Internal capsule | 0 | ++ – CG, Cl, Li |
| Hippocampus | +/++ – FG, CG, PN | ++ – FG, CG, Cl, PN |
| Mesencephalon (midbrain) | ||
| Rostral colliculi | 0 | 0/+ – CG, Cl, Li |
| Caudal colliculi | 0 | 0/+ – CG, Cl, Li |
| Substantia nigra | 0 | ++ – CG, Cl, Li, IN |
| Cuneiform nucleus | 0 | ++ – FG, CG, PN, IN |
| Rostral cerebellar peduncle | 0 | +/++ – FG, CG, Cl, IN |
| Tegmental field | 0 | +/++ – CG, Cl, PN, IN, Li |
| Periaqueductal gray matter | 0 | 0/+ – FG, CG, Cl, IN |
| Metencephalon/myelencephalon | ||
| Nucleus coeruleus | 0 | 0/+ – FG, CG, IN, PN |
| Pontine nuclei | 0 | + – FG, CG, Cl, St, IN, PN |
| Pyramidal tract | 0 | + – CG, Cl, PN, IN |
| Trapezoid body | 0 | + – CG, Cl, St, Li, PN |
| Raphe nucleus | 0 | 0/+ – FG, CG, IN, PN |
| Cochlear nucleus | 0 | ++/+++ – FG, CG, IN, PN |
| Cerebellar nucleus | 0 | +++ – FG, CG, Cl, IN, PN |
| Vestibular nucleus | 0 | +++ – FG, CG, Cl, IN, PN |
| Nucleus of the solitary tract | +/++ – FG, CG, IN | +++ – CG, Cl, IN, PN, Li |
| Parasympathetic nucleus of the vagus | +/++ – FG, CG, IN | +++ – CG, Cl, IN, PN, Li |
| Cerebellum | ||
| White matter | 0 | 0 |
| Molecular layer | 0 | 0/+ – FG, CG, Cl, PN, IN |
| Granular layer | 0 | ++/+++ – CG, Cl, PN, IN |
| Purkinje layer | 0 | 0/+ – FG, CG, PN, IN |
Each neuroanatomic site is followed by the semiquantitative scoring of the severity of the PrPD immunoreactivity (0 = no PrPD identified; + = mild PrPD identified; ++ = moderate PrPD identified, and +++ = marked PrPD identified). In addition, PrPD deposits were classified according to their morphologic characteristics (see text for details) as follows: CG, coarsely granular PrPD; Cl, clumped PrPD; FG, finely granular PrPD; IN, intraneuronal PrPD; Li, linear PrPD; PN, perineuronal PrPD; and St, stellate PrPD.
The examined thalamic nuclei consisted of the anterodorsal, anteroventral, medioventral, mediodorsal, ventromedial, central lateral, paracentral, medial geniculate, and lateral geniculate.
The examined hypothalamic sites consisted of the ventromedial and dorsomedial nuclei and the dorsal and lateral hypothalamic areas.
Figure 3.

Brain; cat, primary and secondary passage FelCWD. In both primary and secondary passage cats, misfolded prion protein (PrPD) was found in 1 of 6 morphologic patterns. (a) Hippocampus, primary passage; fine and coarsely granular (black arrow), perineuronal PrPD. (b) Parasympathetic nucleus of the vagus, primary passage; fine and coarsely granular, intraneuronal PrPD (black arrow). (c) Pontine gray, secondary passage; clumped and intraneuronal PrPD (black arrow). (d) Cerebral cortex, secondary passage; fine (black arrow) and coarsely granular PrPD. (e) Nucleus of the solitary tract, fine and coarsely granular, perineuronal (white arrow) and stellate (black arrow) PrPD. (f) Internal capsule, secondary passage; fine and coarsely granular, linear PrPD (black arrow).
Topography and Morphology of PrPD in Secondary Passage FelCWD
In secondary passage cats, PrPD deposits were identified in all 4 of the evaluated cats with mild to marked accumulations of PrPD found in 31 of 33 (94%) examined areas (Fig. 2b, Table 3). Unless otherwise indicated, for each examined area, PrPD was found in all 4 cats. Specifically, PrPD was found in the cerebral cortex, cerebral white matter, septal nucleus, caudate nucleus, putamen, claustrum, pars supracommissuralis of the hippocampus (2/4 cats), thalamic nuclei, hypothalamic nuclei, internal capsule, hippocampus, rostral colliculus, caudal colliculus, substantia nigra, cuneiform nucleus, rostral cerebellar peduncle, tegmental field, periaqueductal gray matter, nucleus coeruleus, pontine nuclei, pyramidal tract, trapezoid body, raphe nucleus, cochlear nucleus, cerebellar nucleus, vestibular nucleus, nucleus of the solitary tract, DMNV, cerebellar molecular layer, cerebellar granular layer, and cerebellar Purkinje layer.
