

Article first published online 29 April 2015.
Key Words: inflammatory bowel disease, viral metagenomics, microbiome
Abstract:
Inflammatory bowel diseases (IBD), Crohn's disease and ulcerative colitis, are poorly understood disorders affecting the intestinal tract. The current model for disease suggests that genetically susceptible patients develop intolerance to gut microflora, and chronic inflammation develops as a result of environmental insults. Although interest has mainly focused on studying genetic variants and gut bacterial flora, little is known about the potential of viral infection to contribute to disease. Accordingly, we conducted a metagenomic analysis to document the baseline virome in colonic biopsy samples from patients with IBD in order to assess the contribution of viral infection to IBD. Libraries were generated from colon RNA to create approximately 2 GB sequence data per library. Using a bioinformatic pipeline designed to detect viral sequences, more than 1000 viral reads were derived directly from tissue without any coculture or isolation procedure. Herein, we describe the complexity and abundance of viruses, bacteria/bacteriophage, and human endogenous retroviral sequences from 10 patients with IBD and 5 healthy subjects undergoing surveillance colonoscopy. Differences in gut microflora and the abundance of mammalian viruses and human endogenous retroviruses were readily detected in the metagenomic analyses. Specifically, patients with herpesviridae sequences in their colon demonstrated increased expression of human endogenous viral sequences and differences in the diversity of their microbiome. This study provides a promising metagenomic approach to describe the colonic microbiome that can be used to better understand virus–host and phage–bacteria interactions in IBD.
Inflammatory bowel diseases (IBD) encompass 2 chronic inflammatory conditions predominantly involving at the distal gastrointestinal tract, namely Crohn's disease (CD) and ulcerative colitis (UC). Both disorders are genetically complex and thought to arise from a combination of influences that trigger a symptom complex that at times may be indistinguishable from intestinal infection. Adherent–invasive strains of Escherichia coli and Mycobacterium avium paratuberculosis have been studied as putative triggers for IBD, but none of these agents has been causally linked with either disease, and clinical trials using targeted antibiotics have been disappointing.1 One current pathophysiological model suggests that patients develop dysbiosis2; this could then lead to an abnormal mucosal immune response to commensal intestinal bacteria, resulting in chronic inflammation. Several metagenomic studies of gut microflora support this view and have hinted at a reduced microbial diversity in patients with IBD as well as an increased representation of specific lineages of microbes, such as the enterobacteriaceae. The alternative possibility persists that a single infectious agent, or related agents, may trigger the inflammatory response causing an imbalance of gut microbiota associated with a loss of tolerance to the microflora.
There are limited data available concerning the potential role that viruses may play in triggering IBD. Viral infection within the intestines may have a variety of consequences ranging from no obvious pathology to a global disturbance in gut physiology, modulation of the enteric immune system, and the disruption of the biophysical integrity of the bowel. Some viruses, such as cytomegalovirus (CMV), have a complex interaction with the disease process in IBD, where infection can manifest with the primary presentation of colitis or cause superimposed disease with the use of immunosuppressive therapy.3,4 Many viruses, including the herpesviridae, rubella, and measles viruses have been studied both serologically and in tissues, without any clear evidence of causal involvement in IBD.5,6 The potential for viral infection to trigger loss of tolerance to bacteria on a specific genetic background associated with CD has been demonstrated in an animal model and underscores the need to further document viral involvement in patients with IBD.7,8
The study of viruses in IBD has been limited by the lack of systematic methodologies to detect potential agents. The advent of next-generation sequencing has made metagenomic analyses feasible for human studies, and this approach has been used to study viral communities in blood9 and respiratory secretions10 and for cataloging the collective DNA and RNA viral species in stool samples from healthy subjects11,12 and patients with IBD.13 Metagenomic analysis has also served to discover viral agents in idiopathic neoplastic and inflammatory conditions where other approaches were unrevealing.14,15 Herein, we report for the first time the metagenomic analysis of viral sequences in colon RNA derived from patients with either CD or UC. Although these studies lack the ability to distinguish passenger viruses activated by inflammation and immunosuppression from potentially causal agents, our investigation provides a detailed description of the colonic virome in patients with IBD.
MATERIALS AND METHODS
Patients
Colon samples were collected from patients undergoing surgery for IBD or from healthy subjects undergoing colonoscopy surveillance for polyps and cancer. The tissues were de-identified by diagnosis and stored at −80°C. Available patients and control samples were subsequently selected for this descriptive pilot study to determine the feasibility of using a metagenomic approach to describe the microbiome and virome in colon tissues. After metagenomic analyses, patients' charts were reviewed for the use of medications before surgery (see Table, Supplemental Digital Content 1, http://links.lww.com/IBD/A805), the type of surgery performed and stratification of disease using the Montreal classification.16 The University of Alberta Ethics Committee approved this investigation.
