

Article first published online 29 April 2015.
Key Words: IL-12, IL-23, monoclonal antibody
Abstract
Background:
This study assessed the efficacy and safety of briakinumab, a human anti-IL-12/23p40 monoclonal antibody, compared with placebo for the induction and maintenance of remission in patients with moderately to severely active Crohn's disease.
Methods:
In this phase 2b, multicenter, double-blind, parallel group study, 246 patients stratified by prior tumor necrosis factor–antagonist use and response, were randomized (1:1:1:3) to 4 intravenous induction regimens: placebo, 200, 400, or 700 mg briakinumab, at weeks 0/4/8. At week 12, responders in the placebo or 400-mg induction groups entered the maintenance phase with the same regimen, whereas responders in the 700-mg induction group were rerandomized (1:1:1) to receive placebo, 200, or 700 mg briakinumab at weeks 12/16/20. At week 24, patients in remission stopped receiving study drug (withdrawal phase) until relapse. Patients experiencing relapse, nonresponders, and nonremitters could enter the open-label phase.
Results:
The primary end point of clinical remission at week 6 was not met. There were numerically greater rates of remission and response at 6, 12, or 24 weeks in patients treated with briakinumab. The safety and tolerability profile of briakinumab was similar in the induction and maintenance phases of the trial.
Conclusions:
Briakinumab showed a similar safety and tolerability profile to placebo in the induction and maintenance phases, and comparable rates of serious adverse events, adverse events leading to discontinuation, and malignancy. These data provide support for the potential efficacy of briakinumab and other IL-12/23 inhibitors in the treatment of moderate-to-severe Crohn's disease.
Crohn's disease (CD) is a chronic and progressive immune-mediated disease characterized by transmural intestinal inflammation.1 Although the etiology of CD is incompletely understood, a dysregulated immune response to normal intestinal flora plays a key role. Consequently, an exaggerated type 1 T helper (Th1) response occurs, resulting in increased production of proinflammatory cytokines including tumor necrosis factor alpha (TNF-α) and interleukin 12 (IL-12).2,3 IL-12, a heterodimeric protein composed of p35 and p40 subunits, is the major inducer of Th1 proliferation, and elevated levels of IL-12 have been reported in patients with CD.3–5 IL-23 is another heterodimeric cytokine implicated in the pathogenesis of CD3,5 and consists of a p19 subunit and the IL-12 shared subunit, p40. The presence of IL-23 drives differentiation of T cells into Th17 cells, which in turn stimulates production of the inflammatory mediator, IL-17.6 Increased levels of IL-12 and IL-23 are seen in murine models of colitis,7,8 and administration of an anti-IL-12p40 antibody ameliorates colitis in these animals.8
The treatment of CD is evolving. Traditional therapy consists of nonspecific anti-inflammatory and/or immunosuppressive agents, such as mesalamine, corticosteroids, thiopurines, and methotrexate (MTX). Over the past decade, anti-TNF agents have been shown to be effective for the induction and maintenance of remission.9–13 However, over one-third fail to respond to therapy, and an additional one-third of responsive patients become drug intolerant or lose drug response over time.14 Briakinumab, a fully human anti-IL-12/23p40 monoclonal antibody, has been shown in clinical trials to be highly effective and well tolerated for the treatment of moderate-to-severe psoriasis.15–17 More recently, the safety and tolerability of briakinumab was investigated in a phase 2a study of CD.18 The results of this preliminary trial showed that anti-IL-12/23 therapy had a safety profile similar to that of placebo and may be effective for induction of clinical response and remission in CD. Similar to briakinumab, the anti-IL-12/23 antibody ustekinumab has shown positive results for the induction of clinical response in patients with CD.19,20
The aim of this phase 2b study was to assess the efficacy and safety of briakinumab compared with placebo for the induction and maintenance of remission in patients with moderately to severely active CD.
MATERIALS AND METHODS
Patients
This phase 2b, multicenter, randomized, double-blind, parallel group, placebo-controlled, dose-ranging, efficacy, safety, and pharmacokinetic study was designed to evaluate the effectiveness of briakinumab in the induction and maintenance of remission in patients with moderately to severely active CD. The study was conducted at 60 sites in Australia, Canada, Europe, and the United States, from December 2007 to April 2010 (www.clinicaltrials.gov, NCT00562887).
Inclusion Criteria
Adult patients with a diagnosis of CD for >4 months, confirmed by endoscopy or radiologic evaluation, and a Crohn's Disease Activity Index21 (CDAI) score ≥220 and ≤450, were eligible for inclusion. Previous exposure to approved anti-TNF agents (including adalimumab, certolizumab, etanercept, infliximab, certain investigational drugs, and TNF receptor [immunoglobulin G1]) was permitted if discontinued at least 8 weeks before baseline. Secondary nonresponders (who discontinued a prior anti-TNF agent because of loss of response or lack of tolerance) and primary nonresponders to prior anti-TNF agents were eligible. Patients were allowed to continue azathioprine, 6-mercaptopurine (6-MP), or MTX provided they had received these medications for at least 12 weeks with stable doses for at least 4 weeks before week 0 visit.
Patients on stable doses of a corticosteroid (prednisolone ≤40 mg/d or equivalent, or budesonide ≤9 mg/d) for at least 2 weeks before week 0 were also eligible. Patients were not allowed to adjust the corticosteroid dose during the first 12 weeks of the study.
Exclusion Criteria
Patients with colitis other than CD, prior exposure to systemic or biologic anti-IL-12 therapy, receipt of an investigational TNF antagonist at any time, or receipt of any investigational agent within 30 days or 5 half-lives before week 0 visit were not eligible. Additional exclusion criteria were known symptomatic strictures; intestinal resection within the past 6 months; ostomy or ileoanal pouch; short bowel syndrome; evidence of dysplasia or history of malignancy; history of moderate-to-severe congestive heart failure or recent cerebrovascular accident; pregnancy or lactation; infection or risk for severe infections; abscess or suspicion of abscess; positive Clostridium difficile stool assay at the screening visit; receipt of total parenteral nutrition within 2 weeks before week 0 visit; initiation or discontinuation (within 4 wk of week 0 visit) or change in dosage (within 4 wk before week 0 visit) in aminosalicylates, mesalamine, sulfasalazine, or Crohn's-related antibiotics; or use of cyclosporine (intravenous [IV], oral), tacrolimus (any form) or mycophenolate mofetil within 8 weeks of week 0 visit.
