

Abstract
Neurodegenerative diseases ranging from Alzheimer disease and polyglutamine diseases to transmissible spongiform encephalopathies are associated with the aggregation and accumulation of misfolded proteins. In several cases the intracellular and extracellular protein deposits contain a fibrillar protein species called amyloid. However while amyloid deposits are hallmarks of numerous neurodegenerative diseases, their actual role in disease progression remains unclear. Especially perplexing is the often poor correlation between these deposits and other markers of neurodegeneration. As a result the question remains whether amyloid deposits are the disease-causing species, the consequence of cellular disease pathology or even the result of a protective cellular response to misfolded protein species. Here we highlight studies that suggest that accumulation and sequestration of misfolded protein in amyloid inclusion bodies and plaques can serve a protective function. Furthermore, we discuss how exceeding the cellular capacity for protective deposition of misfolded proteins may contribute to the formation of toxic protein species.
Keywords: neurodegenerative disease, amyloid formation, Alzheimer disease, Huntington disease, prion diseases, yeast prion, Rnq1
Introduction
The study of neurodegenerative diseases began over a hundred years ago when Alois Alzheimer identified fibrillar structures within the postmortem brain of a patient who had exhibited progressive cognitive dysfunction and psychosis.1 It is now known that the majority of neurodegenerative diseases characterized by progressive neuronal dysfunction and loss are associated with the deposition of misfolded proteins. These misfolded proteins are frequently found in a β-sheet-rich fibrillar protein conformation known as amyloid2,3 (Fig. 1). For more than forty years amyloid deposits were thought to be causative agents in the degenerative process.4 But the tables have turned. Recent studies suggest instead that a group of still poorly defined pre-amyloid species, rather than the amyloid deposits themselves, are the true toxic conformations5–8 (Fig. 1). These soluble prefibrillar oligomers share conformational characteristics independent of the proteins’ primary amino acid sequences and may share a common mechanism of toxicity.5 Indeed even proteins completely unrelated to disease, such as PI3 kinase and the E. Coli protein HypF-N, can be induced to form such prefibrillar structures in vitro and, when they do, they are toxic when applied extracellularly to cells in culture or injected into rat brains.9,10 The intra- and extracellular conversion of misfolded proteins into highly structured and less reactive amyloid forms may reduce the levels of these toxic protein species and therefore be protective.
Figure 1.

Aggregation of misfolded protein can give rise to oligomers, amorphous aggregates and inclusion bodies. The accumulation of misfolded protein leads to the formation of different protein assemblies. Prefibrillar oligomers formed by different proteins share a common structure and are thought to be the toxic protein species in diseases such as Alzheimer disease and Huntington disease.5,6 Oligomers are conformationally molten and can associate to form amorphous aggregates or convert to an amyloidogenic nucleus to initiate amyloid fibril formation. Amyloid fibrils have a highly organized structure due to repeating β-sheets and are insoluble. Amyloid fibrils are often found in intra- and extracellular inclusions such as inclusion bodies and amyloid plaques. The generation of amyloid fibers and inclusion bodies can protect cells by reducing the formation of highly interactive toxic oligomers and amorphous aggregates.
For the purpose of this perspective, we focus on three neuro-degenerative diseases, Alzheimer disease, Huntington disease and prion disease. We will first present studies in which the formation of inclusion bodies and amyloid plaques protects against proteo-toxicity and then discuss how exceeding the cellular capacity for deposition of misfolded proteins may give rise to toxic protein species. Although these studies do not preclude detrimental effects of amyloid deposits in particular contexts (e.g., obstructive vascular amyloid), they clearly show that amyloid formation can be beneficial.
Pathological Features Associated with Neurodegenerative Diseases
The protein deposits found in Alzheimer disease, Huntington disease and prion disease are formed by completely unrelated proteins. They accumulate in distinct brain regions and have highly characteristic morphologies that form the basis of histological diagnosis. Neurodegeneration also affects distinct regions of the brain in each disease, reflecting a disease-specific vulnerability of particular neurons.11 However, in all three diseases the correlation between the localization of neurodegeneration and protein deposition is weak.
