

Abstract
Angiogenesis remains a sensible target for pancreatic ductal adenocarcinoma (PDAC) therapy. VEGF, PDGF, FGF and their receptors are expressed at high levels and correlate with poor prognosis in human PDAC. Nintedanib is a triple angiokinase inhibitor that targets VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β signaling. We investigated the antitumor activity of nintedanib alone or in combination with the cytotoxic agent gemcitabine in experimental PDAC. Nintedanib inhibited proliferation of cells from multiple lineages found in PDAC, with gemcitabine enhancing inhibitory effects. Nintedanib blocked PI3K/MAPK activity and induced apoptosis in vitro and in vivo. In a heterotopic model, net local tumor growth compared to controls (100%) was 60.8 ± 10.5% in the gemcitabine group, −2.1 ± 9.9% after nintedanib therapy and −12.4 ± 16% after gemcitabine plus nintedanib therapy. Effects of therapy on intratumoral proliferation, microvessel density and apoptosis corresponded with tumor growth inhibition data. In a PDAC survival model, median animal survival after gemcitabine, nintedanib and gemcitabine plus nintedanib was 25, 31 and 38 days, respectively, compared to 16 days in controls. The strong antitumor activity of nintedanib in experimental PDAC supports the potential of nintedanib-controlled mechanisms as targets for improved clinical PDAC therapy.
Keywords: Pancreatic cancer, Nintedanib, Angiogenesis, Chemotherapy, Combination therapy
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States, and is expected to become the second-leading cause of cancer death by 2030 [1]. Despite recent advancements in multimodality strategies and availability of novel and more effective antineoplastic combination treatments, the 5-year survival rate for PDAC overall remains less than 6% [2]. Late-stage diagnosis, early and aggressive invasion with metastatic disease and resistance to conventional cytotoxic therapy are the major factors for dismal outcomes in the majority of patients. Much attention has been placed on systemic treatment options for pancreatic cancer as either definitive or perioperative therapy. Single agent gemcitabine (Gem), a deoxycytidine nucleoside analog, has been the standard of care for advanced PDAC since 1997 after producing a response rate of 5% and a median survival of 5.7 months in a pivotal randomized trial [3]. Greater efficacy against advanced pancreatic cancer has recently been demonstrated when nab-paclitaxel was added to gemcitabine [4]. The more intense cytotoxic regimen FOLFIRINOX (oxaliplatin/irinotecan/5-FU/leucovorin) showed better responses than gemcitabine but it also resulted in increased toxicity [5]. Despite these recent trial-based moderate improvements, there is still an urgent requirement for novel therapeutic strategies to improve overall survival of patients with pancreatic cancer.
Angiogenesis is an essential process for tumor growth and metastasis, and remains a target process with potential for cancer therapy including PDAC. Pathological angiogenesis as encountered in tumor growth represents a highly regulated yet disturbed balance between proangiogenic and antiangiogenic mechanisms [6]. Among several positive regulators of angiogenesis, growth factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) can induce the division of cultured endothelial cells thus indicating a direct action on these cells [6]. VEGF, PDGF, FGF and their receptors are expressed at high levels and correlate with poor prognosis in pancreatic cancer [7,8], providing a strong rationale for exploiting the therapeutic potential of multitarget antiangiogenic agent combinations with an established cytotoxic regimen. Single-target antiangiogenic agents including bevacizumab, a monoclonal antibody against VEGF-A [9,10], the cyclooxygenase inhibitor celecoxib [11] and the PDGF signaling inhibitor imatinib [12] have been studied in combination therapy in PDAC with limited success [13]. A second-class of antiangiogenic agents, small molecules with multi-tyrosine kinase inhibitor (TKI) activity including sunitinib and sorafenib that target several pro-angiogenic receptor tyrosine kinase axes showed promising antitumor response but this did not translate into higher survival rates in PDAC. Most broad-spectrum antiangiogenic TKIs such as sorafenib, sunitinib, pazopanib and vandetanib have complex efficacy data and in many cases the use of these drugs is associated with a significant increase in the incidence and risk of side effects [14–16]. Lower specificity of most of these TKIs toward their therapeutic targets, particularly at the FGF-FGFR axis, is a major factor for risk of increased toxicity [17]. Nintedanib (formally known as BIBF 1120) is a novel, specific and potent triple angiokinase inhibitor of VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β; it also targets other kinases such as RET, FLT-3 and Src in the low nanomolar range. Nintedanib has very high specificity toward its therapeutic targets with significantly lower IC50: VEGFR1/2/3 (IC50 13–34 nM); FGFR1/2/3 (IC50 37–108 nM); and PDGFRα/β (IC50 59–65 nM). Due to its unique targeting profile with high specificity, nintedanib has the potential to effectively prevent tumor growth and metastasis while minimizing toxicity and resistance development across a broad range of cancers. In preclinical animal studies, nintedanib showed antitumor activity in several tumor types [18]. In some clinical studies, nintedanib combination with chemotherapy showed promising antitumor activity where other antiangiogenic agents failed to show a response suggesting nintedanib might be superior [19,20]. Thus, we aimed to determine the therapeutic efficacy of the multi-targeting antiendothelial agent nintedanib in combination with the standard chemotherapy agent gemcitabine in experimental pancreatic cancer.