Sample Processing for Illumina Sequencing
Colonic tissue was homogenized, and total RNA was extracted with TRIzol (Invitrogen, CA). Genomic DNA was removed with DNase I (RNase-free, Qiagen, CA), and total RNA was further purified with RiboMinus (Invitrogen, CA) to remove human ribosomal RNA. Next-generation sequencing library construction was performed with Illumina mRNA-Seq Prep Kit following manufacturer's instructions (Illumina, CA). Approximately 2 GB of pair-end 75 bp to 90 bp sequence data consisting of about 30 million reads were generated per library using the Illumina HiSeq 2000 sequencing platforms.
Bioinformatic Pipeline for Viral Metagenomics
A bioinformatic pipeline was developed to optimize detection of low abundance viral sequences in human RNA and to avoid ambiguous annotation (Fig. 1).9 Raw pair-end reads were first filtered with WindowMasker to remove low-quality low-complexity reads.17 Ribosomal, mitochondrial, and centromere sequences were annotated using NCBI nt/nr with SOAPaligner18,19 and RepeatMasker aligned to RepBase 16.04. To maximize the detection of viral reads and subtract the background of human and bacteria, 3 databases were established from NCBI nt/nr database with the taxon ID including human, bacteria, and virus-specific databases for the multialignment stage.9 The human database included the human genome assembly (GRCh37/hg19, February 2009) and all human transcript refseq from GenBank databases; virus and bacteria databases were constructed using taxon ID to extract all related sequences from nt/nr database.
FIGURE 1.
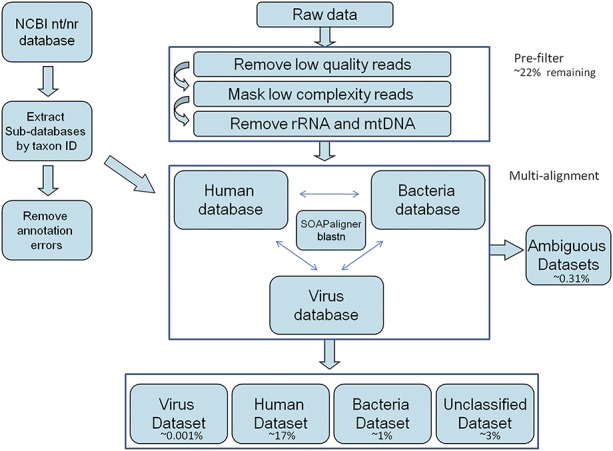
Bioinformatic pipeline for metagenomics. Reads were prefiltered and removed if they were low quality/complexity (∼7%), ribosomal RNA (∼69%), or mitochondrial RNA (∼6%). The remaining reads were assigned as human (∼17%) including HERV (∼0.006%), bacterial (∼1%), viral (∼0.001%), ambiguous (∼0.31%), or unclassified (∼3%) based on SOAPaligner or blastn.
When we aligned reads against human, bacteria, and virus databases separately, we found some reads annotated as “equally best hits” with less than 3 bp variations within matched region from either 2 or all 3 databases. We assigned these reads to an ambiguous data set. Based on the annotation, we further categorized the data into 3 major processed data sets, namely human, bacteria, and virus that contained pair-end reads annotated with best hits only from the single database. The reads with no matches from all the 3 databases were assigned to an unclassified data set. To optimize the computing resources for this multialignment analysis, we first used SOAPaligner with default parameters.18 This software is stringent and consumes less memory than blastn. This was used to acquire nearly perfect hits against human, bacteria, and virus database allowing a maximum variation of 5 bp with no gaps to the subject. After reducing the query size by SOAPaligner, we then used blastn to perform a second round of alignment with the unmatched reads using e-value <1e−5 as the cutoff for all 3 databases. Pair-end relationship of reads and alignment results from both the SOAP and blastn searches were integrated to evaluate the accuracy of annotation for further categorization of different data sets.
We further extracted a subgroup to include the human endogenous retroviruses (HERVs) from the human data set. HERV reads were normalized as transcripts per million with total high-quality reads of each library for relative expression level. Statistical analysis (F-test) has been used to evaluate the significant difference of HERV abundance between samples with and without herpesviruses. The reads were classified into major family members of HERV-K, HERV-H, HERV-W, and a miscellaneous category. This classification is based on the amino acid of each specific transfer RNA used during replication; for example, the betaretrovirus family, HERV-K, uses lysine transfer RNA for priming reverse transcription.20
Verification of Virus Hits
Viral reads were further analyzed by aligning against the whole viral genome using blastn with an e-value <1e−5. To identify the most relevant viral genotypes and strains, the reads were also aligned against related viral genomes when available. Reads specific to 1 viral strain were considered unique reads, whereas reads aligning to multiple related entries were selected by the best alignment result with lowest e-value, longest matched length, and least variation to subject (Fig. 2). Based on the read distribution and coverage, viruses with multiple unique reads distributed along the whole genome were considered to be the most likely viral candidate within each clinical sample.
FIGURE 2.