Alzheimer disease
Alzheimer disease (AD), the most common cause of dementia, most severely affects the temporal pole, hippocampus and amygdala.12 AD is characterized by the accumulation of two very different proteins, each with a distinct distribution. Aβ (amyloid β) peptide accumulates extracellularly in amyloid plaques while hyperphosphorylated tau, a microtubule binding protein, accumulates intracellularly in neurofibrillary tangles. A definitive pathological diagnosis of AD requires the detection of both types of aggregation. Aβ may accumulate in different plaque forms. Neuritic plaques, also referred to as classic or cored plaques, contain a dense amyloid core surrounded in turn by a ring of abnormal cellular processes and a rim of diffuse amyloid.11,12 In these neuritic plaques tau accumulation can also be present in dystrophic neurites surrounding the amyloid core.
It has been hypothesized that Aβ accumulation is the primary cause of pathogenesis in AD, yet there is a weak correlation between Aβ plaque density and the severity of dementia.11 For example, brain samples of aged patients without clinical dementia can display abundant Aβ plaques.11 To some extent this may be the consequence of Aβ accumulating in plaques without any associated neuritic degeneration (such as “burned-out” and “diffuse plaques”). Tau-containing neurofibrillary tangles correlate better with clinical severity of AD than Aβ plaques, but even here, the question remains whether tau aggregation itself is toxic or if it is the result of a protective mechanism.13–16
Huntington disease
Huntington disease (HD), classified as a hyperkinetic movement disorder, tends to affect brain regions distinct from those affected by Alzheimer disease. HD is characterized by atrophy of the cerebral cortex, globus pallidus and striatum, specifically the loss of medium spiny neurons within the neostriatum.17–19 HD is caused by CAG repeat expansions in the huntingtin gene, which lead to the accumulation of polyglutamine-expanded huntingtin protein within intranuclear inclusion bodies or neurites.11 The density of intranuclear inclusions correlates positively with the CAG repeat length present in the huntingtin gene.20 However, neuronal vulnerability does not correspond to the cellular concentration of huntingtin protein nor the distribution of huntingtin inclusions.21 In fact there is a distinct dissociation of inclusion distribution and the selective pattern of striatal neuron loss, as few to no inclusions are detected in the vulnerable striatal neurons.22
Prion diseases or spongiform encephalopathies
Prion diseases or spongiform encephalopathies can present in numerous ways, such as the sporadic versus variant forms of Creutzfeldt-Jakob disease (CJD). While the prion disease subtypes all involve the accumulation of a proteinase-K resistant form of the prion protein PrP (PrPres), they each affect different brain regions and involve distinct patterns of PrP aggregation.11 Sporadic CJD causes spongiform change in the neuropil of the cerebral cortex, subcortical grey matter and cerebellar molecular layer.23 The brainstem and spinal cord do not exhibit spongiform change although PrP deposits can be present. PrPres deposits in sporadic CJD are found in synaptic, perivacuolar, perineuronal and plaque-like patterns.23 Neuronal loss correlates with microglial activation and axonal damage, but not with local deposition of PrPres. In contrast, variant CJD, caused by the consumption of meat from cows with Bovine Spongiform Encephalopathy, leads to the presence of a large number of florid plaques in the cerbral and cerebellar cortex.24 Florid plaques have a dense amyloid core, a pale radiating fibrillar periphery and are surrounded by a halo of spongiform change. Interestingly, spongiform change in variant CJD is most pronounced in the basal ganglia, which contain relatively few amyloid plaques.24
In summary, while the particular misfolded proteins vary in these diseases, in all three cases protein deposits are a poor indicator of neuronal loss. This makes it plausible that structured protein deposits help cells to cope with misfolded proteins. In turn, the failure of particular neurons to create such deposits may cause their disease-specific vulnerability.