Study Design
The original planned recruitment for this study specified a total sample size of 420 patients to be randomly assigned 1:1:1:3 to placebo or 200, 400, or 700 mg IV doses of briakinumab every 4 weeks (q4wk). Because of low recruitment, the 200 mg IV arm was dropped (amendment 3); therefore, a greater proportion of total study subjects were exposed to the 2 highest doses than originally planned. This allowed the investigation of exposure response relationships in CD at higher exposures and did not have a significant impact on the scientific output of the study. The total planned sample size was reduced to 225 patients, with an assumed delta to placebo increase from 25% to 30%. Of the final total sample size of 246 patients (intent-to-treat analysis set), 230 were enrolled on or after protocol amendment 3 (full analysis set [FAS]). See the following text for details regarding the calculation of sample size (Statistical Methods and Sample Size Determination).
In April 2010, after a prespecified analysis, the sponsor terminated the study early, due to a lack of efficacy for induction of remission, while patients were continuing treatment in the open-label (OL) phase. At study termination, 6 of the 246 randomized patients (2.4%) had completed the 2-year study and 128 (52.0%) had discontinued for other reasons. The remaining 112 patients (45.5%) discontinued due to termination of the study by the sponsor.
The planned study duration was 115 weeks and included 6 phases, starting with screening (≤4 wk), induction (12 wk), and maintenance (12 wk). Patients who remained in the study for 24 weeks and achieved remission at that time then entered into a monitored withdrawal phase. Patients without a response during the induction phase, or who relapsed during the maintenance or withdrawal phases, were eligible to enter an OL phase (Fig. 1), and a 45-day (approximately 7 wk) follow-up phase. The duration of the withdrawal phase and the OL phase was ≤92 weeks, but could vary among patients. The screening phase allowed the patients to washout any previous medications that were prohibited during the study. All patients needed to have completed the study after 2 years of treatment (or 104 wk post-week 0).
FIGURE 1.
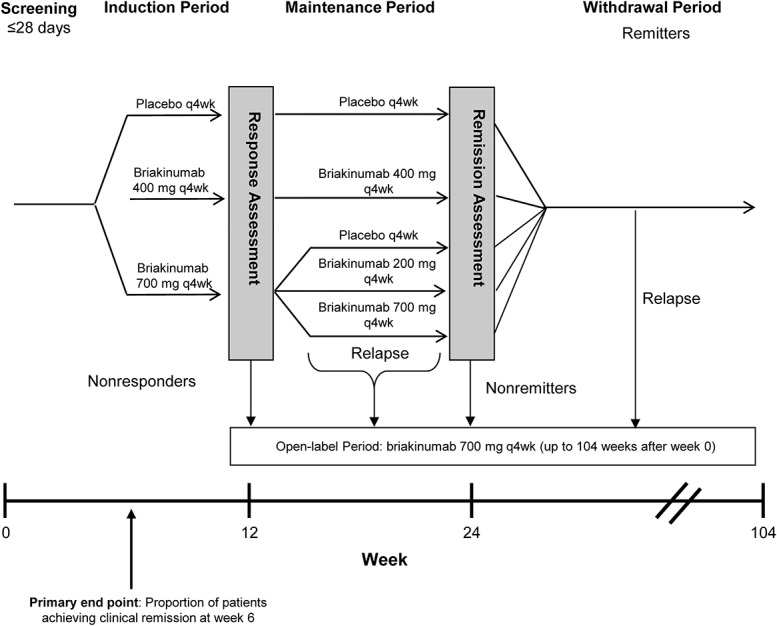
Study design. Patients were randomized to 4 induction groups: placebo, 200, 400, or 700 mg briakinumab. The primary end point was clinical remission at 6 weeks. At week 12, clinical response was assessed and patients in the placebo and 400 mg induction group continued into the maintenance phase on the same regimen, whereas responders in the 700-mg induction group were rerandomized to receive placebo, 200, and 700 mg briakinumab. At week 24, patients in clinical remission stopped receiving the study drug (withdrawal phase) until relapse. Patients with relapse, nonresponse, or nonremission could enter the OL phase.
Patients were randomly assigned 1:1:1:3 to IV infusion induction regimens: placebo, 200 mg briakinumab, 400 mg briakinumab, or 700 mg briakinumab administered at weeks 0, 4, and 8 and stratified at baseline (week 0) by prior TNF antagonist use (TNF-antagonist naive versus TNF-antagonist experienced) and TNF antagonist response (primary nonresponse versus secondary loss of response or secondary nonresponders). At week 12, patients in the placebo and 400-mg induction groups who achieved a clinical response (defined as a decrease in CDAI score of ≥70 points compared with week 0) continued into the maintenance phase, receiving the same treatment and dosage. Patients in the 700 mg induction group who achieved a clinical response were rerandomized 1:1:1 (with stratification by prior anti-TNF use and prior anti-TNF response): placebo, 200 mg briakinumab, or 700 mg briakinumab. Patients received placebo or briakinumab IV at weeks 12, 16, and 20 of the maintenance phase. Randomization (week 0) and rerandomization (week 12) were performed centrally, using randomization schemes generated by the study sponsor before the start of the study. The study sponsor, site personnel, and patients were unaware of the treatment assignments throughout both the induction and maintenance phases.
Patients who lacked a clinical response at week 12 could receive OL therapy at 700 mg briakinumab every 4 weeks. Patients in the maintenance dose groups who relapsed (defined as a CDAI increase ≥70 points compared with week 12 score and a minimum score of ≥220 points) at any time after week 12 could begin OL therapy.
Patients completed daily CDAI diary cards throughout the study, recording number of liquid or very soft stools per day and daily ratings of abdominal pain and general well-being. A CDAI score was calculated at week 0 and every 2 weeks thereafter through week 8, then every 4 weeks during the maintenance phase. At week 24, patients were assessed for clinical response and remission (response defined as a decrease in CDAI score of ≥70 points compared with week 0). Patients in clinical remission (defined as CDAI score <150 points) entered the withdrawal phase and stopped receiving study drug until a relapse (defined as a CDAI increase ≥70 points compared with week 24 score and a minimum score of ≥220 points). After relapse, patients had the option to enter the OL phase, as did nonresponders and nonremitters at week 24.
In the OL phase, all patients received cyclical dosing of 700 mg briakinumab by IV infusion q4wk for 3 doses, followed by a withdrawal phase. This dosing cycle could be repeated at the discretion of the investigator. The OL phase of the study could continue until the patient had completed a total of 2 years of treatment (defined as 104 wk post-week 0).
Interim Analysis
A prespecified interim analysis, conducted after all patients had completed the induction phase and 50% of the patients had completed the maintenance phase, was performed to make decisions about the designs of phase 3 studies.