In secondary passage FelCWD, all 7 patterns of PrPD deposition were identified (Fig. 3, Table 3) and, like the primary passage FelCWD, coarsely granular (31/31, 100%) was the most common deposition pattern, followed by perinuclear (25/31, 81%) and interstitial (24/31, 77%). Also similar to the primary passage animals, the linear (15/31, 48%) and stellate patterns (4/31, 13%) were the least common.
Comparison of the PrPD distribution data between primary and secondary passage FelCWD revealed more widespread PrPD deposition in the second passage cats, with 94% neuroanatomic sites evaluated demonstrating PrPD compared with 24% in the primary passage cats. In addition to more widespread PrPD deposition, the secondary passage cats demonstrated more severe PrPD deposition with increased PrPD in the 6 brain areas in which PrPD was found in both passages. Last, those sites with the most severe PrPD deposition tended to have the most severe vacuolar change (eg, the parasympathetic nucleus of the vagus and the thalamus).
A Comparison of Feline CWD With Cervid CWD and FSE
A comparison of primary and secondary passage FelCWD lesion profile data with previously published FSE data, obtained from the publication by Wells et al,55 revealed only a single point of overlap—namely, the ventromedial nucleus of the thalamus (Table 4, Fig. 1). Using an identical, 0 to 4 semiquantitative scoring system, secondary passage FelCWD and FSE were scored 3.4 and 3.3 at this site, respectively. A thorough statistical comparison of the FelCWD and FSE lesion profile data was not performed since the publication by Wells et al did not include the complete FSE data set.
Table 4.
Misfolded Prion Protein (PrPD) Distribution in the Brains of CWD-Infected Cats, CWD-Infected Cervids, and Cats With Feline Spongiform Encephalopathy (FSE).
| Neuroanatomic Location | First Passage (n = 4) | Second Passage (n = 4) | Cervids With CWDa | Cats With FSEa |
|---|---|---|---|---|
| Telencephalon | ||||
| Cortical gray matter | 0/+ | ++/+++ | +/++ | +++ |
| Cortical white matter | 0 | + | + | NA |
| Septal nuclei | + | +/++ | +/++ | NA |
| Basal nuclei | ||||
| Caudate nucleus | 0/+ | ++ | ++ | NA |
| Putamen | 0/+ | ++ | ++ | NA |
| Diencephalon | ||||
| Corpus callosum | 0 | 0 | + | NA |
| Thalamic nucleib | 0 | ++/+++ | +++ | NA |
| Hypothalamic nucleib | 0 | +/++ | ++/+++ | NA |
| Internal capsule | 0 | ++ | + | NA |
| Hippocampus | +/++ | ++/+++ | + | NA |
| Mesencephalon (midbrain) | ||||
| Rostral colliculi | 0 | 0/+ | ++ | NA |
| Substantia nigra | 0 | ++ | ++ | NA |
| Tegmental field | 0 | +/++ | +/++ | NA |
| Periaqueductal gray matter | 0 | 0/+ | ++ | NA |
| Metencephalon/myelencephalon | +++ | |||
| Pontine nuclei | 0 | + | +/++ | NA |
| Pyramidal tract | 0 | + | 0/+ | NA |
| Raphe nuclei | 0 | 0/+ | +/++ | NA |
| Cochlear nuclei | 0 | ++/+++ | ++ | NA |
| Vestibular nuclei | 0 | +++ | +/++ | NA |
| Parasympathetic nucleus of the vagus | +/++ | +++ | ++ | NA |
| Cerebellum | ||||
| White matter | 0 | 0 | + | 0 |
| Molecular layer | 0 | 0/+ | +/++ | + |
| Granular layer | 0 | ++/+++ | ++ | +++ |
| Purkinje layer | 0 | 0/+ | ++ | NA |
Each neuroanatomic site is scored as described in Table 3. The PrPD distribution data were extrapolated from previously published reports of chronic wasting disease (CWD) infection in mule deer (see text for references). NA = Not Available.
The PrPD distribution data for cervids with CWD and cats with FSE were extrapolated from previously published reports (see text for references).
The examined thalamic and hypothalamic sites are identical to those described in Table 3.