Protein Deposition as a Cellular Response to Misfolded Proteins
One of the first indications that protein inclusions may protect cells from toxic misfolded proteins came from a study investigating the response of tissue culture cells to either proteasome inhibitors or to overexpression of proteins targeted to the proteasome. The Kopito laboratory established that exceeding the proteasome’s capacity to cope with misfolded proteins, by either perturbation, leads to the accumulation of stable aggregates at a distinct structure adjacent to the centrosome.25 This structure was termed the aggresome to emphasize that its formation is a common cellular response to the presence of aggregated misfolded protein.
The aggresome is a highly structured deposit of insoluble protein surrounded by a cage formed by the intermediate filament protein vimentin. Most strikingly, aggresomes are formed near the centrosome through dynein-dependent retrograde transport of protein aggregates along microtubules.25–27 Far from being amorphous protein accumulations, aggresomes are formed through an active and conserved cellular process, that appears to serve a vital purpose: sweeping the cytoplasm clear of potentially toxic forms of misfolded proteins.28
Protein Deposition as a Protective Mechanism
A host of studies involving proteins linked to neurodegenerative diseases and other amyloidogenic proteins have investigated the role of inclusion and plaque formation in pathogenicity. The case is perhaps strongest for Huntington disease, for which it has been postulated that inclusions cause toxicity due to the sequestration of proteins critical for cell homeostasis.29 Inclusions formed by mutant huntingtin protein have been shown to sequester glycer-aldehyde-3-phosphate dehydrogenase, to impair transcription due to sequestration of the transcriptional coactivator CREB binding protein, and to interfere with the function of the ubiquitin-proteasome system.30–32 However smaller oligomeric species and loosely packed amorphous aggregates may be more prone to interact with and sequester proteins than densely packed amyloid deposits.
Indeed, several studies suggest that the formation of tightly packed huntingtin deposits is beneficial for cell survival. The Greenberg laboratory transfected with mutant huntingtin primary striatal neuron to induce the formation of inclusions.33 The inclusions formed resembled protein deposits found in the brains of Huntington patients, as they were intranuclear and ubiquitinylated. But these inclusions were not sufficient to induce apoptosis. On the contrary, inhibition of the ubiquitinylation of mutant Huntingtin prevented the formation of inclusions and actually increased cell death.33
In a complementary study, the Finkbeiner group used time-lapse microscopy to follow the fate of individual huntingtin transfected neurons. The majority of neurons died without the formation of inclusion bodies and the formation of an inclusion body actually increased the probability of neuron survival.34 The formation of inclusion bodies directly correlated with a decrease in soluble huntingtin, suggesting that inclusion bodies protect neurons by decreasing levels of soluble toxic isoforms of huntingtin.34 Inclusion body formation may also serve a protective function by increasing the autophagic degradation of the aggregated protein species.35 Inclusions of mutant huntingtin directly induce autophagy through sequestration of mTOR, a negative regulator of autophagy, and autophagy not only reduces the levels of aggregated but also soluble mutant huntingtin.36,37
Together, these studies suggest that compounds elevating the formation of inclusion bodies, such as aggresomes, could lessen cellular pathology. On the other hand compounds that antagonize the toxicity of mutant huntingtin by reducing its aggregation have been identified.38 On the surface, this appears to conflict with the notion that promoting inclusions may be beneficial, but both, solubilization and inclusion body formation, may diminish the levels of toxic oligomers, the more critical species in pathogenesis. In fact, in one HD model a compound prevented huntingtin-mediated proteasome dysfunction by promoting inclusion formation.39
Although the characteristic protein deposits are found extracellularly in AD and prion disease, not intracellularly as in HD, here too studies suggest that structured protein deposits are less toxic than other conformers. As for HD, amyloid assembly may serve a beneficial function by shifting the equilibrium away from more toxic conformers, such as prefibrillar oligomers.5,6,8
In a collaborative effort, the Kelly and Dillin laboratories investigated the roles of the aging process and the heat shock response in the formation of proteotoxic species in a Caenorhabiditis elegans model of AD. The intracellular expression of Aβ resulted in the formation of Aβ aggregates, but these aggregates did not correlate with toxicity.40 RNAi-mediated repression of the insulin/IGF-1 receptor DAF-2, resulting in increased life span, reduced Aβ-mediated toxicity while slightly increasing the amount of Aβ aggregates. This protection depended on both daf-16 and hsf-1. Interestingly, repression of DAF-16 reduced the number of high molecular weight Aβ aggregates, while repression of HSF-1 increased it. The Kelly and Dillin laboratories concluded that two dichotomous cellular pathways counteract Aβ toxicity: The HSF-1 pathway controls disaggregation, while the DAF-16 pathway transforms toxic Aβ oligomers into larger Aβ aggregates of lower toxicity.40
In a separate study by the Mucke group, a point mutation within Aβ, the Arctic mutation (Aβ E22G), influenced the rate at which Aβ assembled into amyloid fibers. In vitro and in transgenic mice, the Artic mutation enhanced formation of neuritic amyloid plaques and diminished non-amyloid Aβ assemblies.41 As non-amyloid Aβ assemblies correlated with behavioral and neuronal deficits in these transgenic mice, the promotion of Aβ amyloid fibril formation, without a coinciding increase in oligomeric Aβ, may be beneficial.