Viral reads from individual libraries were aligned against reference viral genomes based on blastn to reference genomes, where red bars indicate number of reads that align only to 1 genome, whereas blue bars represent number of reads that align to multiple related genomes. Evidence for combined herpesviridae infection was observed in sample cdC08 with (A) 286 reads matching approximately 19 kB of CMV genome and (B) 195 reads covering 12 kB of EBV genome (CMV, gi 155573622; EBV, gi 139424478). In sample ucC07, 434 reads matched HSV with a coverage of (C) 25,944 bp for the HSV-2 genome and (D) 14081 bp for the HSV-1 genome, suggesting the presence of HSV-2 (HSV-2, gi 9629267; HSV-1, gi 9629378). (E) Torque teno mini virus was also detected in sample ucC07 (gi 295441877). (F), Human parvovirus B19 was detected in sample cdC07 (gi 9632996).
Bacterial Profiling
Reads from all libraries were pooled together for combined assembly. Bacterial data sets were further annotated from contigs/scaffolds using either a blastn or balstx e-value <1e−5 to recover longer bacterial sequences for more accurate classification. Hierarchical clustering and principal component analysis (PCA) were used to further regroup among 15 samples based on the bacterial profiling using software MeV (4.8.1) and scripts written in R. A second data set was created for bacteriophage from the virus data set with taxon ID, and the phage data were stored with the bacterial data set.
RESULTS
Viral Sequences in Colon Samples
RNA libraries were made from colon samples derived from 10 patients with CD and UC (see Table, Supplemental Digital Content 1, http://links.lww.com/IBD/A805) and 5 subjects undergoing colonoscopy for colon cancer surveillance. Each colon library was sequenced on to a depth of approximately 30 million 75 bp to 90 bp pair-end reads that were subjected to analysis by the viral metagenomic pipeline (Fig. 1). The prefiltration process removed ribosomal sequences and low-quality reads, leaving approximately 7 million reads for further multialignment identification. On average, human sequences constituted 71% of reads in clinical tissue samples, bacteria accounted for 1% of reads, and viruses for less than 1% of reads. The number of verified viral reads varied among the 10 IBD libraries and ranged from 50 to 1000 (see Table, Supplemental Digital Content 2, http://links.lww.com/IBD/A806). Of the remaining reads, 3% were ambiguous and about 25% of reads were unclassified in need of further analysis.
A complexity of viruses was detected in the colon libraries (Fig. 3). Sequences aligning to these 15 different viruses were identified and verified by the detection of the same contiguous genomic sequence in the accompanying paired read. Among the 10 IBD samples, a relatively high diversity was observed ranging from 2 to 5 viral strains per library (Fig. 3). Human adenovirus was found in all IBD patient libraries (and none of the non-IBD patients), but the reads aligned to a conserved region of 1.2 kB, limiting our ability to distinguish specific adenovirus strains (Fig. 2). Herpesviridae sequences were observed in cdC08, ucC06, and ucC07 (Fig. 2) but not in non-IBD patients. Initially, it was challenging to discriminate between the 2 strains of herpes simplex virus (HSV) in sample ucC07, as genetically both HSV-1 and HSV-2 genomes share 83% similarity. However, the HSV-1–like reads covered a shorter region of the genome and demonstrated a greater variance with HSV-1 (8.6%), and none were unique. Accordingly, the presence of HSV-2 could be inferred based on the higher genome coverage and unique read count (compare Fig. 2C and D). A complex infection with evidence of both Epstein–Barr virus (EBV) and CMV was observed in cdC08 matching approximately 19 kB and 12 kB of each viral genome, respectively (Fig. 2A and B).
FIGURE 3.

Diversity of virome in IBD and cancer surveillance colon samples. Mammalian viruses and human pathogens were found with higher abundance in IBD colon libraries, whereas plant and other viruses were mainly observed in colon samples from patients undergoing colon cancer surveillance without documented disease (x-axis: patient samples, y-axis: viral families, z-axis: log plot of number of reads).
In total, we observed a 1.0% variance from reference genomes for EBV, 1.6% for CMV, and 0.4% for HSV-2, suggesting the presence of these viruses in the colon samples. Other mammalian viral sequences were observed in the IBD libraries with nucleotide similarity to torque teno mini virus, parvovirus, and porcine endogenous retrovirus B. Furthermore, various plants and insects were detected in both IBD and control colon samples. In 1 CD library (cdC07), viral sequences resembling a nucleocytoplasmic large double-stranded DNA virus were detected that were related to paramecium bursaria chlorella virus. Although these megaviruses have not been previously reported in humans, the acanthocystis turfacea chlorella virus has recently been found in the oropharynx of humans with diminished performance on neurocognitive tests.21
HERV in IBD Colon Samples
A small proportion of the human sequences detected in the IBD libraries were HERV, which constitute approximately 8% of the human genome. These elements are molecular fossils of ancient germ line retrovirus infection that have accumulated in the human genome throughout evolution. Although HERV expression does not indicate viral infection per se, the expression of HERV proteins has been linked with inflammatory and neoplastic conditions.22–24 For example, reports have revealed that HERV expression can be activated by exogenous viral infection,25 and expression of structural proteins can be cytotoxic in specific conditions.22 In our initial evaluation, we observed that the HERV-K and HERV-H reads were far more abundant than HERV-W by more than 30-fold average transcripts per million. Two of the IBD libraries, cdC08 and ucC07, had a very high relative abundance of HERV and also a higher diversity and abundance of viral strains as compared with the other samples (Fig. 2). Furthermore, the libraries containing herpesviruses had a 5- to 10-fold higher abundance of HERV as compared with those without (Fig. 4; see Table, Supplemental Digital Content 3, http://links.lww.com/IBD/A807). This is of potential interest because herpesviruses, such as EBV, have been reported to increase the transcriptional activity of HERV-K family members.25
FIGURE 4.