The primary efficacy analysis tested whether at least 1 treatment group would show significant improvement over placebo by comparing the proportion of patients achieving clinical remission, defined as CDAI score of <150 points at week 6. Data for the primary end point were collected at week 6, well before the time of the interim analysis. Therefore, the interim analysis was the only and final analysis of the primary end point. Accordingly, no adjustment of the alpha error for this comparison was necessary. Nominal P values for key efficacy and key safety end points were also generated in the interim analysis.
Efficacy and Safety Evaluations
The primary end point was the proportion of patients with clinical remission (CDAI <150 points) at week 6. Secondary end points included clinical remission at week 12, clinical remission at week 24 among patients who responded at week 12, CDAI 100 clinical response (decrease in CDAI ≥100 points versus week 0) at weeks 6, 12, and 24, mean change from baseline in Inflammatory Bowel Disease Questionnaire scores at weeks 12 and 24, and the proportion of remitters who relapsed by week 24. Response, remission, and relapse at week 24 were assessed in the rerandomized group and in the patients who continued on their induction dose regimen. Subgroup analyses included clinical remission at week 6 stratified by baseline C-reactive protein (CRP ≥1 mg/dL or CRP <1 mg/dL) or by prior anti-TNF exposure. In addition, the mean decrease in CDAI score from baseline was assessed during the withdrawal and OL phases, as was the percentage of patients relapsing and time to relapse during withdrawal.
Vital signs, physical examinations, laboratory profiles, and reporting of adverse events were used to assess safety. Treatment-emergent adverse events were analyzed up to 45 days after the last dose date.
Statistical Methods and Sample Size Determination
All randomized patients who received at least 1 dose of study drug were included in the intent-to-treat analysis. However, as per protocol amendment 3, the efficacy analysis population excluded patients who were randomized to treatment group 200 mg briakinumab at week 0. The primary population used for the efficacy analyses was the FAS population. The maintenance population included all patients in the FAS population who achieved a clinical response at week 12 and received at least 1 dose of study drug between weeks 12 and 24. The safety population included all patients who received at least 1 dose of study drug at any time.
The primary efficacy analysis in the FAS population compared the difference between each briakinumab treatment group and placebo in the proportion of patients with clinical remission at week 6, controlling for stratification, using the Cochran–Mantel–Haenszel test. Patients with missing data at week 6 were considered not in clinical remission (nonresponder imputation method).
Secondary outcomes were assessed in the FAS population during the induction phase and in the maintenance population during the maintenance phase (weeks 12–24), using the chi-square test or the Fisher's exact test as appropriate, for subgroup analyses of TNF antagonist stratification status. For overall comparisons, the Cochran–Mantel–Haenszel test was used controlling for TNF antagonist stratification status to generate nominal P values. No adjustments for multiple comparisons were performed. The software package SAS version 8.2 was used for all statistical analyses.
Assuming a 20% clinical remission in the placebo group at week 6 and a 50% clinical remission in an active group at week 6, a sample of 45 patients per treatment group provided 80% power for comparing 2 treatment groups with a two-sided alpha of 0.05. To ensure a reasonable number of patients were available for evaluation in the 3 rerandomized arms of the maintenance phase, initially 135 patients (3 × 45) were planned to be randomized in the 700 mg briakinumab group in the induction phase at week 0. Therefore, a 1:1:3 allocation ratio was used to randomize a planned total of 225 (45 + 45 + 135) patients into 3 treatment arms at week 0. Sample size estimates were performed using nQuery Advisor 6.0.
Ethical Considerations
The study protocol was approved by the independent ethics committee or the institutional review board of each study site. All patients provided written informed consent before undergoing any study-related screening procedures.
All authors had access to the data, contributed to the development of the content, reviewed each draft of the article, and approved the final content.
RESULTS
Patient Disposition
Recruitment of patients began in December 2007. Low total enrollment in the study resulted in changes to the study design reflected in protocol amendment 3 (21 July 2008) with the removal of the 200 mg briakinumab arm and the reduction of total sample size to 225. Full enrollment of a total of 246 patients was achieved by March 2009. The 16 patients previously enrolled in the 200-mg briakinumab arm were not included in the efficacy analyses (FAS, N = 230), but were included in the safety analysis (intent-to-treat, N = 246). At the end of the induction phase, 14 of the 46 patients randomized to placebo and 21 of the 45 patients randomized to briakinumab 400 mg entered the maintenance phase. Sixty-four of the 139 patients randomized to 700 mg entered the maintenance phase after being rerandomized to either 700 mg or 200 mg briakinumab, or placebo. In April 2010, after the interim analysis, the study was terminated due to a lack of efficacy for induction of remission. At the time of study termination, 6 of the 246 randomized patients (2.4%) had completed the 2-year study and 128 (52.0%) had discontinued for other reasons (Fig. 2). The remaining 112 patients (45.5%) discontinued because of the termination of the study by the sponsor.
FIGURE 2.

Patient disposition during entire study. AE, adverse event; MP, maintenance phase; WP, withdrawal phase.