Most recently, in a follow-up study of a clinical trial, immunization of AD patients with the full length Aβ peptide reduced Aβ immunostaining and amyloid plaques.42 Unfortunately, immunization neither slowed nor stopped the progression of neurodegeneration. As immunization with Aβ peptide may not have reduced the levels of toxic oligomeric Aβ species, the authors suggest, that immunization specifically against oligomeric Aβ species may be more successful at halting neurodegeneration.
Plaque formation may also prove beneficial in the case of prion disease. PrPres isoforms, the protease resistant forms of PrP that include amyloid, are not toxic on their own. Mice that do not express their own PrP protein (Prn-p0/0) are completely resistant to intracerebral injection of even very high doses of PrPres.43 Equally striking, mice producing a secreted form of PrP, GPI anchor-less PrP, accumulated massive plaque-like amyloid deposits, yet had no clinical manifestations of prion disease.44 Some brain lesions were present, but less neurodegeneration was associated with these amyloid plaques than with diffuse wild type PrPres deposits. These results were especially significant as transgenic mice had up to 40% more PrPres than mice with WT PrP.44 Using human tissue samples, the Barron laboratory showed that the accumulation of certain forms of PrPres fails to result in spongiform degeneration.45 Brain extracts from two cases of familial prion disease were used to test the transmission of disease to transgenic mice. One of the samples exhibited PrPres deposits and spongiform change, while the other presented with PrPres deposits and no spongiform change. Brain extract from the patient without spongiform degeneration did not result in disease transmission but elicited PrPres deposition in large multicentric plaques. Therefore, PrPres would appear to be rendered nonpathogenic by its sequestration in amyloid plaques.45
Lessons from a Yeast Model
Yeast prion proteins, just as PrP, can adopt self-perpetuating conformational states. In yeast, however, prions do not cause disease, but rather serve as heritable genetic elements, perpetuated by the transfer of the prion template from mother to daughter cells.46 The heritable protein conformation of the yeast prions is amyloid in nature and, as for Aβ, Huntingtin and PrP, amyloid formation by the yeast prion proteins proceeds through intermediate oligomeric protein species.47,48 In fact, the observation that prefibrillar oligomers are intermediates in amyloid formation was first made for the yeast prion protein Sup35.47,49 Oligomers formed by the yeast prion protein Sup35 share structural features with the oligomers formed by disease-related amyloidogenic proteins, including recognition by anti-oligomeric antibodies and interaction with specific small compounds.49,50 Thus the study of yeast prions can provide insight into amyloid formation and cellular responses to the presence of amyloid.