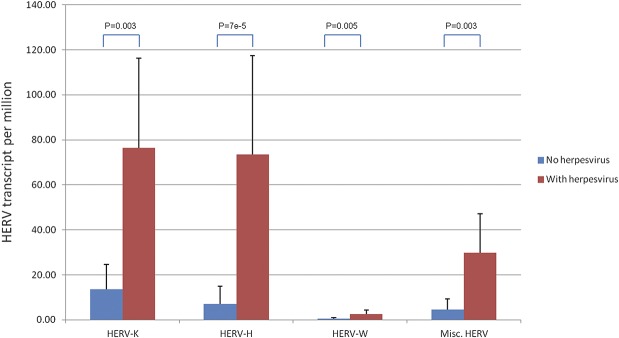
Abundance of HERVs (transcripts per million) in samples with herpesviruses (cdC08, ucC06, and ucC07) and without herpesviruses (P < 0.01, F-test).
Diversity of Bacteria and Phages
Bacterial sequences were observed in approximately 1% of reads, providing the opportunity to investigate the colonic microbiome within each library. To accomplish this, a combined assembly of all the bacterial reads from the 15 libraries was performed to recover longer contigs, which improved our ability to more closely identify related bacteria. We conducted a hierarchical clustering analysis using the abundance of bacteria in each library and then a PCA with scripts written in R language to determine the potential relationship of each library. Most of the IBD samples are clustered together (Fig. 5), and we observed a higher abundance of bacterial families that included the bradyrhizobiaceae, enterobacteriaceae, comamonadaceae, and moraxellaceae in the IBD samples, as previously reported for IBD.1,26–31 The PCA was consistent with the hierarchical clustering analysis and showed 2 predominant clusters with IBD and control subjects. Of interest, 3 samples with high viral diversity, including the 2 with predominant herpesvirus infection, appeared to disperse separately and did not fall into the 2 predominant clusters (Fig. 6).
FIGURE 5.
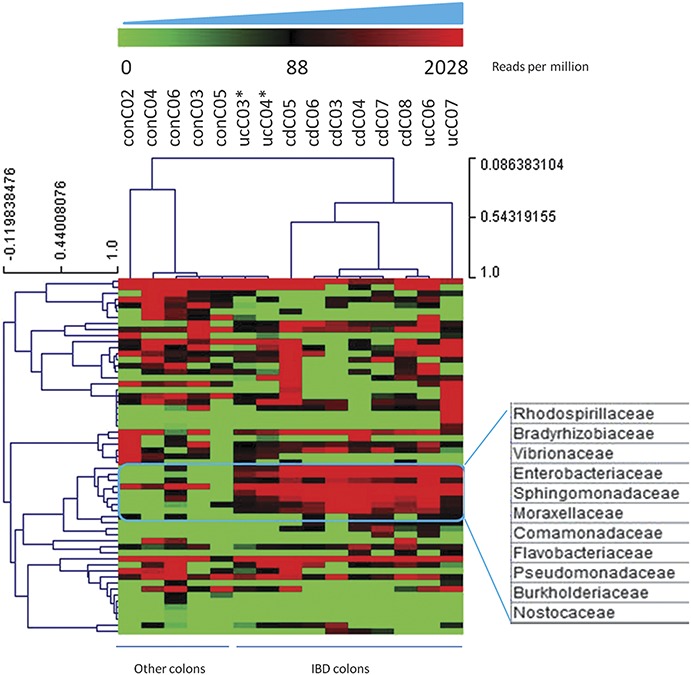
Hierarchical clustering of bacterial data sets based on transcripts per million (per sample counts ranging from 0 to about 2000). Clusters were identified from the bacterial profiling patterns on a per-bacterial-family (rows with distance values ranging from −0.12 to 1) and a per-sample (columns with distance values ranging from 0.08 to 1) basis. Most of the IBD samples clustered together except 2 UC colon samples marked with asterisks (ucC03 and ucC04 with limited viral sequences/diversity). Cluster of bacteria expressed at consistently higher levels in IBD samples as strongly associated with IBD in previous publications.
FIGURE 6.