Materials and methods
Cell culture and reagents
The human pancreatic cancer cell lines AsPC-1, BxPC-3, Panc-1, MIA PaCa-2, the human umbilical vein endothelial cells (HUVEC), and the human fibroblast cell line WI-38 were all purchased from the American Type Culture Collection (ATCC, Rockville, MD). Pancreatic cancer associated stromal cells were kindly provided by Dr. Melissa Fishel at the Indiana University Pancreatic Cancer Signature Center, Indianapolis. The murine pancreatic cancer cell line PanO2 was obtained from the National Cancer Institute (Bethesda, MD). Cells were initially grown and multiple aliquots were cryopreserved. All the cell lines were used within 6 months after culture start. AsPC-1 and BxPC-3 cells were grown in RPMI 1640 medium; Panc-1, MIA PaCa-2, PanO2, WI-38 and pancreatic cancer stromal cells were grown in DMEM (Sigma Chemical Co. St. Louis, MO), both media were supplemented with 10% FBS. HUVECs were grown in EndoGRO-LS medium containing endothelial cell growth supplements (Millipore Corp., Billerica, MA). Gemcitabine was purchased from Eli Lilly Corporation (Indianapolis, IN). Nintedanib was purchased from LC Laboratories (Woburn, MA). The cell proliferation reagent WST-1 was purchased from Roche Diagnostic Corporation (Indianapolis, IN).
Cell viability assay
Cell viability assays were performed in 96-well plates using the colorimetric WST-1 reagent as previously described [21]. Briefly, cells were plated in a 96-well plate and treated with nintedanib and gemcitabine. After a 72-hour incubation of cells with nintedanib and gemcitabine, 10 μl WST-1 reagent was added in each well, and absorbance at 450 nm was measured after 2 hours using a microplate reader. Drug sensitivity graphs were plotted using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA).
In vitro scratch (wound-healing) assay
The in vitro scratch assays were performed to measure cell migration in 12-well plates. Monolayers of PanO2 cells or HUVECs in low-serum media were scratched once per well with a 200 μl pipette tip to create an artificial in vitro cell-free wound. Cells were washed twice to remove non-adherent cells and then treated with 10 μM nintedanib. The scratch closure (wound healing) was measured as a percentage of original scratch area after 24-hour incubation of cells with nintedanib and gemcitabine.
Western blot analysis
Cells were plated in T25 flasks and sub-confluent monolayers were treated with nintedanib and gemcitabine and lysed after 16 hours. Tumor tissue lysates were prepared as previously described [22]. Briefly, tumor tissues were immediately snap-frozen in liquid nitrogen and stored at −80 °C. These tumor tissues were suspended in lysis buffer and homogenized using the Bullet Blender Homogenizer (Next Generation, Averill Park, NY), and extracts were sonicated. Proteins in supernatants were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, Hercules, CA). Membranes were incubated overnight at 4 °C with the following antibodies: total AKT, phospho-AKT (Ser473), total ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), cleaved caspase-3 (all from Cell Signaling Technology, Beverly, MA), α-tubulin and GAPDH (both from Sigma). The membranes were then incubated with the corresponding HRP-conjugated secondary antibodies (Pierce Biotechnologies, Santa Cruz, CA) for 1-2 hours. Specific bands were detected using the enhanced chemiluminescence reagent (ECL, Bio-Rad) on autoradiographic film and quantitated by densitometry.
Tumor implantation and in vivo tumor growth experiment
Animal experiments were performed according to the guidelines and approved Institutional Animal Care and Use Committee protocols of the University of Texas Southwestern Medical Center (Dallas, TX) (Animal Protocol Number 2012-0081) and the Indiana University School of Medicine (South Bend, IN) (Animal Protocol Number 16–023). Female athymic nu/nu mice (aged 4–6 weeks) were used to establish a subcutaneous xenograft model as previously described [23]. Mice were injected with AsPC-1 cells (0.75 × 106), randomly grouped and intraperitoneal therapy started after two weeks with PBS (control), nintedanib (25 mg/kg, 5× a week) and gemcitabine (50 mg/kg, 2× a week). The tumor size was measured twice weekly and tumor volume (V) was calculated by using the formula [V = ½ (L × (W)2], where L= length and W = width. After completion of the 2 week therapy, the animals were euthanized, tumors were removed, weighed, dissected and processed for histological or immunohistochemical analysis.
Immunohistochemical analysis
Tumor tissues fixed in 4% paraformaldehyde were embedded in paraffin. Intratumoral proliferative activity was measured by using Ki67 nuclear antigen staining as per manufacturer's protocol (Abcam, Cambridge, MA). Briefly, tissue sections (5 μm) were deparaffinized and rehydrated followed by heat-mediated antigen retrieval using citrate buffer. The tissue sections were incubated with CAS blocking buffer followed by 1-hour incubation with anti-Ki67 antibody (1:200) and 40 minutes incubation with Cy3 (1:200) secondary antibody. Slides were mounted with DAPI containing mounting solution (Invitrogen, Carlsbad, CA). Proliferative activity was evaluated by calculating Ki67-positive cells from five different high-power fields (HPF) in a blinded manner. Intratumoral apoptosis was analyzed by staining tissue sections with “Apoptag Apoptosis Detection Kit” according to the manufacturer's (Millipore) instructions. For evaluating intratumoral microvessel density (MVD), paraffin-embedded tissues were sectioned (5 μm), deparaffinized and rehydrated followed by heat-mediated antigen retrieval using citrate buffer. The tissue sections were incubated for 20 minutes in CAS blocking buffer followed by overnight incubation at 4 °C with anti-endomucin clone V.5C7 (1:100 dilution in blocking solution) antibody (Millipore; MAB2624). Subsequently, the tissue sections were incubated in 1:200 Cy3-labeled secondary antibody at room temperature for 40 minutes. Tissues were then washed and mounted with DAPI containing mounting solution. Endomucin positive vessels were calculated within a microscopic HPF in a blinded manner. Fluorescence microscopy was used to detect fluorescent signals using IX81 Olympus microscope and images were captured with a Hamamatsu Orca digital camera (Hamamatsu Corporation, Bridgewater, NJ) with a DSU spinning confocal unit using Slidebook software (Intelligent Imaging Innovations, Philadelphia, PA).