The yeast prion [RNQ+] is formed by the Rnq1 protein (The cytoplasmic inheritance of yeast prions is designated by [ ]). Rnq is nonessential and has no known biological function, except when it is in the prion state.51 The [RNQ+] prion interacts with other amyloidogenic proteins in vivo and enables them to adopt their amyloid conformation. For example, [RNQ+] facilitates the de novo induction of the [PSI+] prion state by enhancing the amyloid conversion of the yeast prion protein Sup35.52
We recently reported that moderate ectopic overexpression of Rnq1 is extremely toxic if endogenous Rnq1 is in the [RNQ+] prion conformation.53 While overexpression of Rnq1 did result in the formation of amyloid inclusions, as assessed by Thioflavin-T staining, semi-denaturing agarose gels and in vitro seeding assays, the amyloid conformation did not represent the toxic species. In fact, co-expression of an Hsp40 chaperone, Sis1, known to interact with the prion form of Rnq1,54 suppressed the toxicity elicited by Rnq1 overexpression by promoting Rnq1 assembly into amyloid. Mutants of Rnq1, impaired in their interaction with the chaperone and their ability to readily form amyloid, exhibited enhanced toxicity. Chaperones have been shown to antagonize toxicity associated with protein misfolding before, but in those cases overexpressed chaperones either decreased protein aggregation55 or appeared to have no observable effect on protein aggregation.56 Our study presents the first instance in which a chaperone antagonizes the toxicity of a misfolding protein by facilitating its deposition into an amyloid inclusion. It clearly demonstrates that actively promoting the formation of inclusion bodies and even amyloid plaques may prove beneficial in protein misfolding pathologies.
Formation of Toxic Protein Species Due to Non-Productive Templating
While the formation of aggresomes and extracellular amyloid plaques appears to serve a protective function, they could be associated with toxicity if their assembly is overwhelmed by the amount of protein damage or impeded by other molecular and cellular factors. As shown by the Kampinga group, aggresome formation by mutant huntingtin in tissue culture cells did not affect the cellular progression through mitosis. However, when the mutant huntingtin formed scattered secondary inclusions, the completion of mitosis was delayed or even failed completely.57 The Kampinga group speculated that these secondary inclusions, distinct from aggresomes, form when the process of aggresome formation is saturated. These results are reminiscent of our studies in which overexpression of the yeast prion protein Rnq1 resulted in toxicity when it exceeded the cellular capacity to efficiently assemble the prion protein into amyloid. The toxicity of Rnq1 overexpression was exacerbated by factors interfering with amyloid assembly, such as repression of Sis1, the chaperone required for Rnq1 amyloid formation, or mutations within Rnq1, which reduce its interaction with the chaperone.53
Importantly, Rnq1 overexpression only resulted in toxicity if the endogenous Rnq1 protein was in its [RNQ+] prion conformation, making the otherwise benign prion state a prerequisite for Rnq1 mediated toxicity. Interestingly, the Rnq1 prion state is also required for toxicity of mutant huntingtin exon 1 in yeast models of Huntington disease.58 While the Rnq1 prion conformation usually acts as a template for the conversion of soluble Rnq1 protein into benign amyloid conformers, we hypothesize that this process can also result in the formation of toxic protein species. We refer to this as non-productive templating, which occurs when the cellular capacity to facilitate amyloid formation is exceeded or impeded (Fig. 2).
Figure 2.
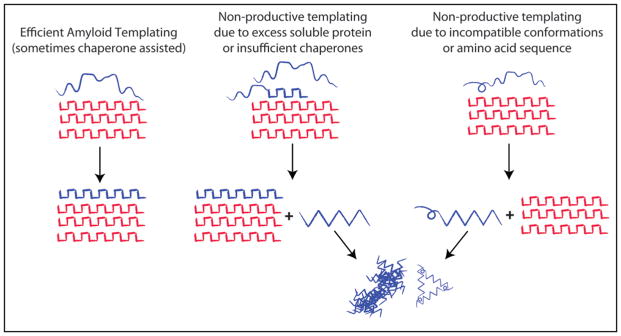
Amyloid and Non-productive templating. Amyloid fibrils grow by causing proteins of the same amino-acid sequence to adopt the same amyloid conformation. This is referred to as amyloid templating and involves the efficient addition of monomers or oligomeric species to the amyloid fiber, which maybe assisted by specific chaperones (e.g., Rnq1 and the Hsp40 Sis1, Sup35NM and Hsp104,49). If the amount of substrate exceeds the cellular capacity for amyloid conversion or if amino acid sequences are incompatible, the interaction of substrate with the amyloid fibrils may give rise to other abnormal conformational species, which may go on to form toxic oligomers and amorphous aggregates (Rnq1, PrP and Het-s/S). We refer to this as non-productive templating.