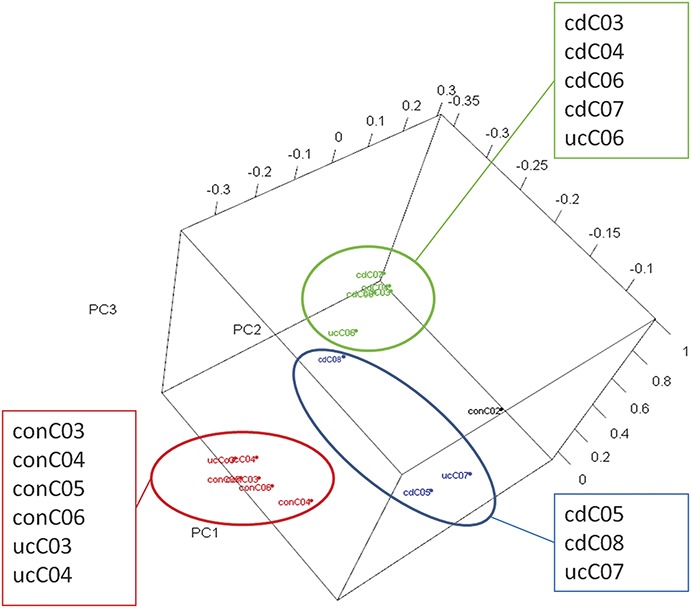
Three-dimensional PCA analysis on bacterial data sets subcategorized into 3 major groups: (1) the majority of the IBD samples (in green), (2) samples from patients undergoing colonic surveillance and 2 UC samples with limited viral sequences (in red: ucC03 and ucC04), and (3) outliers of 3 IBD colon samples with relatively higher abundance of human viruses (in blue: cdC05, parvovirus B19; cdC08, CMV and EBV; ucC07, HSV-2 and torque teno virus).
We observed that the majority of DNA viruses within the gut virome were bacteriophages. These viruses are reported to maintain a stable population within the gut microbiome over time with less than 20% differences in strains recorded over a 2-year period.32 To study the bacteriaphage populations, we tabulated all bacteriophages and identified 119 different strains with unique reads that have been associated with 49 different bacterial host strains (see Table, Supplemental Digital Content 4, http://links.lww.com/IBD/A808). We studied the more diverse IBD colon samples and found the highly abundant phage groups in only 1 or 2 colon samples. For example, the ucC07 library contained sequences homologous to several Enterococcus phages, Streptococcus phage phi-m46.1, Escherichia phage K1ind3, and others. In contrast, most of the other phages were observed in the multiple colon samples. This observation implies that a common phage–host population may be discovered in IBD colon samples along with the expected differences one would expect to be observed in individual patient samples.13 Furthermore, we found that nearly half of the bacteriophages were associated with bacterial strains identified in the colon samples. Indeed, 5 strains were related to bacteria found in probiotics, like Lactococcus lactis, which may be linked with VSL#3 probiotic therapy used to treat 2 of our patients (see Table, Supplemental Digital Content 1, http://links.lww.com/IBD/A805).
DISCUSSION
In this study, we developed a bioinformatic pipeline for defining the virome in colon samples processed by deep sequencing of total RNA depleted of ribosomes. This process was performed without any coculture or isolation procedure to provide data suggesting the presence of multiple virus strains. We observed a marked difference in both abundance of viruses and diversity within the virome of colons derived from IBD patients undergoing colonic surveillance for colon cancer. However, it is duly recognized that one of the weaknesses of this study was the comparison of surgical samples from patients with IBD and colonoscopic biopsies from our control subjects.
With this metagenomic approach of studying RNA from colon tissue, there was a paucity of viral reads (∼10 per million). In contrast, we observed that approximately 1 in 4 reads was unclassified, and these reads likely represent novel bacterial, viral, or other sequences that require further characterization, as noted by other studies.9,12,33 However, the most striking finding was that the viral abundance and diversity were associated with differences in the bacterial composition within the colon. These data are consistent with the idea that viruses may contribute to the dysbiosis observed in patients with IBD, as recently demonstrated in an animal model of IBD,7 by disturbing specific immune responses.8 Moreover, samples with abundant herpesviridae also expressed elevated levels of HERV sequences that have been associated with pathology in other inflammatory disorders.22,25,34,35
Within the known virus data set, we found strains that have been observed previously in the colon of patients with IBD. EBV has been found in previous case–control studies on IBD36–38 and observed infecting intestinal lymphocytes in patients with UC and to a lesser extent those with CD.39,40 Intestinal CMV infection has been linked with exacerbation of IBD in extensive case–control studies.39,41–44 Human herpesvirus 2 has been associated with exacerbation of UC and may complicate disease in immunocompromised individuals.45 Human papillomavirus has been linked with squamous cell carcinoma in patients with IBD.46–48 Previous studies have reported serological evidence of parvovirus B19 in patients with IBD on immunosuppressive therapy49 and B19 infection in the diseased intestinal mucosa of a patient with poorly controlled UC.50 Accordingly, our metagenomic approach of using next-generation sequencing to discover viral sequences had the capacity to detect viruses in an unbiased fashion, as observed by other investigators using more conventional techniques.