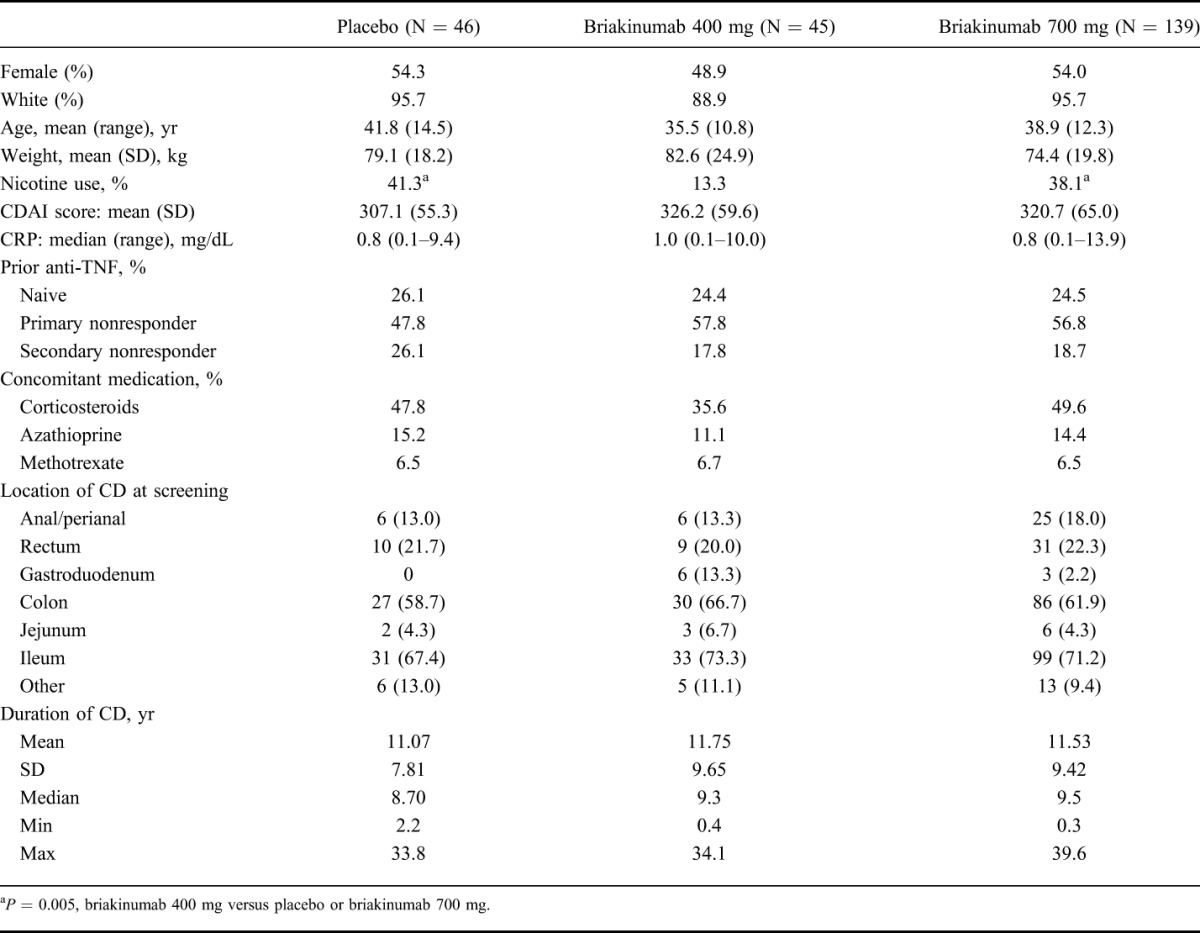
Baseline Demographic and Clinical Characteristics
The study groups were similar with respect to baseline demographic and clinical characteristics (Table 1), with the exception of smoking status. The briakinumab 400 mg group had a lower proportion of patients who were current smokers compared with the placebo and briakinumab 700 mg groups.
TABLE 1.
Baseline Demographics and Clinical Characteristics
Efficacy Outcomes
Induction Phase
At week 6, a greater number of patients in both the briakinumab 400 mg (6 patients, 13.3%) and 700 mg (24 patients, 17.3%) groups achieved remission compared with placebo (4 patients, 8.7%); however, statistical significance was not present and the primary end point was not met with an observed difference of 4.6% (P = 0.455) in the 400 mg group and 8.6% (P = 0.157) in the 700 mg group (Fig. 3A).
FIGURE 3.

Efficacy of briakinumab during the induction phase. A, Rates of clinical remission at weeks 6 and 12. Clinical remission was defined as CDAI score <150 points (*statistically significant versus placebo at P <0.05). B, Rates of clinical response at weeks 6 and 12 (defined as a decrease in CDAI score ≥100 points compared with week 0; *statistically significant versus placebo at P <0.05). C, Rates of clinical remission at week 6, stratified by baseline CRP levels (CRP ≥1 mg/dL or CRP <1 mg/dL). D, Rates of clinical remission at week 6, stratified by baseline history of anti-TNF treatment N.S., not statistically significant versus placebo at P <0.05.
At week 12, a significantly greater proportion of patients in the briakinumab 400 mg but not the 700 mg group were in remission compared with patients in the placebo group (Fig. 3A). The proportion of patients with CDAI 100 clinical response was significantly higher in patients treated with 700 mg briakinumab at both week 6 and week 12 compared with placebo-treated patients (Fig. 3B). There were no significant differences between the placebo and the briakinumab treatment groups in the proportion of patients achieving remission at week 6 or week 12 (data not shown) in subgroups of patients with an elevated CRP at baseline (Fig. 3C), or those with prior exposure to TNF antagonists (Fig. 3D).
At week 12, the improvement in Inflammatory Bowel Disease Questionnaire score was significantly greater for patients in the 400 mg and 700 mg briakinumab groups as compared with placebo-treated patients (mean change ± SD: 400 mg, 33.5 ± 39.45; 700 mg, 28.6 ± 36.63; placebo: 10.4 ± 30.19; P = 0.002, for both comparisons).
Maintenance Phase
During the maintenance phase, there were no significant differences in the proportion of patients with clinical remission (Fig. 4A) or CDAI 100 clinical response (Fig. 4B) at week 24 as assessed in the rerandomized groups (i.e., patients randomized to 700 mg briakinumab in the induction phase, who were in clinical response at week 12, and were rerandomized to placebo, 200 mg briakinumab, or 700 mg briakinumab). There were greater proportions of patients achieving clinical remission (Fig. 4A) or CDAI 100 response (Fig. 4B) at week 24 for patients who continued their briakinumab induction dose during the maintenance phase compared with patients who continued their placebo induction dose (i.e., those randomized to placebo or briakinumab 400 mg during the induction phase, and patients in the 700 mg induction group who were rerandomized to 700 mg briakinumab at week 12). However, these differences were not statistically significant.
FIGURE 4.