To compare patterns of PrPD distribution between FelCWD, CerCWD, and FSE, we used previously published data from Spraker et al50 and Hilbe et al21 (Table 4). No areas of matching PrPD intensity were identified between primary passage FelCWD and CerCWD, whereas in secondary passage FelCWD, there were 6 brain areas in which the severity of PrPD accumulation was identical to CerCWD—the cortical white matter, the septal nuclei, the 2 basal nuclei, the substantia nigra, and the tegmental field. Although there are limited published data on the distribution of PrPD in FSE,21 there appears to be a slightly different pattern of PrPD between FSE and FelCWD. Notably, cats with FSE accumulate slightly higher levels of cerebral cortical and cerebellar granular layer PrPD, in contrast to the lesser amounts of PrPD seen in both primary and secondary passage FelCWD (Table 4).
Cellular and Subcellular Localization of PrPD
To determine the phenotype of brain cells harboring PrPD, we used multilabel immunofluorescence to simultaneously label the prion protein and glial fibrillary acidic protein (GFAP; astrocytes), NFM (neurons), Iba1 (microglia), LAMP1 (lysosomes), or synaptophysin in secondary passage FelCWD. Sparse association (2/4 cats) was identified between PrPD and GFAP+ astrocytes (Fig. 4), whereas substantial (4/4) association was found between PrPD and Iba1+ microglia (Fig. 5). In addition, confirming the light microscopic morphologic classification of PrPD deposition, we identified large aggregates of clumped PrPD within the cytoplasm of large, NFM+ neurons (4/4 cats; Fig. 6). Anatomically, the association between PrPD and both microglia and neurons was most evident in the areas of highest PrPD accumulation (eg, within nuclei of the myelencephalon). We also identified substantial association between PrPD and Lamp1+ lysosomes (4/4 cats; Fig. 7) and synaptophysin (4/4 cats; Fig. 8). Similar to the cell phenotype studies, these associations between PrPD and lysosomes and synaptophysin were most common in the nuclei of the myelencephalon. However, the association between PrPD and synaptophysin was most common in the cerebellar nuclei, whereas the accumulation of PRPD within NFM+ neurons was most evident in the nucleus of the solitary tract and the parasympathetic nucleus of the vagus.
Figure 4.
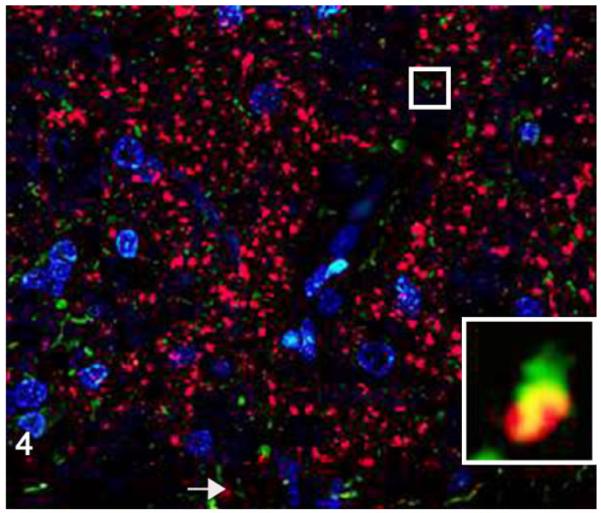
Medulla oblongata; cat, secondary passage FelCWD, case No. 1. Sparse misfolded prion protein (PrPD) is seen within astrocytes as evident by dual immunofluorescence using anti–prion protein (red) and anti–glial fibrillary acidic protein (GFAP; green) antibodies. The yellow color represents the overlapping of red and green immunofluorescent staining, and nuclei are stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (blue). Inset: higher magnification of confocal orthogonal view illustrates the rarity of the association between PrPD and GFAP-positive astrocytes.
Figure 5.
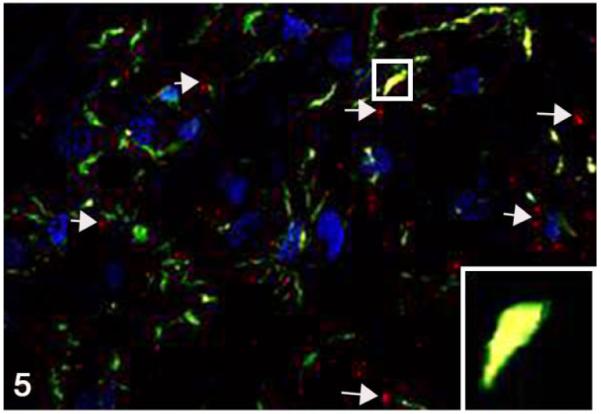
Medulla oblongata; cat, second passage FelCWD, case No. 1. Linear to clumped misfolded prion protein (PrPD) (red, white arrows) is seen throughout the tissue and in association with microglia (yellow) as evident by co-localization with the anti–Iba1 antibody (green). The yellow color represents the overlapping of red and green immunofluorescent staining, and nuclei are stained with DAPI (blue). Inset: higher magnification of an orthogonal view further illustrates the association between PrPD and Iba1-positive microglia.