Animal survival analysis
Animal survival studies were performed using 6- to 8-week-old female nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice [24]. The mice were intraperitoneally injected with AsPC-1 (0.75 × 106) cells. Two weeks later the animals were randomly grouped (n = 6–8 per group) and treated intraperitoneally with PBS (control), nintedanib (25 mg/kg, 5× a week) and gemcitabine (50 mg/kg, 2× a week) for a duration of two weeks. Mice were euthanized when moribund according to predefined criteria including rapid weight loss or gain (>15%), tumor size, lethargy, inability to remain upright and lack of strength. Animal survival was evaluated from the first day of treatment until death.
Statistical analysis
In vitro cell proliferation data are expressed as mean ± standard deviation. Statistical significance was analyzed by the two-tailed Student's t-test using GraphPad Prism 6.0 Software (GraphPad Software, San Diego, CA) for individual group comparison. Statistical analysis for in vivo tumor growth studies was performed by oneway ANOVA for multiple group comparison and Student's t-test for the individual group comparison. Survival study statistics were evaluated using logrank group comparison (GraphPad Prism 6.0). Values of p < 0.05 were considered to represent statistically significant group differences.
Results
Nintedanib inhibits PDAC related cell proliferation and migration, and enhances gemcitabine response
Analysis of human PDAC cells AsPC-1, BxPC-3, MIA PaCa-2, Panc-1, murine PDAC cells PanO2, PDAC associated stromal cells, HUVECs endothelial cells and WI-38 fibroblasts revealed that nintedanib dose-dependently inhibited proliferation of all these PDAC related cells. Importantly, combination of nintedanib with gemcitabine had enhancing inhibitory effects. At 1 μM concentration, percent inhibition in cell proliferation by gemcitabine, nintedanib and gemcitabine plus nintedanib was 54%, 56%, 74% for AsPC-1, 46%, 45%, 69% in Panc-1, 90%, 62%, 94% for PanO2, 23%, 38%, 48% in PDAC stromal cells, 92%, 72%, 99% for HUVECs and 87%, 45%, 99% in WI-38 cells (Fig. 1A and B). Nintedanib and gemcitabine also inhibited proliferation of the other PDAC cell lines BXPC-3 and MIA PaCa-2, and combination of nintedanib and gemcitabine had additive effects (data not shown). The effect of nintedanib on cell migration was evaluated by an in vitro scratch (wound healing) assay. Nintedanib treatment caused a 75% inhibition in wound healing in PanO2 cells (p = 0.0002) and a 83% inhibition in HUVECs as compared with controls (p = 0.022) (Supplemental Fig. S1).
Fig. 1.
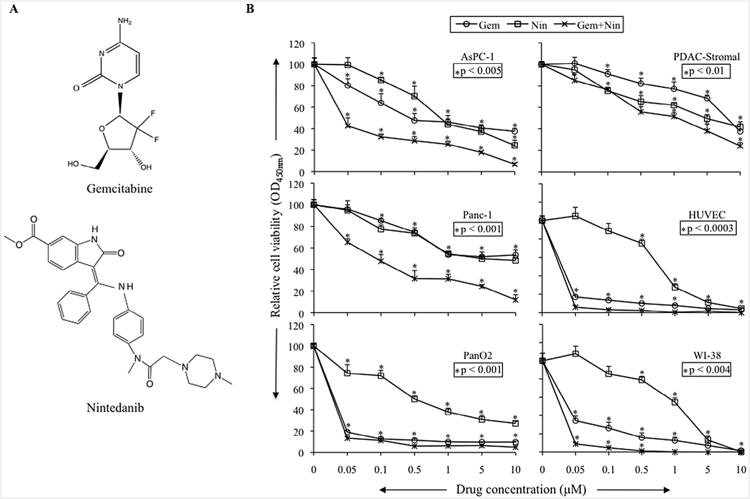
Nintedanib inhibits in vitro cell proliferation of PDAC related cell types and had additive effects in combination with gemcitabine. (A) Chemical structures of gemcitabine and nintedanib. (B) Cells were plated on 96-well plates and treated with 50 nM, 100 nM, 500 nM, 1 μM, 5 μM and 10 μM concentrations of gemcitabine and nintedanib. After 72 hours, 10 μl WST-1 reagent was added in each well and incubated for 2 additional hours. The absorbance at 450 nm was measured using a microplate reader. The resulting number of viable cells was calculated by measuring absorbance of color produced in each well. Data are the mean ± standard deviation of triplicate determinations.
Nintedanib blocks PI3K/MAPK activity and induces apoptosis in PDAC associated stromal cells
Activated signaling of VEGF/FGF/PDGF pathways, the main inducers of tumor angiogenesis and growth, has been shown to converge on induced expression and signaling of PI3K/AKT and MAPK/ERK pathways [25]. This well-defined effect of nintedanib on targeting VFGF/FGF/PDGF signaling was investigated by examining phospho-AKT, phospho-ERK and apoptosis-related cleaved caspase-3 protein expression in PDAC relevant cells. Immunoblot analysis revealed that nintedanib, either alone or in combination with gemcitabine, caused a decrease in the levels of phospho-AKT but no significant effect on phospho-ERK in AsPC-1 cells; decrease in phospho-AKT, phospho-ERK and increase in apoptosis-related protein cleaved caspase-3 in PDAC stromal cells and HUVECs; decrease in phospho-AKT, no change in phospho-ERK and increase in cleaved caspase-3 for WI-38 cells (Fig. 2). In other PDAC cells, nintedanib decreased p-AKT levels in all three cells tested (BxPC-3, MIA PaCa-2 and Panc-1), while decreasing p-ERK levels in BxPC-3 and MIA PaCa-2 cells but not in Panc-1 cells (Supplemental Fig. S2).
Fig. 2.