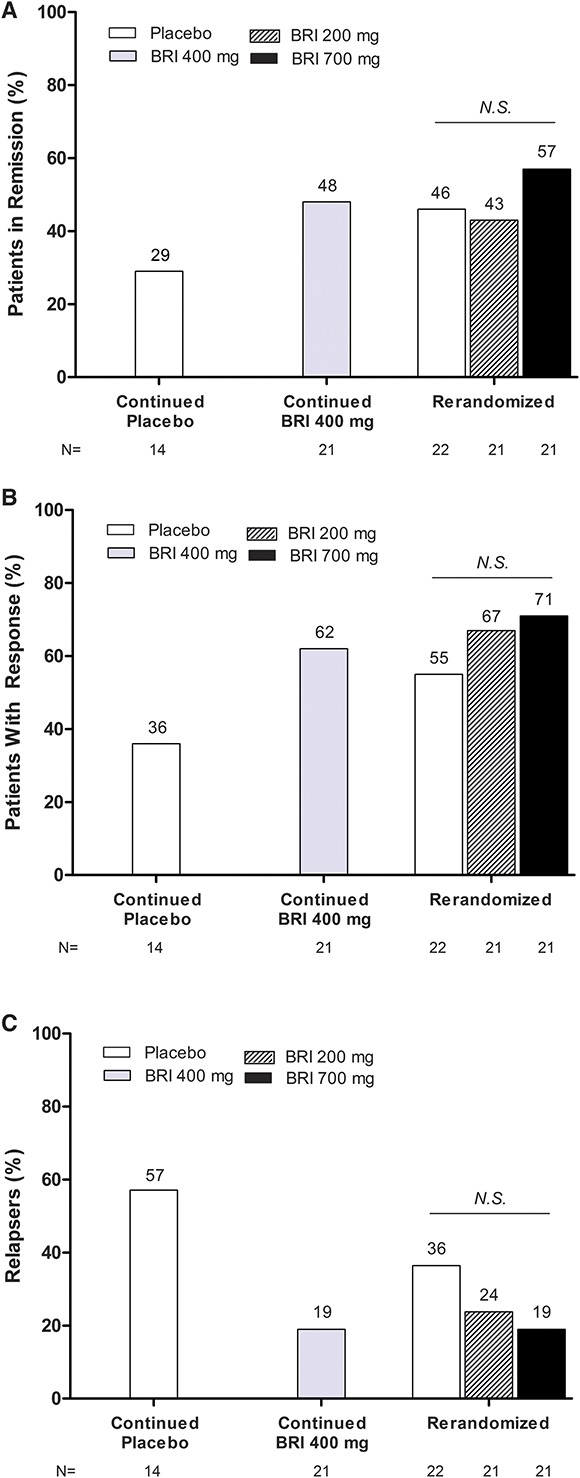
Efficacy of briakinumab during the maintenance phase. A, Rates of clinical remission at week 24 (defined as CDAI score <150 points). B, Rates of clinical response at week 24 (defined as a decrease in CDAI score ≥100 points compared with week 0). C, Relapse rate at week 24 (defined as a CDAI increase ≥70 points compared with week 24 score and a minimum score of ≥220 points). BRI, briakinumab; N.S., not statistically significant versus placebo at P <0.05.
During the maintenance phase through week 24, a greater percentage of patients who continued to receive placebo relapsed as compared with patients who received either 400 mg or 700 mg of briakinumab (Fig. 4C). For rerandomized patients, the greatest relapse rate was seen in patients rerandomized to placebo, but the difference in relapse rate was not significant between patients receiving placebo compared with patients rerandomized to 400 mg or 700 mg. However, there was a statistically significant difference in the time to relapse in patients assigned to either briakinumab 400 mg or 700 mg compared with patients who received placebo (median time to relapse: 400 mg, 12 wk, P = 0.045; 700 mg, >12 wk, P = 0.016; placebo, 8 wk).
Because only a small percentage of patients entered the withdrawal phase (Fig. 2), no inferences could be made about rates of relapse.
Safety
The adverse event profile of briakinumab was similar to that of placebo during both the induction and maintenance phases (Table 2). The occurrence of adverse events leading to discontinuation of the study drug was low and similar across groups. During the induction phase, serious adverse events were reported in 4 (8.7%) placebo patients (1 case of left chest basal cell carcinoma, 2 cases of abdominal pain, and 1 patient had invasive ductal carcinoma of the left breast and ductal carcinoma in situ of the right breast), 3 (18.8%) briakinumab 200 mg patients (CD exacerbation, herpes zoster, and ruptured ovarian cyst), 2 (4.4%) briakinumab 400 mg patients (asthma exacerbation and CD exacerbation), and 4 (2.9%) briakinumab 700 mg patients (drug dependence, 2 patients with CD exacerbation, and 1 elective abortion).
TABLE 2.
Adverse Events in the Induction and Maintenance Phases
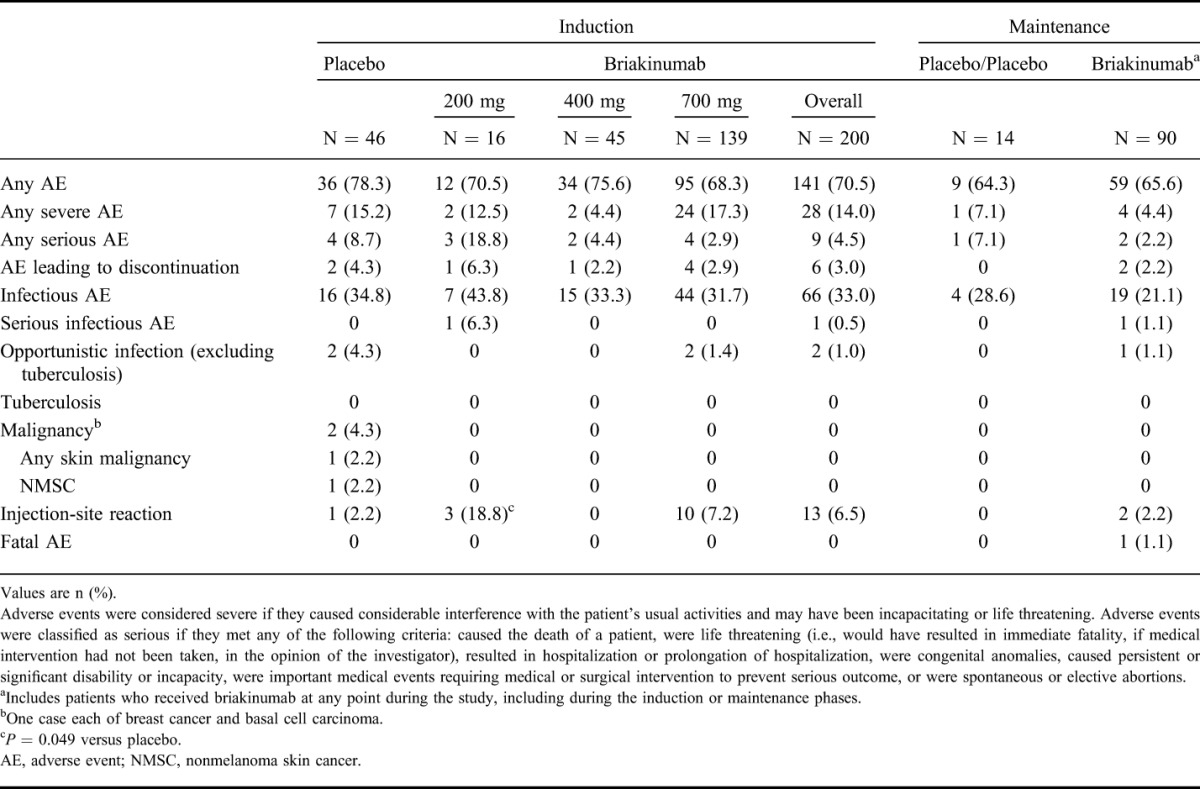
During the maintenance phase, serious adverse events were reported in 1 (7.1%) placebo patient (small bowel obstruction) and 2 (2.2%) briakinumab patients (1 patient experienced deep vein thrombosis in the right leg after prolonged flight, and 1 patient died of respiratory distress syndrome during hospitalization for pancreatitis and sepsis. This patient, a 65-year-old white male who received briakinumab 200 mg in both the induction and maintenance phases, had a relevant medical history of comedication with drugs known to have the potential to induce pancreatitis, obesity, diabetes mellitus, chronic obstructive pulmonary disease [COPD], sleep apnea, hypertension, and hyperlipidemia. The patient died 66 days after the last dose of the study drug). No malignancies were reported in patients exposed to briakinumab. One serious infection each was reported during the induction and maintenance phases. During induction, a case of herpes zoster was observed in the briakinumab 200 mg group; during maintenance, 1 case of sepsis was reported (in the above patient hospitalized for pancreatitis) in the briakinumab 200 mg group.