Figure 6.
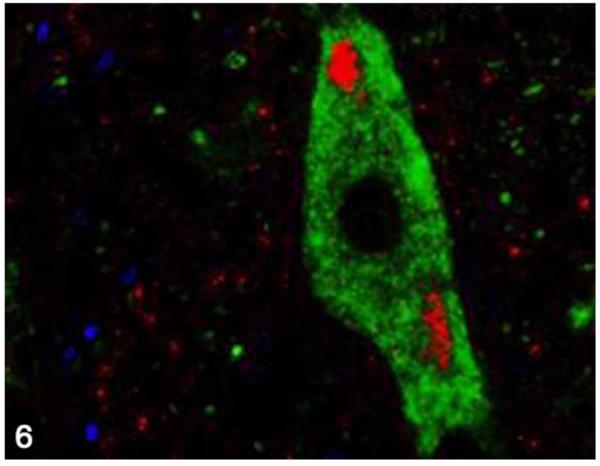
Nucleus of the solitary tract; cat, secondary passage FelCWD, case No. 2. Clumped misfolded prion protein (PrPD) deposits (red) are found within the tissue, and dual immunofluorescence with the anti–neuronal antibody NFM (green) reveals the accumulation of PrPD within the cell body of large neurons. The yellow color represents the overlapping of red and green immunofluorescent staining, and nuclei are stained with DAPI (blue).
Figure 7.

Parasympathetic nucleus of the vagus; cat, secondary passage FelCWD, case No. 1. Finely granular to clumped misfolded prion protein (PrPD) deposits (red) are seen dispersed through the tissue, and dual immunofluorescence with the anti–lysosome antibody Lamp1 (green) reveals extensive accumulation of PrPD with lysosomes (yellow). The yellow color represents the overlapping of red and green immunofluorescent staining, and nuclei are stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; blue). Inset: higher magnification further illustrates the association between PrPD and Lamp1-positive lysosomes.
Figure 8.

Cerebellar nucleus; cat, secondary passage FelCWD, case No. 1. Linear to clumped misfolded prion protein (PrPD) (red, white arrows) deposits are seen throughout the tissue, and dual immunofluorescence with the anti–synaptophysin antibody (green) reveals extensive association of PrPD with synaptophysin (yellow). The yellow color represents the overlapping of red and green immunofluorescent staining, and nuclei are stained with DAPI (blue). Inset: higher magnification further illustrates the association between PrPD and synaptophysin.
Discussion
In the studies presented here, we complement a recent publication confirming the infectability of domestic cats with cervid-origin CWD prions with descriptive data regarding the neuropathology of such an infection.37 These results of these studies (1) provide morphologic evidence of serial passage–associated prion adaptation, (2) illustrate a unique phenotype of cellular and subcellular brain PrPD accumulation, and (3) generate a novel FelCWD neuropathologic profile that could be used to discriminate FelCWD from FSE should natural prion infection be detected in a domestic felid.