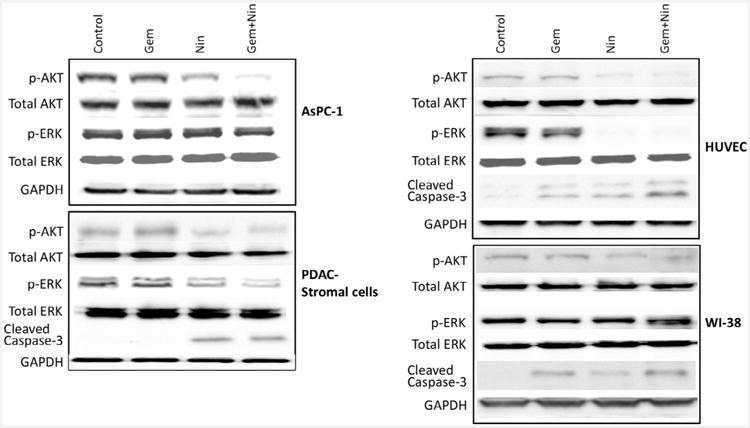
Nintedanib effect on PI3K/MAPK activity and apoptosis in four different PDAC related cells. A sub-confluent monolayer of cells was treated with nintedanib (10 μM) and gemcitabine (10 μM), either alone or in combination for 16 hours. Total protein lysates were prepared and analyzed by immunoblotting for p-AKT, total AKT, p-ERK, total ERK, cleaved caspase-3 and GAPDH (loading control) proteins. Data are representative of two independent experiments with similar results.
Nintedanib inhibits local tumor growth and enhances gemcitabine antitumor response
In AsPC-1 subcutaneous xenografts, treatment of tumor-bearing mice with nintedanib (25 mg/kg, 5× a week for 2 weeks), either alone or in combination with gemcitabine (50 mg/kg, 2× a week for 2 weeks), resulted in statistically significant net tumor growth inhibition (Fig. 3A and B). In this study, compared to controls (100 ± 29), the percent net local tumor growth was 60.8 ± 10.5 (p = 0.02) in the gemcitabine group, −2.1 ± 9.9 (p = 0.0001) after nintedanib therapy and −12.4 ± 16 (p = 0.0001) after gemcitabine plus nintedanib therapy, respectively (Fig. 3A and B). Furthermore, the net tumor growth inhibition in nintedanib monotherapy and gemcitabine plus nintedanib groups was statistically different compared to gemcitabine alone. Therapy with gemcitabine, nintedanib and the combination resulted in decreased mean tumor weight compared to control treated animals. Mean tumor weight in different treatment groups was as follows: control 0.32 ± 0.05 g, gemcitabine 0.23 ± 0.06 g (p = 0.028 vs. control), nintedanib 0.14 ± 0.04 g (p = 0.0003 vs. control), and gemcitabine plus nintedanib 0.12 ± 0.04 g (p = 0.0002 vs. control) (Fig. 3C).
Fig. 3.
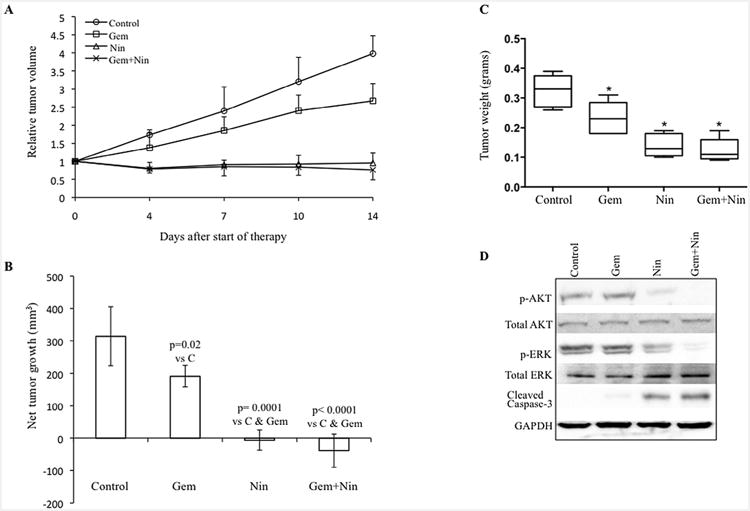
Nintedanib and gemcitabine inhibit growth of established local PDAC tumor. Nude mice were subcutaneously injected with AsPC-1 cells and treated with nintedanib and gemcitabine for 2 weeks. Tumor growth was measured twice a week using calipers. (A) Relative tumor volume is calculated by dividing the tumor volume on any day by the tumor volume at the start of therapy (day 0). (B) Net tumor growth was calculated by subtracting tumor volume on the first treatment day from that on the final day. Data are representative of mean values ± standard deviation from 6 to 8 mice per group. (C) Mean tumor weight was calculated from final day tumor weights in each group and is presented as a box plot. Box height denotes interquartile range; horizontal line within the box denotes median; and whiskers represent minimum and maximum values. Symbols * represent significant difference (p < 0.03) versus controls. (D) Nintedanib blocks PI3K/MAPK signaling proteins and induces apoptosis-related proteins. Tumor lysates were prepared from tumor tissue samples obtained from AsPC-1 tumor bearing mice and were analyzed by immunoblotting. The intensity of bands was quantitated by densitometry and is represented in the bar graph after normalizing values with α-tubulin expression. Data are representative of three independent experiments with similar results.
No significant change in mouse body weight was observed in the nintedanib, gemcitabine, or gemcitabine plus nintedanib therapy groups, and there was no other discernible treatment-related toxicity (Supplemental Fig. S3A).
Nintedanib downregulates signaling proteins, induces intratumoral apoptosis, inhibits proliferation and reduces microvessel density
The effects of nintedanib on the downstream signaling proteins PI3K/AKT and MAPK/ERK were also examined by Western blot analysis of protein lysates from AsPC-1 tumor xenografts. A significant decrease in the expression of phospho-AKT and phospho-ERK was observed in the nintedanib treated groups (Fig. 3D). Evaluation of intratumoral apoptosis by measuring levels of cleaved caspase-3 protein revealed that gemcitabine and nintedanib both induced cleavage of caspase-3 and that the combination of these agents had additive effects on this apoptosis-related protein (Fig. 3D). Intratumoral apoptosis was further confirmed by immunohistochemical analysis of tumor tissues. TUNEL assay revealed that the gemcitabine monotherapy group displayed a 3-fold increase (p = 0.002), while nintedanib monotherapy caused a 7-fold increase (p < 0.003) in apoptotic index as compared with the control group. The combination treatment of gemcitabine and nintedanib generated a 9-fold increase in the apoptotic index compared to controls (p < 0.003). In addition, induction in intratumoral apoptosis in the nintedanib-monotherapy and combination therapy groups was significantly higher than that for the gemcitabine monotherapy group (p < 0.0002) (Fig. 4A).