The most frequently reported treatment-emergent adverse events for patients who received any briakinumab during the study were upper respiratory tract infection (20.7%), nausea (17.3%), abdominal pain (14.3%), and headache (14.3%). No clinically meaningful changes in hematology, clinical chemistry, urinalysis parameters, or vital signs were observed.
During the OL phase (N = 202), 33 patients (16.3%) experienced serious adverse events. These serious adverse events included 1 patient with anaphylaxis that occurred during infusion of briakinumab, 7 patients with infusion-related reaction reported as serious adverse events, 1 patient with basosquamous carcinoma requiring excision, and 1 patient with acute pancreatitis. The 8 serious infusion-related reactions all resolved without sequelae. The case of pancreatitis resolved after 3 days of hospitalization. Approximately, 49% of patients experienced infectious adverse events during the OL phase with a maximum exposure of 2 years (median exposure to study drug was 337.5 d for all patients). The incidence of other adverse events of special interest was low throughout the study. There were no reports of tuberculosis, lymphoma, or melanoma, and no reports of major adverse cardiovascular or cerebrovascular accidents during the study.
DISCUSSION
This phase 2b, double-blind, placebo-controlled, dose-ranging, randomized trial assessed the efficacy of briakinumab for the treatment of moderate-to-severe CD. The primary end point of clinical remission at week 6 was not met. There were numerically greater rates of remission and response at 6, 12, or 24 weeks in patients treated with briakinumab. Briakinumab was well tolerated, with a safety profile in this study similar to that of placebo in the induction and maintenance phases. These findings provide some support for anti-IL-12/23 antibodies as treatment for moderate-to-severe CD.
The safety and efficacy of anti-IL-12/23 antibodies had been shown in previous clinical trials. Both briakinumab and ustekinumab18–20 yielded encouraging results for the induction of clinical remission and response in patients with CD. In addition, similar to the current trial, the results of the previous briakinumab trial demonstrated the durability of the clinical response and remission over time.18,19
The results of the current trial are informative and suggest that a prolonged induction phase may be required to realize the benefit of anti-IL-12/23 antibodies. A recent trial of similar design with ustekinumab in moderate-to-severe CD also failed to achieve statistical significance for the primary end point at week 6.20 Therefore, for briakinumab, up to 12 to 24 weeks may be required to show the maximal clinical benefit in patients with moderate-to-severe CD, especially in a refractory population (75.2% of patients in this study had prior TNF antagonist exposure). Although a significant difference in clinical remission was not seen at week 6 between the briakinumab and placebo groups, by week 24, differences in clinical remission rates were seen among patients who received continuous briakinumab 400 mg and 700 mg treatment compared with patients who received continuous placebo treatment. At week 6, clinical remission rates were 9%, 13%, and 17% for patients receiving placebo, briakinumab 400 mg, and briakinumab 700 mg, respectively. By week 12, clinical remission rates were 11%, 29%, and 22% for the same groups. Among patients continuing on the same dose during the maintenance phase, at week 24, 29% of patients continuing placebo, 48% of patients continuing on briakinumab 400 mg, and 57% of patients continuing on briakinumab 700 mg achieved clinical remission. Similar results were seen in clinical response when defined as a decrease in CDAI score of 100 points (CDAI 100 clinical response).
Although no significant difference was seen for rates of clinical remission or clinical response between patients rerandomized to briakinumab or placebo treatment during the maintenance phase, this may have been due to a carryover effect in the patients rerandomized to placebo who had received briakinumab 700 mg during induction. For patients receiving placebo during both induction and maintenance phases, at week 24, 29% had clinical remission and 36% had a clinical response. For patients rerandomized to placebo (from briakinumab 700 mg) at week 12, week 24 clinical remission rates were 46% and clinical response rates were 55%. This carryover effect could be the result of pharmacokinetics (28-d half-life), pharmacodynamics, and/or the mechanism of action of briakinumab. Of note, in a phase 2 trial of ustekinumab in CD, a similar carryover benefit was seen for up to the 8 weeks studied postcrossover in patients transitioned from ustekinumab to placebo.19 In light of this potential carryover effect, the duration of more than 12 weeks may have been necessary to see the maximal clinical benefit in patients rerandomized to placebo.
Although half of the patients were on concomitant steroids at baseline, steroid tapering was allowed after week 12, and at weeks 12 and 24, the percentage of patients who were steroid free was similar across all the groups, including placebo. A specific analysis for patients achieving steroid-free remission at week 24 was not performed; thus, it is unclear whether differences in tapered steroid doses could partially account for the similar remission rates observed for patients rerandomized to briakinumab versus placebo during the maintenance period.
The majority of patients in this study had prior anti-TNF exposure (75.2%). Of these patients previously treated with TNF antagonists, approximately 50% of patients in each induction treatment group were secondary nonresponders. Subgroup analyses suggested trends toward a worse efficacy outcome in the briakinumab 700 mg group with induction treatment at week 6 and week 12 for patients who were primary anti-TNF therapy nonresponders (week 6, 3.2% over placebo, week 12, 2.6% over placebo) as compared with patients who were responders with secondary loss of response (week 6, 10.6% over placebo, week 12, 11.2% over placebo) or who were anti-TNF naive (week 6, 9.8% over placebo, week 12, 21.1% over placebo) or responders with secondary loss of response (week 6, 10.6% over placebo, week 12, 11.2% over placebo). However, this study lacked statistical power to draw any valid conclusions regarding the efficacy of briakinumab in these subgroups of patients.