Prion adaptation denotes the phenomenon by which a particular TSE isolate exhibits increased pathogenicity upon in vivo serial passage, including an increased attack rate, a shortened incubation period, and more severe neuropathology.41 These studies, which illustrate serial passage–associated worsening of both spongiosis and PrPD deposition, provide morphologic evidence of the successful adaptation of FelCWD. Moreover, these findings complement the initial report by Mathiason et al,37 which provided clinical evidence of FelCWD adaptation (ie, an increased attack rate and shortened incubation period). These findings mimic previous serial passage interspecies CWD studies conducted in ferrets and outbred mice.45,46 While the biochemical underpinnings of the process of prion adaptation have yet to be elucidated, previous work suggests that changes in the PrPD conformation underlie the process.52 Finally, in light of recent work demonstrating host range expansion following either in vivo or in vitro interspecies CWD prion adaptation, including the first evidence of CWD-induced conversion of human PrPC to PrPD, future experiments examining the host range of this newly derived FelCWD are in preparation.3,4
One goal of these studies was to compare the neuropathology of FelCWD with TSEs from a matching host (FSE) and a matching origin (CerCWD) so as to both provide insights into the influence of host and strain on TSE neuropathology and to generate a neuropathologic template that could be used to discriminate infectious etiologies in the event of natural felidprion disease. This approach complements previous work in cattle, hamsters, and sheep, which has illustrated the utility of both the lesion profile and PrPD distribution in the experimental identification of unique host/strain-relationships and in diagnostically distinguishing between sheep infected with scrapie or BSE.6,15,16,24,40,54
Using both lesion profile and PrPD distribution data, the present study illustrates the uniqueness of FelCWD. Comparing our FelCWD lesion profile data with previously published FSE lesion profile data, only a single matching point (the ventromedial nucleus of the thalamus) was identified. Unfortunately, differences in the scoring rubrics used in these studies and by Spraker et al49–51 prevented a comparison of CerCWD and FelCWD lesion profiles. Further evidence of the uniqueness of FelCWD is illustrated by a side-by-side comparison of the FelCWD PrPD distribution data with previously published correlative CerCWD data, in which only 6 of 24 (25%) of the evaluated brain regions demonstrate matching severity. Moreover, the limited available information on the neural distribution of PrPD in FSE suggests that there may be slight differences with it and FelCWD. Diagnostically, these findings confirm that combined usage of the lesion profile and the pattern of PrPD deposition could be used to aid in the etiologic characterization of a natural feline prion disease, similar to their usage in the demonstration of the BSE origin of vCJD and FSE.
The morphology of the PrPD deposits in FelCWD simultaneously mimics the morphology of the PrPD in both CerCWD and FSE. The fine and coarsely granular intraneuronal deposits, seen primarily in the primary passage cats, are strongly reminiscent of those seen in FSE.21 Interestingly, this staining pattern is very similar to that described in CWD-infected ferrets.46 In contrast, the secondary passage cats were dominated by coarsely granular and interstitial PrPD deposits, which is similar to that reported in CerCWD.25 We speculate that this alteration of the PrPD morphologic profile may be, in part, further evidence of prion adaptation. However, it is important to note that without subjecting each of these model systems to intense examination using the identical IHC protocol, it is impossible to draw any firm conclusions regarding the significance of the PrPD morphology. Overall, the sum of these findings, including the lesion profile, PrPD distribution data, and the PrPD morphology data, implicate, at the minimum, the dual contribution of both host and strain as key determinants in TSE neuropathology and support previous work concluding the same.25
It is important to note that there are shortcomings in comparing our FelCWD lesion profile and PrPD distribution data with historical CerCWD and FSE studies. Chief among these is that a number of mismatched variables between the studies likely affect any possible comparisons, including differences in route of inoculation, inoculum dosage, observer, tissue treatment protocols, inoculum strain, and host genotype.2,5 In addition, although we used the original data from previously published studies, we did not have access to the raw data and thus were unable to conduct any traditional statistical analysis. However, it is worth noting that all lesion profile studies shared a common scoring protocol—namely, a semiquantitative, 0 to 4 scoring rubric using HE-stained sections based on the original work by Fraser and Dickinson.13 Even in light of these pitfalls, we feel that these comparative analyses provide insights into the determinants of prion neuropathology and provide practical neuropathologic data in the event of the natural interspecies spread of CWD into either domestic or wild cats.