We also identified additional viral strains in patients with IBD such as torque teno mini virus, which is commonly observed in humans without serious pathology.51 As discussed, the detection of porcine endogenous retrovirus B sequences may have been related to the ingestion of pork. The detection of plant and insect viruses would also be expected from a dietary source, such as cassava vein mosaic virus, prunus necrotic ringspot virus, cauliflower mosaic virus, cucumber green mottle mosaic virus, and chilo iridescent virus.
Our pipeline also separated HERV and phage subsets from our virus data set to avoid ambiguous annotations of exogenous viral strains. We observed a marked diversity in both data sets from the IBD samples, suggesting a potential for these agents to impact on the complexity of the pathogenesis of IBD. For example, the diversity and abundance of HERV among IBD colon samples imply that infection of specific virus strains, such as herpesviridae, may trigger the expression of HERV in the colon,34,52,53 consistent with previous reports in patients with autoimmune disorders and specific cancers.35,54,55 However, it remains to be determined whether HERVs have any impact on the disease process in patients with IBD.
It is now appreciated that the high diversity of bacteria in the human gastrointestinal tract impacts on both health and disease, especially in patients with IBD.30,31,56–61 Based on the hierarchical clustering and PCA analysis on bacterial data sets in colon biopsies, we identified a cluster of bacteria (Fig. 5) that have been consistently shown to have a higher abundance in IBD samples as compared with healthy subjects.1,26–31 The variety and abundance of phages found in colon samples are tightly associated with diversity of their bacterial hosts; accordingly, the bacteriophages detected within the colon samples likely highlight the bacterial host strains located in proximity to mucosal epithelial cells with relative strong adherent–invasive ability.62–65 Indeed, we identified multiple phage strains associated with major pathogens associated with diarrhea or other intestinal symptoms.39,66,67
The presence of mammalian viral sequences also seemed to have a marked impact on the microbial diversity (Fig. 6). The interaction of viral infection with enteric microbiota has recently shown to be an important driver in models of IBD. For example, enteric bacteria enable norovirus infection of intestinal B lymphocytes,8 suggesting a complex interaction of the microbiota and virus infection as highlighted by the Atg16L1-deficient mouse model. This mouse develops a CD-like disease after murine norovirus infection, which is preventable by administrating broad-spectrum antibiotics. The viral infection clearly alters the gut microflora because the disease can be passaged to healthy Atg16L1-deficient mice even after the viral infection apparently cleared.7 The mechanism of viral–bacterial interaction in IBD requires further investigation, as discussed.8 Accordingly, the findings reported in this study underscore the importance and clinical utility of a model where viral infection may alter the gut microbiome. Our data also emphasize the utility of using viral metagenomics to detect the potential of viral infection as a complicating factor in the pathogenesis and potential management of IBD. In future, additional studies will be required to more firmly link the metagenomic findings with clinical features of disease.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.ibdjournal.org).
Supported in part by the Alberta Innovates Health Solutions (A.L.M., L.A.D., and W.W.), The Broad Foundation (A.L.M. and G.K.-S.W.), the Canadian Crohn's and Colitis Foundation (A.L.M.), the Canadian Institutes for Health Research (L.A.D.), and Alberta Innovates Technology Futures (G.K.-S.W.). E.W. has been an advisory board member for AbbVie and Janssen and received research support from Janssen; R.F. has been an advisory board member for AbbVie, Ferring, Janssen, Shire, VSL#3, and Celltrion and received research support from AbbVie, Alba, Bristol-Myers Squibb, Centocor, GSK, Genentech, Janssen, Merck, Millennium, Novartis, Pfizer, Procter & Gamble, Roche, VSL#3, and Celltrion. L.A.D. has been an advisory board member for AbbVie, Ferring, Janssen, and Shire and received research support from Beneo-Orafti. A.L.M. has been an advisory board member for Intercept and Novartis and received research support in kind from Abbott and Gilead. The remaining authors have no conflicts of interest to disclose.