Concentrations of CRP correlate with endoscopic activity of disease, with elevated concentrations observed in more severe cases of CD.22 More than half of the patients in this study had baseline CRP levels ≥1 mg/dL, indicating the presence of more inflammation-driven disease in most of these patients. For patients treated with briakinumab, no statistically significant difference in clinical remission at week 6 or week 12 was seen in a subgroup analysis by CRP levels (above and below 1 mg/dL), suggesting that the effectiveness of briakinumab was similar across the spectrum of disease severity.
It should also be noted that nicotine use was significantly lower in the 400 mg treatment group as compared with the other treatment groups. Although randomization should theoretically “equalize” study arms, this chance chronic treatment imbalance could have biased the 400-mg group as a lower-severity group of patients.
Overall, briakinumab was well tolerated, with a safety profile in this study similar to that of placebo in the induction and maintenance phases. Rates of serious adverse events, adverse events leading to discontinuation, and malignancy were low and similar across the placebo and overall briakinumab groups. Of note, infusion reactions occurred in a greater percentage of briakinumab-treated patients than placebo patients during the induction phase. One patient who received 200 mg briakinumab during both the induction and maintenance phases died during the trial. Sixty-six days after the final dose of drug in the maintenance phase, this patient experienced a fatal adverse event of acute respiratory distress syndrome secondary to acute pancreatitis. This pattern of adverse events is similar to what was seen during other clinical trials with anti-IL-12/23 therapy in CD.18,19
During the OL phase, serious adverse events occurred at a higher rate than observed during the induction and maintenance phases. The increased rate was mostly due to an increase in serious infusion reactions; 8 of these types of adverse events occurred during the OL phase. All 8 serious infusion reactions were resolved.
In conclusion, although the primary end point was not met, the data from this phase 2b study provide preliminary evidence for the potential efficacy of briakinumab and other IL-12/23 inhibitors in the treatment of moderate-to-severe CD. Given the results of this study and previous studies, close attention needs to be paid to future study designs evaluating this class to ensure that suitable end points are chosen to evaluate the true therapeutic potential in CD.
ACKNOWLEDGMENTS
The authors thank Amy Gamelli, PhD, Elaine Hanna, Laurel Farmer, PhD, and Huzefa Photowala, PhD, of AbbVie, for providing medical writing and editing support.
R. Panaccione has received consultant and/or lecture fees from AbbVie, Amgen, AstraZeneca, Axcan Pharma (now Aptalis), Biogen Idec, Bristol-Myers Squibb, Centocor, ChemoCentryx, Eisai Medical Research Inc., Elan Pharmaceuticals, Ferring, Genentech, GlaxoSmithKline, Janssen, Merck Sharp and Dohme Corp., Millennium Pharmaceuticals Inc. (now Takeda), Ocera Therapeutics Inc., Otsuka America Pharmaceutical, Pfizer, Shire Pharmaceuticals, Prometheus Labs, Schering-Plough, Synta Pharmaceuticals Corp., Teva, UCB Pharma, and Warner Chilcott. W. J. Sandborn: Consultant—AbbVie, ActoGeniX NV, AGI Therapeutics, Inc., Alba Therapeutics Corporation, Albireo, Alfa Wasserman, Amgen, AM-Pharma BV, Anaphore, Astellas, Athersys, Inc., Atlantic Healthcare Limited, Aptalis, BioBalance Corporation, Boehringer Ingelheim Inc., Bristol-Myers Squibb, Celgene, Celek Pharmaceuticals, Cellerix SL, Cerimon Pharmaceuticals, ChemoCentryx, CoMentis, Cosmo Technologies, Coronado Biosciences, Cytokine Pharmasciences, Eagle Pharmaceuticals, Eisai Medical Research Inc., Elan Pharmaceuticals, EnGene, Inc., Eli Lilly, Enteromedics, Exagen Diagnostics, Inc., Ferring Pharmaceuticals, Flexion Therapeutics, Inc., Funxional Therapeutics Ltd., Genzyme Corporation, Genentech, Gilead Sciences, Given Imaging, GlaxoSmithKline, Human Genome Sciences, Ironwood Pharmaceuticals, Janssen, KaloBios Pharmaceuticals, Inc., Lexicon Pharmaceuticals, Lycera Corp., Meda Pharmaceuticals, Merck Research Laboratories, MerckSerono, Merck & Co., Millennium, Nisshin Kyorin Pharmaceuticals Co., Ltd., Novo Nordisk A/S, NPS Pharmaceuticals, Optimer Pharmaceuticals, Orexigen Therapeutics, Inc., PDL Biopharma, Pfizer, Procter and Gamble, Prometheus Laboratories, ProtAb Limited, Purgenesis Technologies, Inc., Receptos, Relypsa, Inc., Salient Pharmaceuticals, Salix Pharmaceuticals, Inc., Santarus, Shire Pharmaceuticals, Sigmoid Pharma Limited, Sirtris Pharmaceuticals, Inc. (a GSK company), S.L.A. Pharma (United Kingdom) Limited, Targacept, Teva Pharmaceuticals, Therakos, Tillotts Pharma AG, TxCell SA, UCB Pharma, Viamet Pharmaceuticals, Vascular Biogenics Limited (VBL), Warner Chilcott UK Limited; Speakers fees: AbbVie, Bristol-Myers Squibb, and Janssen. Financial support for research: AbbVie, Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Janssen, Millennium, Novartis, Pfizer, Procter and Gamble Pharmaceuticals, Shire Pharmaceuticals, and UCB Pharma. G. L. Gordon: Consultant—AbbVie. Support for travel to meetings: AbbVie. Grant/grant pending: AbbVie, Aptalis, Janssen, Falk, Furlex, GSK, Lexicon, Pfizer, Prometheus, Salix, Shire, Tranzyme, UCB, Tioga, Amgen, Sanofi-Aventis, and Coronado. Speakers fees: AbbVie, Apthalis, Janssen, Prometheus, Salix, Shire, Takeda, UCB, Warner Chilcott. S. D. Lee: Consultant—Abbott Pharmaceuticals, UCB Pharma, Janssen Pharmaceuticals Inc., Takeda Pharmaceuticals. Grant and research support: Abbott Pharmaceuticals, UCB Pharma, Janssen Pharmaceuticals Inc., Salix Pharmaceuticals, Millennium Pharmaceuticals, Prometheus Laboratories, GlaxoSmithKline. A. Safdi: Speaker: AbbVie, Janssen Pharmaceuticals, and Salix Pharmaceuticals. Research support: AbbVie, Janssen Pharmaceuticals, and