Fig. 4.
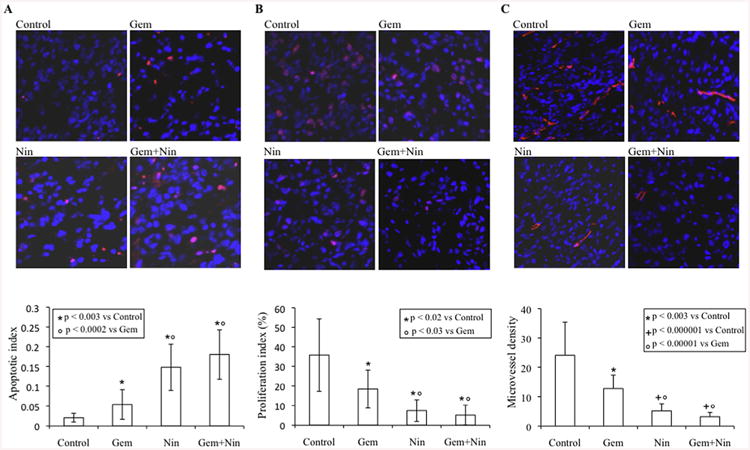
Effects of nintedanib and gemcitabine therapy on intratumoral apoptosis, proliferation and microvessel density. Nude mice were subcutaneously injected with AsPC-1 cells (0.75 × 106) and treated with nintedanib and gemcitabine, either alone or in combination, for 2 weeks. (A) Intratumoral apoptosis was measured by staining the tumor tissue section with TUNEL procedure. (B) Intratumoral proliferation was measured by immunostaining with Ki67 nuclear antigen. The proliferation index was calculated by dividing Ki67 positive cells from the total number of cells per HPF. (C) Microvessel density was evaluated by staining tumor tissue sections with endomucin antibody. For all three immunostaining experiments, slides were photographed under a fluorescent microscope and counting was performed in five different high power fields. The data are expressed as the mean ± standard deviation.
Additional mechanisms of in vivo antitumor activity of nintedanib were examined by determining intratumoral proliferation in tumor tissues. The tumors of nintedanib treated mice presented a decreased tumor cell proliferation rate. A 79% decrease (p = 0.0008) in intratumoral proliferative index was observed in the nintedanib-treated group as compared with controls. Gemcitabine monotherapy caused a 49% decrease in intratumoral proliferation as compared with controls (p = 0.019). The combination treatment group of gemcitabine and nintedanib caused a 86% decrease in proliferative activity as compared with controls (p = 0.00001). The proliferative index in the nintedanib-monotherapy and combination therapy groups was also significantly reduced as compared with the gemcitabine-monotherapy group (p < 0.03) (Fig. 4B).
The effect of nintedanib and gemcitabine treatment on tumor vasculature was measured through endomucin staining of tumor tissue sections. Nintedanib and gemcitabine both reduced intratumoral microvessel counts. Microvessel density in nintedanib monotherapy and gemcitabine plus nintedanib combination therapy groups was significantly lower than in the gemcitabine monotherapy group (Fig. 4C). Furthermore, the combination treatment group gemcitabine plus nintedanib showed some additive effects on decreasing microvessel counts, but these were not significantly lower than in the nintedanib alone group. Mean microvessel counts per HPF were 24 ± 11 (control), 12.7 ± 4.6 (Gemcitabine), 5.1 ± 2.4 (nintedanib) and 3.2 ± 1.4 (gemcitabine plus nintedanib) (Fig. 4C).
Nintedanib prolongs animal survival and enhances gemcitabine survival benefit
The effect of nintedanib and gemcitabine on animal survival was evaluated in an intraperitoneal murine xenograft model [26]. After 2-weeks of therapy, the median animal survival was 16 days in the control group and 25 days after single agent gemcitabine (a 56% increase compared with controls, p = 0.036). The median animal survival was significantly improved by single agent nintedanib (31 days, a 94% increase over controls, p = 0.0004; and a 24% increase compared with single agent gemcitabine, p = 0.042) and the combination of gemcitabine plus nintedanib (38 days, a 138% increase compared with controls, p = 0.001). Median survival in the combination therapy group was also significantly greater than that in the monotherapy groups (52% increase over gemcitabine, p = 0.001; and 23% increase compared with nintedanib, p = 0.01) (Fig. 5). There was no obvious treatment-related toxicity in this study and no significant change in mouse body weight during the 2-week therapy with nintedanib (25 mg/kg, 5×/week) and gemcitabine (50 mg/kg, 2×/week) (Supplemental Fig. S3B).
Fig. 5.