The notion of non-productive templating offers a unifying explanation for the observation that the presence of amyloid formation is sometimes associated with toxicity even when the amyloid form itself is benign. We discuss two cases in point: As mentioned earlier, the expression of GPI-anchorless PrP resulted in the formation of amyloid plaques but was not overtly toxic. However, when GPI-anchorless PrP was expressed together with WT PrP, deposits of both amyloid and non-amyloid PrPres formed and the clinical manifestations of prion disease were enhanced.44 Furthermore, it has been suggested that PrPres subverts a stress protective function of PrP into an apoptotic signal.59 The toxic signal elicited is dependent on the presence of PrPres and the expression of GPI-anchored PrP.59 PrPres may influence the folding state of the GPI-anchored PrP through incomplete templating and thus cause the induction of a toxic signal.
The second case in point involves the fungal prion [Het-s]. Non-productive templating may explain how the [Het-s] prion mediates heterokaryon incompatibility in the fungus Podospora anserina.60 Heterokaryon incompatability, a type of programmed cell death, results when two cells with incompatible geneotypes, het-s and het-S, fuse to form a mixed cytoplasm. The two alleles encode distinct sequence variants of the Het-s protein. One, HET-s, is able to form the [Het-s] prion, where as the other, HET-S, cannot adopt an amyloid conformation. The prion form of the HET-s allele by itself is completely benign. However, if the HET-S allele is expressed in the presence of the [Het-s] prion form it results in cell death. The interaction of HET-s protein with the [Het-s] prion form leads to the templated formation of additional non-toxic prion amyloid. On the other hand, we speculate that non-productive templating of the HET-S protein variant, which cannot form amyloid, by the [Het-s] prion form leads to the formation of a toxic misfolded species resulting in cell death (Fig. 2).
Conclusions
The protein deposits that are the hallmark of neurodegenerative diseases are now seen in a different light. Formerly viewed as the cause of cellular dysfunction and neuronal loss, especially intracellular protein deposits may be the product of a cellular process enabling cells to cope with the accumulation of misfolded and damaged proteins. This notion is supported by studies demonstrating that the deposition of damaged and misfolded proteins in inclusions enables mitotic cells, ranging from the unicellular organism Escherichia coli to human embryonic stem cells, to asymmetrically segregate the accumulated damage to the daughter cell with the shorter life expectancy.57,61–64 Based on our results in yeast, we suggest that inefficiencies in inclusion body and plaque formation, arising with accumulating protein damage, can result in the inception of toxic protein species due to non-productive templating. Further studies are needed to elucidate which types of protein deposits in the individual diseases are protective, how their formation is controlled and how they circumvent the formation of more toxic species, such as prefibrillar oligomers.
Acknowledgments
We would like to thank Peter Douglas, Vikram Khurana, Kent Matlack and Chris Pacheco for critical reading of the manuscript. We also thank Tom DiCesare for help with the illustrations. This work was supported by the National Institutes of Health (D.M.C. and S.L.) and a Howard Hughes Medical Institute Investigatorship (to S.L.).