The immunohistochemical detection of PrPD in paraffin-embedded tissue sections can be hindered by the masking of antigens by fixation in aldehyde fixatives. To overcome this problem, we modified our previous protocol, which combined PLP fixation, 88% FA immersion, and HIER, to include proteinase digestion, based on a recent publication describing amyloid β detection in a mouse model.26 Among the aldehyde fixatives, PLP has previously been shown to enhance the sensitivity of IHC-based prion detection techniques.36,43 The antigen retrieval benefits of FA are believed to be the result of acidic hydrolysis, which is proposed to result in the unfolding of amyloid polymers.28 The addition of a PK-mediated protein digestion step greatly enhanced the sensitivity of our PrPD IHC. Mechanistically, it has been proposed that the proteolytic digestion of blocking proteins that surround amyloid fibrils might occur and thereby enhance subsequent antigen retrieval and antibody binding steps.26 Our demonstration of improved IHC PrPD detection using a combination of FA, HIER, and PK is similar to reports using the L42 antibody in scrapie-infected sheep.20
The PrPD co-localization studies enabled us, for the first time in any CWD model system, to detect PrPD in direct association with microglia and astrocytes. While this is the first CWD work to document this association, previous work in both prion and nonprion neurodegenerative diseases, including Alzheimer and Parkinson diseases, has revealed a similar association between the requisite misfolded protein and microglia and astrocytes.7,30,33 Moreover, although multilabeling studies have not been performed, a prion-microglia association is suspected in FSE. Both astrocytes and microglia are 2 resident CNS cell populations commonly implicated in the atypical inflammatory response in prion disease and in neuronal degeneration and cell death.8,9 It is very likely that these cells are similarly involved in the neuropathogenesis of FelCWD, although complete elucidation of their exact role would require additional studies. We also demonstrated an association between PrPD and lysosomes. While this is also a first in any CWD model system, these results confirm findings previously reported in ovine and murine scrapie.27 While the downstream effect of lysosomal accumulation in FelCWD is uncertain, the role of lysosomes in prion and nonprion neurodegenerative diseases is being reevaluated, with lysosomal-mediated conversion of PrPC to PrPD, lysosomal-induced spongiosis, and lysosomal permeabilization all implicated in neuropathogenesis.27,29,32 We are currently examining the functional effects of intralysosomal PrPD accumulation in our FelCWD model. Last, we identified a significant association between PrPD and the presynaptic protein synaptophysin, which, while also unique in any CWD model system, mimics previous studies of scrapie, CJD, and BSE.30,48,53 Although we did not investigate the consequences of this PrPD-synaptophysin association, these heavy accumulations, which parallel the severity seen in other TSEs, suggest that the synaptic disturbances described in scrapie and other TSEs are very likely present in FelCWD.12,48
In conclusion, these studies illustrate a novel neuropathologic profile of FelCWD and provide strong circumstantial evidence implicating unique interactions between host and strain in the development and progression of TSE neuropathology following interspecies infection. The lesion profile and the PrPD characteristics of primary and secondary passage FelCWD, which are distinct from yet share certain characteristics with both CerCWD or FSE, suggest that neither the infecting strain (CWD) nor the host (the cat) acts as the singular dominant determinant in FelCWD neuropathology. Rather, it is likely a combination of these and probably more factors that underlie the progressive neuropathology in this system. Moreover, through confocal analysis, we demonstrate substantial PrPD association with microglia, lysosomes, and synaptophysin, implicating each of these in the neuropathology of FelCWD. Finally, through the description of a unique FelCWD-specific neuropathologic profile, these studies will ensure the ability to distinguish between FSE and FelCWD should a natural felid TSE be detected in a CWD-endemic area.
Acknowledgments
Funding The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Supported by grants from the National Institutes of Health (NIH): RO1-NS-061902 and 1K01RR031488-01A2.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Afanasieva EG, Kushnirov VV, Ter-Avanesyan MD. Interspecies transmission of prions. Biochemistry (Mosc) 2011;76:1375–1384. doi: 10.1134/S0006297911130013. [DOI] [PubMed] [Google Scholar]
- 2.Angers RC, Kang HE, Napier D, et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328:1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barria MA, Telling GC, Gambetti P, et al. Generation of a new form of human PrP(Sc) in vitro by interspecies transmission from cervid prions. J Biol Chem. 2011;286:7490–7495. doi: 10.1074/jbc.M110.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartz JC, Marsh RF, McKenzie DI, et al. The host range of chronic wasting disease is altered on passage in ferrets. Virology. 1998;251:297–301. doi: 10.1006/viro.1998.9427. [DOI] [PubMed] [Google Scholar]