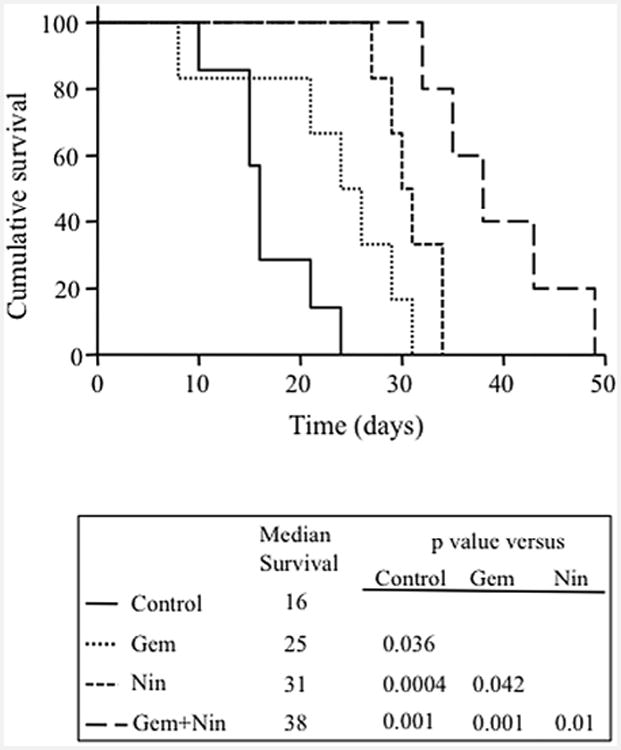
Effects of nintedanib and gemcitabine therapy on animal survival. AsPC-1 cells (0.75 × 106) were injected intraperitoneally into NOD/SCID mice and treatment started after 2 weeks with nintedanib (25 mg/kg, 5 times a week) and gemcitabine (50 mg/kg, 2 times a week), either alone or in combination, for 2 weeks. The graph represents the animal survival time from the beginning of therapy. Statistical group differences in survival time were calculated using logrank testing (GraphPad Prism 6.0).
In a separate experiment with higher doses of nintedanib (50 mg/kg) and gemcitabine (100 mg/kg), there was some drug related toxicity as observed by a decrease in mouse body weight. In this experiment, mice were only treated for one week; the resulting animal survival in the control group was 18 days, after gemcitabine 25 days and after nintedanib 34 days. However, animal survival in the combination therapy group (35 days) was not significantly longer than that in the nintedanib monotherapy group (Supplemental Fig. S4).
Discussion
In PDAC, high expression of several growth factors and their receptors is a major promoter of angiogenesis and metastasis [27]. Therefore, antiangiogenic therapy is a sensible and promising therapeutic avenue due to its potential for synergistic or additive effects with other antitumor agents. However, given the complexity of several aberrant angiogenic pathways and regulators, it is unlikely that any single agent will be effective for all PDAC patients. Therefore, targeting multiple pathways that are most commonly expressed in the progression of pancreatic cancer will be a critical step in developing novel therapeutic strategies to achieve a meaningful clinical benefit.
Several antiangiogenic agents have been evaluated in pancreatic cancer, while these agents showed significant preclinical effects, only a modest effect was observed in clinical studies, probably due to induction of tumor escape mechanisms [28,29]. Two major challenges in the success of antiangiogenic therapy are development of resistance in the primary tumor and induction of metastasis [30]. Sunitinib, a multitarget angiogenic inhibitor, has been shown to inhibit local tumor growth but it also increased VEGF expression and metastatic burden, resulting in no survival benefit [31,32]. This has been corroborated in our murine PDAC models, in which we also observed that single agent sunitinib effectively blocked local tumor growth but was unable to create any significant survival benefit [33,34]. The single-target anti-VEGF agent bevacizumab has not shown any significant clinical activity in pancreatic cancer in combination with gemcitabine [15]. However, our preclinical studies demonstrated the benefits of combining polymechanistic, multi-targeting antiangiogenic agents beyond bevacizumab with cytotoxic therapy [33,34]. In the present study, we evaluated the efficacy of nintedanib, a triple angiokinase inhibitor that acts by blocking not only VEGFR, but also FGFR and PDGFR, which are involved in the development of resistance to anti-VEGF therapy and induction of metastasis.
The dense desmoplastic stroma surrounding malignant epithelial cells that is composed of several cellular and acellular components including fibroblasts, stellate cells and endothelial cells creates a complex tumor microenvironment that plays a major role in PDAC development, invasion, metastasis and resistance to chemotherapy [35]. Targeting fibroblasts and endothelial cells for solid tumor treatment has been shown to be a potentially effective strategy [36,37]. We observed that nintedanib inhibited proliferation of pancreatic tumor stromal cells and representative fibroblast WI-38 cells. Pancreatic stromal cells were less sensitive to nintedanib as compared with WI-38 cells. As expected, nintedanib had a significant antiproliferative effect on representative endothelial cells HUVECs. In addition, combination of nintedanib with gemcitabine had additive effects. Importantly, the nintedanib effect was not limited to stromal cells but it also differentially inhibited the proliferation of human PDAC cells and murine PDAC cells PanO2. As our results with both epithelial and nonepithelial cell type responses in vitro show, nintedanib also appears to induce its strong in vivo effects through affecting multiple cell types that are involved in tumor progression beyond those partaking in angiogenesis. Recently, consistent with our findings, Tai et al. [38] demonstrated that nintedanib treatment caused significant antiproliferative effect in a panel of four hepatocellular carcinoma cell lines and one hepatoblastoma cell line. However, previously Kutluk Cenik et al. [39] showed that nintedanib has no antiproliferative effect on selected lung and pancreatic cancer cell lines. This difference can most likely be attributed to differential expression of nintedanib target receptors, the concentration of nintedanib used and the assay incubation time. Of note, nintedanib not only has high inhibitory activity toward VEGFR, FGFR and PDGFR, but it also inhibits Flt-3, Lck, Lyn and Src induced tumorigenic signaling [17]. Previous studies in the literature showed differential expression of VEGFR/FGFR/PDGFR in PDAC cell lines at the transcript level but due to non-detectable levels of phospho-forms of these receptors, most of these studies focused on PI3K and MAPK signaling for evaluating antitumor mechanisms. We also failed to detect p-VEGFR2, p-FGFR2 or p-PDGFRβ in AsPC-1, Panc-1, MIA PaCa-2 and BxPC-3 cells by Western blot analysis. Furthermore, most of the nintedanib targetable signaling pathways converge on induced PI3K and MAPK signaling. Additionally, the PI3K and MAPK pathways are involved in intensive well-defined crosstalk at multiple points [40]. Importantly, this crosstalk leads to the activation of compensatory signaling which can result in activation of one pathway upon inhibition of the other, thus conferring resistance to single agent treatment. Combined inhibition of the PI3K and MAPK pathways has shown to be superior to single agent therapy in K-ras mutant breast, lung and colorectal cancers [41,42]. Therefore, as previously reported [43], we observed that nintedanib differentially affected levels of downstream signaling proteins phospho-AKT, phospho-ERK and cleaved caspase-3 protein in different PDAC associated cell lines. In our studies nintedanib blocked phospho-ERK levels in all cell types tested and in tumor lysates but not in the two PDAC lines AsPC-1 and Panc-1. ERK activation in cancer-associated cells is regulated by a complex system of functionally diverse proteins including downstream signaling of several growth factors such as VEGF, FGF, HGF, IGF, PDGF, TGFβ and EGF. A possible explanation for nintedanib to not block ERK-phosphorylation in some PDAC cell lines is that the predominant ERK activation pathways in these cells might not be targeted by nintedanib. Furthermore, nintedanib blocked ERK-phosphorylation in AsPC-1 tumor xenografts but not in the AsPC-1 cell line, probably due to the significant activity of stromal cells within tumors, where ERK-phosphorylation has been shown to be blocked by nintedanib. Therefore, our findings suggest that these molecular signaling changes are likely operational in the nintedanib induced antitumor effects, and that they seem to represent good candidate markers of in vivo activity testing and for clinical validation.