References
- 1.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caughey B, Lansbury PT. Protofibrils, pores, fibrils and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 4.Roth M, Tomlinson BE, Blessed G. Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature. 1966;209:109–10. doi: 10.1038/209109a0. [DOI] [PubMed] [Google Scholar]
- 5.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 6.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 7.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, et al. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci. 2006;26:8160–7. doi: 10.1523/JNEUROSCI.4809-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 11.Ellison D, Love S, Chimelli L, Harding BN, Lowe J, Vinters HV. Neuropathology: A reference text of CNS pathology. St Louis: MOSBY; 2004. [Google Scholar]
- 12.Duyckaerts C, Dickson D. Neuropathology of Alzheimer’s Disease. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 47–65. [Google Scholar]
- 13.Hernandez F, Avila J. Tau aggregates and tau pathology. J Alzheimers Dis. 2008;14:449–52. doi: 10.3233/jad-2008-14414. [DOI] [PubMed] [Google Scholar]
- 14.Gotz J, Ittner LM, Fandrich M, Schonrock N. Is tau aggregation toxic or protective: a sensible question in the absence of sensitive methods? J Alzheimers Dis. 2008;14:423–9. doi: 10.3233/jad-2008-14410. [DOI] [PubMed] [Google Scholar]
- 15.Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14:453–7. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- 16.Bretteville A, Planel E. Tau aggregates: toxic, inert or protective species? J Alzheimers Dis. 2008;14:431–6. doi: 10.3233/jad-2008-14411. [DOI] [PubMed] [Google Scholar]
- 17.Ross CA. Introduction to Trinucleotide Repeat Disorders. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 226–8. [Google Scholar]
- 18.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–47. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 19.Cummings CJ, Zoghbi HY. Trinucleotide repeats: mechanisms and pathophysiology. Annu Rev Genomics Hum Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 20.Hedreen JC, Roos RAC. Huntington’s Disease. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 229–41. [Google Scholar]
- 21.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, et al. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–9. [PubMed] [Google Scholar]
- 23.Budka H, Head MW, Ironside JW, Gambetti P, Parchi P, Zeidler M, Tagliavini F. Sporadic Creutzfeldt-Jakob Disease. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 287–97. [Google Scholar]
- 24.Ironside JW, Head MW, Will RG. Variant Creutzfeldt-Jakob Disease. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 310–7. [Google Scholar]
- 25.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146:1239–54. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 28.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 29.Preisinger E, Jordan BM, Kazantsev A, Housman D. Evidence for a recruitment and sequestration mechanism in Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:1029–34. doi: 10.1098/rstb.1999.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, et al. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996;2:347–50. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 31.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 32.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 33.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 34.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 35.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–57. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 36.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 37.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, et al. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum Mol Genet. 2006;15:2743–51. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 39.Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci USA. 2006;103:4246–51. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 41.Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–28. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 42.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 43.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–47. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 44.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–9. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 45.Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci USA. 2007;104:4712–7. doi: 10.1073/pnas.0609241104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 47.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–21. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 48.Chiti F, Dobson CM. Protein misfolding, functional amyloid and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 49.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–7. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Duennwald ML, Roberts BE, Rozeboom LM, Zhang YL, Steele AD, et al. Direct and selective elimination of specific prions and amyloids by 4,5-dianilinophthalimide and analogs. Proc Natl Acad Sci USA. 2008;105:7159–64. doi: 10.1073/pnas.0801934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–72. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 52.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)] Cell. 2001;106:171–82. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 53.Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA. 2008;105:7206–11. doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sondheimer N, Lopez N, Craig EA, Lindquist S. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 2001;20:2435–42. doi: 10.1093/emboj/20.10.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–54. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 56.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–8. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 57.Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ, et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 2006;4:417. doi: 10.1371/journal.pbio.0040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rambold AS, Muller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–84. doi: 10.1038/emboj.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coustou-Linares V, Maddelein ML, Begueret J, Saupe SJ. In vivo aggregation of the HET-s prion protein of the fungus Podospora anserina. Mol Microbiol. 2001;42:1325–35. doi: 10.1046/j.1365-2958.2001.02707.x. [DOI] [PubMed] [Google Scholar]
- 61.Erjavec N, Larsson L, Grantham J, Nystrom T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21:2410–21. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindner AB, Madden R, Demarez A, Stewart EJ, Taddei F. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc Natl Acad Sci USA. 2008;105:3076–81. doi: 10.1073/pnas.0708931105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–3. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 64.Fuentealba LC, Eivers E, Geissert D, Taelman V, De Robertis EM. Asymmetric mitosis: Unequal segregation of proteins destined for degradation. Proc Natl Acad Sci USA. 2008;105:7732–7. doi: 10.1073/pnas.0803027105. [DOI] [PMC free article] [PubMed] [Google Scholar]