- 5.Beck KE, Sallis RE, Lockey R, et al. Ovine PrP genotype is linked with lesion profile and immunohistochemistry patterns after primary transmission of classical scrapie to wild-type mice. J Neuropathol Exp Neurol. 2010;69:483–497. doi: 10.1097/NEN.0b013e3181db2497. [DOI] [PubMed] [Google Scholar]
- 6.Begara-McGorum I, Gonzalez L, Simmons M, et al. Vacuolar lesion profile in sheep scrapie: factors influencing its variation and relationship to disease-specific PrP accumulation. J Comp Pathol. 2002;127:59–68. doi: 10.1053/jcpa.2002.0558. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007;114:231–241. doi: 10.1007/s00401-007-0244-3. [DOI] [PubMed] [Google Scholar]
- 8.Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 9.Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 2010;41:242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- 10.Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 11.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 12.Ferrer I. Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease. Cerebellum. 2002;1:213–222. doi: 10.1080/14734220260418448. [DOI] [PubMed] [Google Scholar]
- 13.Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 14.Fraser H, Pearson GR, McConnell I, et al. Transmission of feline spongiform encephalopathy to mice. Vet Rec. 1994;134:449. doi: 10.1136/vr.134.17.449. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez L, Martin S, Begara-McGorum I, et al. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol. 2002;126:17–29. doi: 10.1053/jcpa.2001.0516. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez L, Martin S, Jeffrey M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: implications for differential cell targeting and PrP processing. J Gen Virol. 2003;84:1339–1350. doi: 10.1099/vir.0.18800-0. [DOI] [PubMed] [Google Scholar]
- 17.Haley NJ. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE. 2009;4(3):e4848. doi: 10.1371/journal.pone.0004848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haley NJ, Mathiason CK, Zabel MD, et al. Detection of subclinical CWD infection in conventional test-negative deer long after oral exposure to urine and feces from CWD+ deer. PLoS One. 2009;4:e7990. doi: 10.1371/journal.pone.0007990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamir AN, Kunkle RA, Miller JM, et al. Experimental second passage of chronic wasting disease (CWD(mule deer)) agent to cattle. J Comp Pathol. 2006;134:63–69. doi: 10.1016/j.jcpa.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Hardt M, Baron T, Groschup MH. A comparative study of immunohistochemical methods for detecting abnormal prion protein with monoclonal and polyclonal antibodies. J Comp Pathol. 2000;122:43–53. doi: 10.1053/jcpa.1999.0343. [DOI] [PubMed] [Google Scholar]
- 21.Hilbe MM, Soldati GG, Zlinszky KK, et al. Immunohistochemical study of PrP(Sc) distribution in neural and extraneural tissues of two cats with feline spongiform encephalopathy. BMC Vet Res. 2009;5:11. doi: 10.1186/1746-6148-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–450. 526. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 23.Imran M, Mahmood S. An overview of animal prion diseases. Virol J. 2011;8:493. doi: 10.1186/1743-422X-8-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeffrey M, Martin S, Gonzalez L, et al. Differential diagnosis of infections with the bovine spongiform encephalopathy (BSE) and scrapie agents in sheep. J Comp Pathol. 2001;125:271–284. doi: 10.1053/jcpa.2001.0499. [DOI] [PubMed] [Google Scholar]
- 25.Jeffrey M, McGovern G, Siso S, et al. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–134. doi: 10.1007/s00401-010-0700-3. [DOI] [PubMed] [Google Scholar]
- 26.Kai H, Shin RW, Ogino K, et al. Enhanced antigen retrieval of amyloid beta immunohistochemistry: re-evaluation of amyloid beta pathology in Alzheimer disease and its mouse model. J Histochem Cytochem. 2012;60:761–769. doi: 10.1369/0022155412456379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenward N, Laszlo L, Landon M, et al. A role for lysosomes in scrapie pathogenesis. Biochem Soc Trans. 1992;20:265S. doi: 10.1042/bst020265s. [DOI] [PubMed] [Google Scholar]
- 28.Kitamoto T, Ogomori K, Tateishi J, et al. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- 29.Kovacs GG, Gelpi E, Strobel T, et al. Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2007;66:628–636. doi: 10.1097/nen.0b013e318093ecc7. [DOI] [PubMed] [Google Scholar]
- 30.Kovacs GG, Preusser M, Strohschneider M, et al. Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol. 2005;166:287–294. doi: 10.1016/S0002-9440(10)62252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krumm CE, Conner MM, Hobbs NT, et al. Mountain lions prey selectively on prion-infected mule deer. Biol Lett. 2010;6:209–211. doi: 10.1098/rsbl.2009.0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laszlo L, Lowe J, Self T, et al. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J Pathol. 1992;166:333–341. doi: 10.1002/path.1711660404. [DOI] [PubMed] [Google Scholar]
- 33.Lee HJ, Suk JE, Bae EJ, et al. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun. 2008;372:423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 34.Lezmi S, Seuberlich T, Oevermann A, et al. Comparison of brain PrPd distribution in ovine BSE and scrapie. Vet Pathol. 2011;48:1101–1108. doi: 10.1177/0300985810395784. [DOI] [PubMed] [Google Scholar]