AsPC-1 PDAC cells (containing K-Ras activating mutation, p53 inactivating mutation and p16 frameshift mutation) are very aggressive and highly resistant to gemcitabine. We therefore used this cell line in murine xenograft studies of experimental therapeutic evaluation of nintedanib as previously reported [26]. The AsPC-1 intraperitoneal xenograft model for animal survival is highly reproducible, well characterized and closely resembles the metastatic progression pattern of the clinical disease. We observed that in our murine PDAC models, the single-agent nintedanib not only inhibited local tumor growth but, unlike many other antiangiogenic therapies, also significantly enhanced animal survival. Enhancement in animal survival by single agent nintedanib, in contrast to other antiangiogenic agents used in our model [23,33,34,44], emphasize either the advantages of its triple angiokinase spectrum through preventing resistance and further tumor progression through the FGF/PDGF pathways [45], or the benefits of an even broader target range relevant for inhibition of epithelial–mesenchymal pathways [39]. In survival studies, combination of nintedanib with gemcitabine showed maximum survival benefit at optimized, i.e. reduced doses. Although previous clinical studies of nintedanib in combination with chemotherapy suggest a favorable toxicity profile and long-term efficacy [19,20], in our study with higher nintedanib doses the reduction in animal body weight indicated that nintedanib doses need to be selected more cautiously. Importantly, all nintedanib induced antitumor effects and in vivo marker responses showed additive effects in combination with gemcitabine. Gemcitabine itself has antiproliferative and proapoptotic effects on tumor cells as well as endothelial cells and fibroblasts. The exact mechanisms for the enhancement in antitumor activity of gemcitabine by nintedanib addition remains unclear; however, some possible mechanisms include normalization of tumor microvessels, increased gemcitabine delivery into the tumor microenvironment due to reduced interstitial pressure, reduced desmoplastic stromal density or direct augmentation of gemcitabine antitumor effects [34,46,47].
In summary, the results of the present study demonstrate that the triple angiokinase inhibitor nintedanib has strong antitumor activity as a single agent in two murine PDAC xenograft models. Furthermore, nintedanib combination with the cytotoxic agent gemcitabine caused significant additive antitumor effects. These findings suggest that the multifactorial nature of pancreatic cancer progression may be more effectively approached through combinations of cytotoxic agents with antitumor agents that specifically target several redundant or parallel molecular pathways. This multitargeting profile, the in vivo benefit and the potential for a low toxicity profile provide a strong rationale for clinical evaluation of nintedanib in combination with effective cytotoxic agents.
Supplementary Material
Acknowledgments
This work was financially supported in part by the Division of Surgical Oncology, Department of Surgery, Simmons Comprehensive Cancer Center, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center funds; and Department of Surgery, Indiana University School of Medicine funds.
Footnotes
Conflict of interest: None.