REFERENCES
- 1.Vanderploeg R, Panaccione R, Ghosh S, et al. Influences of intestinal bacteria in human inflammatory bowel disease. Infect Dis Clin North Am. 2010;24:977–993, ix. [DOI] [PubMed] [Google Scholar]
- 2.Tamboli CP, Neut C, Desreumaux P, et al. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hussein K, Hayek T, Yassin K, et al. Acute cytomegalovirus infection associated with the onset of inflammatory bowel disease. Am J Med Sci. 2006;331:40–43. [DOI] [PubMed] [Google Scholar]
- 4.Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol. 2006;101:2857–2865. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein CN, Rawsthorne P, Blanchard JF. Population-based case-control study of measles, mumps, and rubella and inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:759–762. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein CN, Blanchard JF. Viruses and inflammatory bowel disease: is there evidence for a causal association? Inflamm Bowel Dis. 2000;6:34–39. [DOI] [PubMed] [Google Scholar]
- 7.Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones MK, Watanabe M, Zhu S, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346:755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Law J, Jovel J, Paterson J, et al. Identification of hepatotropic viruses from plasma using deep sequencing: a new generation diagnostic tool. PLoS One. 2013;8:e60595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet. 2004;38:525–552. [DOI] [PubMed] [Google Scholar]
- 11.Breitbart M, Hewson I, Felts B, et al. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003;185:6220–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang T, Breitbart M, Lee WH, et al. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol. 2006;4:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Brocal V, Garcia-Lopez R, Vazquez-Castellanos JF, et al. Study of the viral and microbial communities associated with Crohn's disease: a metagenomic approach. Clin Transl Gastroenterol. 2013;4:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palacios G, Druce J, Du L, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–998. [DOI] [PubMed] [Google Scholar]
- 16.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19(suppl A):5A–36A. [DOI] [PubMed] [Google Scholar]
- 17.Morgulis A, Gertz EM, Schaffer AA, et al. WindowMasker: window-based masker for sequenced genomes. Bioinformatics. 2006;22:134–141. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Li Y, Kristiansen K, et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. [DOI] [PubMed] [Google Scholar]
- 19.Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. [DOI] [PubMed] [Google Scholar]
- 20.Larsson E, Kato N, Cohen M. Human endogenous proviruses. Curr Top Microbiol Immunol. 1989;148:115–132. [DOI] [PubMed] [Google Scholar]
- 21.Yolken RH, Jones-Brando L, Dunigan DD, et al. Chlorovirus ATCV-1 is part of the human oropharyngeal virome and is associated with changes in cognitive functions in humans and mice. Proc Natl Acad Sci U S A. 2014;111:16106–16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antony JM, van Marle G, Opii W, et al. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. [DOI] [PubMed] [Google Scholar]
- 23.Antony JM, Zhu Y, Izad M, et al. Comparative expression of human endogenous retrovirus-W genes in multiple sclerosis. AIDS Res Hum Retroviruses. 2007;23:1251–1256. [DOI] [PubMed] [Google Scholar]
- 24.Mason AL, Xu L, Guo L, et al. Retroviruses in autoimmune liver disease: genetic or environmental agents? Arch Immunol Ther Exp (Warsz). 1999;47:289–297. [PubMed] [Google Scholar]
- 25.Sutkowski N, Conrad B, Thorley-Lawson DA, et al. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001;15:579–589. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Butcher J, Mack D, et al. Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:139–153. [DOI] [PubMed] [Google Scholar]
- 27.Davenport M, Poles J, Leung JM, et al. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricanek P, Lothe SM, Frye SA, et al. Gut bacterial profile in patients newly diagnosed with treatment-naive Crohn's disease. Clin Exp Gastroenterol. 2012;5:173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minot S, Bryson A, Chehoud C, et al. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 2013;110:12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepage P, Leclerc MC, Joossens M, et al. A metagenomic insight into our gut's microbiome. Gut. 2013;62:146–158. [DOI] [PubMed] [Google Scholar]
- 34.Kwun HJ, Han HJ, Lee WJ, et al. Transactivation of the human endogenous retrovirus K long terminal repeat by herpes simplex virus type 1 immediate early protein 0. Virus Res. 2002;86:93–100. [DOI] [PubMed] [Google Scholar]
- 35.Sicat J, Sutkowski N, Huber BT. Expression of human endogenous retrovirus HERV-K18 superantigen is elevated in juvenile rheumatoid arthritis. J Rheumatol. 2005;32:1821–1831. [PubMed] [Google Scholar]
- 36.Sankaran-Walters S, Ransibrahmanakul K, Grishina I, et al. Epstein-Barr virus replication linked to B cell proliferation in inflamed areas of colonic mucosa of patients with inflammatory bowel disease. J Clin Virol. 2011;50:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spieker T, Herbst H. Distribution and phenotype of Epstein-Barr virus-infected cells in inflammatory bowel disease. Am J Pathol. 2000;157:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakefield AJ, Fox JD, Sawyerr AM, et al. Detection of herpesvirus DNA in the large intestine of patients with ulcerative colitis and Crohn's disease using the nested polymerase chain reaction. J Med Virol. 1992;38:183–190. [DOI] [PubMed] [Google Scholar]