- 35.Ligios C, Jeffrey M, Ryder SJ, et al. Distinction of scrapie phenotypes in sheep by lesion profiling. J Comp Pathol. 2002;127:45–57. doi: 10.1053/jcpa.2002.0589. [DOI] [PubMed] [Google Scholar]
- 36.Liu WG, Brown DA, Fraser JR. Immunohistochemical comparison of anti–prion protein (PrP) antibodies in the CNS of mice infected with scrapie. J Histochem Cytochem. 2003;51:1065–1071. doi: 10.1177/002215540305100810. [DOI] [PubMed] [Google Scholar]
- 37.Mathiason CK, Nalls AV, Seelig DM, et al. Susceptibility of domestic cats to chronic wasting disease. J Virol. 2013;87:1947–1956. doi: 10.1128/JVI.02592-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathiason CK, Powers JG, Dahmes SJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–136. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- 39.McGovern G, Jeffrey M. Scrapie-specific pathology of sheep lymphoid tissues. PLoS One. 2007;2:e1304. doi: 10.1371/journal.pone.0001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meade-White KD, Barbian KD, Race B, et al. Characteristics of 263K scrapie agent in multiple hamster species. Emerg Infect Dis. 2009;15:207–215. doi: 10.3201/eid1502.081173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Race R, Meade-White K, Raines A, et al. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J Infect Dis. 2002;186(suppl 2):S166–S170. doi: 10.1086/344267. [DOI] [PubMed] [Google Scholar]
- 42.Saunders SE, Bartelt-Hunt SL, Bartz JC. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg Infect Dis. 2012;18:369–376. doi: 10.3201/eid1803.110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seelig DM, Mason GL, Telling GC, et al. Chronic wasting disease prion trafficking via the autonomic nervous system. Am J Pathol. 2011;179:1319–1328. doi: 10.1016/j.ajpath.2011.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sigurdson CJ, Barillas-Mury C, Miller MW, et al. PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol. 2002;83:2617–2628. doi: 10.1099/0022-1317-83-10-2617. [DOI] [PubMed] [Google Scholar]
- 45.Sigurdson CJ, Manco G, Schwarz P, et al. Strain fidelity of chronic wasting disease upon murine adaptation. J Virol. 2006;80:12303–12311. doi: 10.1128/JVI.01120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sigurdson CJ, Mathiason CK, Perrott MR, et al. Experimental chronic wasting disease (CWD) in the ferret. J Comp Pathol. 2008;138:189–196. doi: 10.1016/j.jcpa.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Sigurdson CJ, Williams ES, Miller MW, et al. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus) J Gen Virol. 1999;80(pt 10):2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- 48.Siso S, Gonzalez L, Blanco R, et al. Neuropathological changes correlate temporally but not spatially with selected neuromodulatory responses in natural scrapie. Neuropathol Appl Neurobiol. 2011;37:484–499. doi: 10.1111/j.1365-2990.2010.01152.x. [DOI] [PubMed] [Google Scholar]
- 49.Spraker TR, Miller MW, Williams ES, et al. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J Wildl Dis. 1997;33:1–6. doi: 10.7589/0090-3558-33.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Spraker TR, Zink RR, Cummings BA, et al. Distribution of protease-resistant prion protein and spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. Vet Pathol. 2002;39:546–556. doi: 10.1354/vp.39-5-546. [DOI] [PubMed] [Google Scholar]
- 51.Spraker TR, Zink RR, Cummings BA, et al. Comparison of histological lesions and immunohistochemical staining of proteinase-resistant prion protein in a naturally occurring spongiform encephalopathy of free-ranging mule deer (Odocoileus hemionus) with those of chronic wasting disease of captive mule deer. Vet Pathol. 2002;39:110–119. doi: 10.1354/vp.39-1-110. [DOI] [PubMed] [Google Scholar]
- 52.Ushiki-Kaku Y, Endo R, Iwamaru Y, et al. Tracing conformational transition of abnormal prion proteins during interspecies transmission by using novel antibodies. J Biol Chem. 2010;285:11931–11936. doi: 10.1074/jbc.M109.058859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vidal E, Marquez M, Tortosa R, et al. Immunohistochemical approach to the pathogenesis of bovine spongiform encephalopathy in its early stages. J Virol Methods. 2006;134:15–29. doi: 10.1016/j.jviromet.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 54.Wells GA, Wilesmith JW. The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol. 1995;5:91–103. doi: 10.1111/j.1750-3639.1995.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 55.Wells GAH, Hawkins SAC, Cunningham AA, et al. Comparative pathology of the new transmissible spongiform encephalopathies. In: Bradley R, Marchant B, editors. A Consultation on BSE With the Scientific Veterinary Committee of the Commission of the European Communities. European Commission; Brussels, Belgium: 1993. pp. 327–345. [Google Scholar]
- 56.Williams ES, Young S. Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and elk (Cervus elaphus nelsoni) Vet Pathol. 1993;30:36–45. doi: 10.1177/030098589303000105. [DOI] [PubMed] [Google Scholar]
- 57.Wyatt JM, Pearson GR, Smerdon TN, et al. Naturally occurring scrapie-like spongiform encephalopathy in five domestic cats. Vet Rec. 1991;129:233–236. doi: 10.1136/vr.129.11.233. [DOI] [PubMed] [Google Scholar]