Salix Pharmaceuticals. S. Sedghi: Speaker—AbbVie. B. G. Feagan: Consultant and/or advisory board—AbbVie, ActoGeniX, Albireo, Amgen, AstraZeneca, Avaxia Biologics Inc., Axcan, Baxter Healthcare Corp., Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Centocor, Elan/Biogen, EnGene, Ferring, Roche/Genentech, GICare, Gilead, Given Imaging Inc., GSK, Ironwood, Johnson & Johnson/Janssen, Lexicon, Lilly, Merck, Millennium, Nektar, Novartis Novo Nordisk, Prometheus Therapeutics & Diagnostics, Pfizer, Receptos, Salix, Serono, Shire, Sigmoid, Synergy, Takeda, Teva, Tillotts, UCB, Warner-Chilcott, Wyeth, Zealand, Zyngenia; Speakers fees: AbbVie, Johnson & Johnson/Janssen, Warner-Chilcott, UCB; Financial support for research: AbbVie, ActoGeniX, Bristol-Myers Squibb, Centocor, CombinatoRx, Elan/Biogen, Johnson & Johnson/Janssen, Roche/Genentech, Merck, Millennium, Novartis, Protein Design Labs, Tillotts, UCB, and Wyeth. S. Hanauer: Consultant—Bristol-Myers Squibb, Novartis, Procter and Gamble, Salix Pharmaceuticals, Inc. Advisory committees or review panels: Abbott, Caremark, Centocor, Elan Pharmaceuticals, McNeal Pharma, Millennium Pharma, Procter and Gamble, Wyeth, Salix Pharmaceuticals, Inc. Grant/Research support: Elan Pharmaceuticals. W. Reinisch has served as a speaker, a consultant, and/or an advisory board member for AbbVie, Aesca, Amgen, Astellas, AstraZeneca, Biogen Idec, Bristol-Myers Squibb, Cellerix, Chemocentryx, Celgene, Janssen, Danone Austria, Elan, Ferring, Genentech, Grünenthal, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, Millennium, Mitsubishi Tanabe Pharma Corporation, MSD, Novartis, Ocera, Otsuka, PDL, Pharmacosmos, Pfizer, Procter & Gamble, Prometheus, Robarts Clinical Trial, Schering-Plough, Setpointmedical, Shire, Takeda, Therakos, Tigenix, UCB, Vifor, Yakult, Zyngenia, Austria and 4SC. J. F. Valentine: Consultant—AbbVie, Genentech, UCB. Support for research: Millennium Pharmaceuticals, Merck, AbbVie, UCB, Bristol-Myers Squibb, Genentech, Celgene Cellular Therapeutics, Janssen, and Pfizer. B. Huang: AbbVie employee, may own AbbVie stock and/or options. R. Carcereri: AbbVie employee, may own AbbVie stock and/or options.
Author Contributions: All authors contributed to the planning and conduct of the study, the collection and interpretation of the data, and the drafting of the article. All of the authors approved the final draft.
Footnotes
Supported by AbbVie. The authors and AbbVie scientists designed the study and analyzed and interpreted the data. AbbVie provided writing support and reviewed and approved the publication. All authors had access to the data, contributed to the development of the content, reviewed each draft of the article, and approved the final content.
When this study was conducted, W. J. Sandborn was affiliated with the Mayo Clinic, Rochester, Minnesota; W. Reinisch was affiliated with the Medical University Vienna, Vienna, Austria; and J. F. Valentine was affiliated with the University of Florida, Gainesville, Florida.
Author disclosures are available in the Acknowledgments.
REFERENCES
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. [DOI] [PubMed] [Google Scholar]
- 2.Baert FJ, Rutgeerts PR. Anti-TNF strategies in Crohn's disease: mechanisms, clinical effects, indications. Int J Colorectal Dis. 1999;14:47–51. [DOI] [PubMed] [Google Scholar]
- 3.Sarra M, Pallone F, Macdonald TT, et al. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–1813. [DOI] [PubMed] [Google Scholar]
- 4.Fuss IJ, Becker C, Yang Z, et al. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. [DOI] [PubMed] [Google Scholar]
- 5.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veldman G. Targeting the p40 cytokines interleukin (IL)-12 and IL-23 in Crohn's disease. Drug Discov Today Ther Strateg. 2006;3:375–379. [Google Scholar]
- 7.Hue S, Ahern P, Buonocore S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. [DOI] [PubMed] [Google Scholar]
- 10.Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549. [DOI] [PubMed] [Google Scholar]
- 11.Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–333; quiz 591. [DOI] [PubMed] [Google Scholar]
- 12.Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132:52–65. [DOI] [PubMed] [Google Scholar]
- 13.Sandborn WJ, Feagan BG, Stoinov S, et al. Certolizumab pegol for the treatment of Crohn's disease. N Engl J Med. 2007;357:228–238. [DOI] [PubMed] [Google Scholar]
- 14.Peyrin-Biroulet L, Desreumaux P, Sandborn WJ, et al. Crohn's disease: beyond antagonists of tumour necrosis factor. Lancet. 2008;372:67–81. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb AB, Leonardi C, Kerdel F, et al. Efficacy and safety of briakinumab vs. etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol. 2011;165:652–660. [DOI] [PubMed] [Google Scholar]
- 16.Strober BE, Crowley JJ, Yamauchi PS, et al. Efficacy and safety results from a phase III, randomized controlled trial comparing the safety and efficacy of briakinumab with etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol. 2011;165:661–668. [DOI] [PubMed] [Google Scholar]
- 17.Reich K, Langley RG, Papp KA, et al. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N Engl J Med. 2011;365:1586–1596. [DOI] [PubMed] [Google Scholar]
- 18.Mannon PJ, Fuss IJ, Mayer L, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. [DOI] [PubMed] [Google Scholar]
- 19.Sandborn WJ, Feagan BG, Fedorak RN, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–1141. [DOI] [PubMed] [Google Scholar]
- 20.Sandborn WJ, Gasink C, Gao LL, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012;367:1519–1528. [DOI] [PubMed] [Google Scholar]
- 21.Best WR, Becktel JM, Singleton JW, et al. Development of a Crohn's disease activity index. National Cooperative Crohn's Disease Study. Gastroenterology. 1976;70:439–444. [PubMed] [Google Scholar]
- 22.Vermeire S, Van Assche G, Rutgeerts P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:661–665. [DOI] [PubMed] [Google Scholar]