- 39.Lidar M, Langevitz P, Shoenfeld Y. The role of infection in inflammatory bowel disease: initiation, exacerbation and protection. Isr Med Assoc J. 2009;11:558–563. [PubMed] [Google Scholar]
- 40.Yanai H, Shimizu N, Nagasaki S, et al. Epstein-Barr virus infection of the colon with inflammatory bowel disease. Am J Gastroenterol. 1999;94:1582–1586. [DOI] [PubMed] [Google Scholar]
- 41.Criscuoli V, Rizzuto MR, Cottone M. Cytomegalovirus and inflammatory bowel disease: is there a link? World J Gastroenterol. 2006;12:4813–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kambham N, Vij R, Cartwright CA, et al. Cytomegalovirus infection in steroid-refractory ulcerative colitis: a case-control study. Am J Surg Pathol. 2004;28:365–373. [DOI] [PubMed] [Google Scholar]
- 43.Kim JJ, Simpson N, Klipfel N, et al. Cytomegalovirus infection in patients with active inflammatory bowel disease. Dig Dis Sci. 2010;55:1059–1065. [DOI] [PubMed] [Google Scholar]
- 44.Kojima T, Watanabe T, Hata K, et al. Cytomegalovirus infection in ulcerative colitis. Scand J Gastroenterol. 2006;41:706–711. [DOI] [PubMed] [Google Scholar]
- 45.Schunter MO, Walles T, Fritz P, et al. Herpes simplex virus colitis complicating ulcerative colitis: a case report and brief review on superinfections. J Crohns Colitis. 2007;1:41–46. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg R, Greenwald B, Roth JS, et al. Squamous dysplasia of the rectum in a patient with ulcerative colitis treated with 6-mercaptopurine. Dig Dis Sci. 2008;53:760–764. [DOI] [PubMed] [Google Scholar]
- 47.Kong CS, Welton ML, Longacre TA. Role of human papillomavirus in squamous cell metaplasia-dysplasia-carcinoma of the rectum. Am J Surg Pathol. 2007;31:919–925. [DOI] [PubMed] [Google Scholar]
- 48.Saul SH. Inflammatory cloacogenic polyp: relationship to solitary rectal ulcer syndrome/mucosal prolapse and other bowel disorders. Hum Pathol. 1987;18:1120–1125. [DOI] [PubMed] [Google Scholar]
- 49.Montanari M, Cortelezzi C, Parravicini M, et al. Parvovirus B19 infection and immunosuppressed IBD-affected pediatricians. Am J Gastroenterol. 2009;104:537. [DOI] [PubMed] [Google Scholar]
- 50.Pironi L, Bonvicini F, Gionchetti P, et al. Parvovirus b19 infection localized in the intestinal mucosa and associated with severe inflammatory bowel disease. J Clin Microbiol. 2009;47:1591–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernardin F, Operskalski E, Busch M, et al. Transfusion transmission of highly prevalent commensal human viruses. Transfusion. 2010;50:2474–2483. [DOI] [PubMed] [Google Scholar]
- 52.Lee WJ, Kwun HJ, Kim HS, et al. Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol Cells. 2003;15:75–80. [PubMed] [Google Scholar]
- 53.Nellaker C, Yao Y, Jones-Brando L, et al. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology. 2006;3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruprecht K, Obojes K, Wengel V, et al. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: implications for multiple sclerosis. J Neurovirol. 2006;12:65–71. [DOI] [PubMed] [Google Scholar]
- 55.Stauffer Y, Theiler G, Sperisen P, et al. Digital expression profiles of human endogenous retroviral families in normal and cancerous tissues. Cancer Immun. 2004;4:2. [PubMed] [Google Scholar]
- 56.Dimitrov DV. The human gutome: nutrigenomics of the host-microbiome interactions. OMICS. 2011;15:419–430. [DOI] [PubMed] [Google Scholar]
- 57.Gosalbes MJ, Durban A, Pignatelli M, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One. 2011;6:e17447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sleator RD, Shortall C, Hill C. Metagenomics. Lett Appl Microbiol. 2008;47:361–366. [DOI] [PubMed] [Google Scholar]
- 59.Tuohy KM, Gougoulias C, Shen Q, et al. Studying the human gut microbiota in the trans-omics era—focus on metagenomics and metabonomics. Curr Pharm Des. 2009;15:1415–1427. [DOI] [PubMed] [Google Scholar]
- 60.Vacharaksa A, Finlay BB. Gut microbiota: metagenomics to study complex ecology. Curr Biol. 2010;20:R569–R571. [DOI] [PubMed] [Google Scholar]
- 61.Vaishampayan PA, Kuehl JV, Froula JL, et al. Comparative metagenomics and population dynamics of the gut microbiota in mother and infant. Genome Biol Evol. 2010;2:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chichlowski M, Hale LP. Bacterial-mucosal interactions in inflammatory bowel disease: an alliance gone bad. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1139–G1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knight P, Campbell BJ, Rhodes JM. Host-bacteria interaction in inflammatory bowel disease. Br Med Bull. 2008;88:95–113. [DOI] [PubMed] [Google Scholar]
- 64.Macfarlane S, Steed H, Macfarlane GT. Intestinal bacteria and inflammatory bowel disease. Crit Rev Clin Lab Sci. 2009;46:25–54. [DOI] [PubMed] [Google Scholar]
- 65.Tieng V, Le Bouguenec C, du Merle L, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A. 2002;99:2977–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matricon J, Barnich N, Ardid D. Immunopathogenesis of inflammatory bowel disease. Self Nonself. 2010;1:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. [DOI] [PubMed] [Google Scholar]