Appendix: Supplementary material: Supplementary data to this article can be found online at doi:10.1016/j.canlet.2014.12.027.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 6.Al-Husein B, Abdalla M, Trepte M, Deremer DL, Somanath PR. Antiangiogenic therapy for cancer: an update. Pharmacotherapy. 2012;32(12):1095–1111. doi: 10.1002/phar.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujimoto K, Hosotani R, Wada M, Lee JU, Koshiba T, Miyamoto Y, et al. Expression of two angiogenic factors, vascular endothelial growth factor and platelet-derived endothelial cell growth factor in human pancreatic cancer, and its relationship to angiogenesis. Eur J Cancer. 1998;34(9):1439–1447. doi: 10.1016/s0959-8049(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 8.Wagner M, Lopez ME, Cahn M, Korc M. Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 1998;114(4):798–807. doi: 10.1016/s0016-5085(98)70594-3. [DOI] [PubMed] [Google Scholar]
- 9.Ko AH, Dito E, Schillinger B, Venook AP, Xu Z, Bergsland EK, et al. A phase II study evaluating bevacizumab in combination with fixed-dose rate gemcitabine and low-dose cisplatin for metastatic pancreatic cancer: is an anti-VEGF strategy still applicable? Invest New Drugs. 2008;26(5):463–471. doi: 10.1007/s10637-008-9127-2. [DOI] [PubMed] [Google Scholar]
- 10.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28(22):3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dragovich T, Burris H, 3rd, Loehrer P, Von Hoff S, Chow S, Stratton S, et al. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: results of a phase II trial. Am J Clin Oncol. 2008;31(2):157–162. doi: 10.1097/COC.0b013e31815878c9. [DOI] [PubMed] [Google Scholar]
- 12.Moss RA, Moore D, Mulcahy MF, Nahum K, Saraiya B, Eddy S, et al. A multi-institutional phase 2 study of imatinib mesylate and gemcitabine for first-line treatment of advanced pancreatic cancer, Gastrointest. Cancer Res. 2012;5(3):77–83. [PMC free article] [PubMed] [Google Scholar]
- 13.Assifi MM, Hines OJ. Anti-angiogenic agents in pancreatic cancer: a review. Anticancer Agents Med Chem. 2011;11(5):464–469. doi: 10.2174/187152011795677463. [DOI] [PubMed] [Google Scholar]
- 14.Iacovelli R, Palazzo A, Procopio G, Santoni M, Trenta P, De Benedetto A, et al. Incidence and relative risk of hepatic toxicity in patients treated with anti-angiogenic tyrosine kinase inhibitors for malignancy. Br J Clin Pharmacol. 2014;77(6):929–938. doi: 10.1111/bcp.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabebe E, Fisher GA. Clinical trials of VEGF receptor tyrosine kinase inhibitors in pancreatic cancer. Expert Opin Investig Drugs. 2007;16(4):467–476. doi: 10.1517/13543784.16.4.467. [DOI] [PubMed] [Google Scholar]
- 16.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315(3):971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 17.Capdevila J, Carrato A, Tabernero J, Grande E. What could nintedanib (BIBF 1120), a triple inhibitor of VEGFR, PDGFR, and FGFR, add to the current treatment options for patients with metastatic colorectal cancer? Crit Rev Oncol Hematol. 2014;92:83–106. doi: 10.1016/j.critrevonc.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 19.Ledermann JA, Hackshaw A, Kaye S, Jayson G, Gabra H, McNeish I, et al. Randomized phase II placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer. J Clin Oncol. 2011;29(28):3798–3804. doi: 10.1200/JCO.2010.33.5208. [DOI] [PubMed] [Google Scholar]
- 20.Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15(2):143–155. doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- 21.Awasthi N, Schwarz MA, Verma V, Cappiello C, Schwarz RE. Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling. Lab Invest. 2009;89(1):38–46. doi: 10.1038/labinvest.2008.106. [DOI] [PubMed] [Google Scholar]
- 22.Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther. 2012;11(12):2644–2653. doi: 10.1158/1535-7163.MCT-12-0447. [DOI] [PubMed] [Google Scholar]
- 23.Awasthi N, Schwarz MA, Schwarz RE. Enhancing cytotoxic agent activity in experimental pancreatic cancer through EMAP II combination therapy. Cancer Chemother Pharmacol. 2011;68(3):571–582. doi: 10.1007/s00280-010-1514-7. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz RE, McCarty TM, Peralta EA, Diamond DJ, Ellenhorn JD. An orthotopic in vivo model of human pancreatic cancer. Surgery. 1999;126(3):562–567. [PubMed] [Google Scholar]
- 25.Gavalas NG, Liontos M, Trachana SP, Bagratuni T, Arapinis C, Liacos C, et al. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci. 2013;14(8):15885–15909. doi: 10.3390/ijms140815885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarz RE, Awasthi N, Konduri S, Cafasso D, Schwarz MA. EMAP II-based antiangiogenic-antiendothelial in vivo combination therapy of pancreatic cancer. Ann Surg Oncol. 2010;17(5):1442–1452. doi: 10.1245/s10434-009-0879-5. [DOI] [PubMed] [Google Scholar]
- 27.Ozawa F, Friess H, Tempia-Caliera A, Kleeff J, Buchler MW. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen. 2001;21(1):27–44. doi: 10.1002/1520-6866(2001)21:1<27::aid-tcm4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2(3):270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM, et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol. 2008;26(22):3743–3748. doi: 10.1200/JCO.2007.15.5416. [DOI] [PubMed] [Google Scholar]
- 32.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. Evaluation of poly-mechanistic antiangiogenic combinations to enhance cytotoxic therapy response in pancreatic cancer. PLoS ONE. 2012;7(6):e38477. doi: 10.1371/journal.pone.0038477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Awasthi N, Zhang C, Schwarz AM, Hinz S, Schwarz MA, Schwarz RE. Enhancement of nab-paclitaxel antitumor activity through addition of multitargeting antiangiogenic agents in experimental pancreatic cancer. Mol Cancer Ther. 2014;13(5):1032–1043. doi: 10.1158/1535-7163.MCT-13-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayes AJ, Li LY, Lippman ME. Anti-vascular therapy: a new approach to cancer treatment. West J Med. 2000;172(1):39–42. doi: 10.1136/ewjm.172.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 38.Tai WT, Shiau CW, Li YS, Chang CW, Huang JW, Hsueh TT, et al. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J Hepatol. 2014;61(1):89–97. doi: 10.1016/j.jhep.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Kutluk Cenik B, Ostapoff KT, Gerber DE, Brekken RA. BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol Cancer Ther. 2013;12(6):992–1001. doi: 10.1158/1535-7163.MCT-12-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans. 2012;40(1):139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- 41.Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci USA. 2009;106(43):18351–18356. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiang QF, Wang F, Su XD, Liang YJ, Zheng LS, Mi YJ, et al. Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance. Cell Oncol (Dordr) 2011;34(1):33–44. doi: 10.1007/s13402-010-0003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Awasthi N, Zhang C, Hinz S, Schwarz MA, Schwarz RE. Enhancing sorafenib-mediated sensitization to gemcitabine in experimental pancreatic cancer through EMAP II. J Exp Clin Cancer Res. 2013;32:12. doi: 10.1186/1756-9966-32-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernando NT, Koch M, Rothrock C, Gollogly LK, D'Amore PA, Ryeom S, et al. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin Cancer Res. 2008;14(5):1529–1539. doi: 10.1158/1078-0432.CCR-07-4126. [DOI] [PubMed] [Google Scholar]
- 46.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 47.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312(5777):1171–1175. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.