


Insight from Rejane Rua (left) and Dorian McGavern (right)
The mechanisms regulating wound injury and tissue repair involve complex enzymatic cascades and inflammatory processes, and new components are still being discovered. In this issue, Doni et al. report that the innate immune pattern recognition molecule (PRM), pentraxin 3 (PTX3), has a novel and nonredundant role in the orchestration of tissue repair and remodeling during wound healing.
The innate immune system is equipped with an array of PRMs that detect conserved microbial motifs and mobilize early defense against foreign invaders. This pattern recognition system is multifunctional and can also detect endogenous alarmins that are released from damaged tissue. Detection of alarmins, such as extracellular matrix components and nucleic acids, is known to facilitate wound-healing reactions, in part through the induction of inflammation. PTX3 is a secreted, fluid-phase PRM that is rapidly produced by stromal and myelomonocytic cells. Its functions include opsonization, regulation of the complement cascade, and antimicrobial resistance, among others.
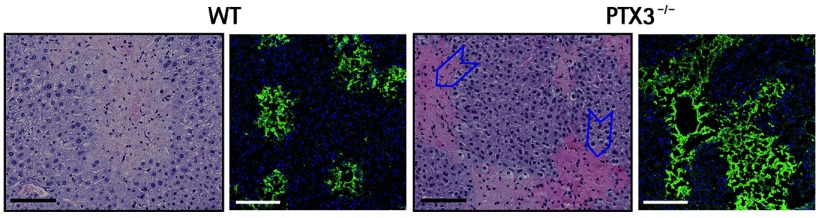
Hematoxylin-eosin and confocal images (green = fibrin; blue = nuclei) of fibrin in liver sections of wild-type (left) and Ptx3−/− (right) mice. Blue arrowheads indicate deposits of fibrin in necroinflammatory areas in the lobular spaces 48 hours after chronic injury. Bars, 100 µm.
Using four different models of tissue damage (including skin damage and liver and lung sterile tissue injury), the authors showed that PTX3 deficiency caused marked alterations in wound healing, including excessive fibrin accumulation, enhanced collagen deposition, epithelial hyperplasia, and defective formation of mature tissue. Interestingly, PTX3 expression was associated with wound-healing cells such as macrophages within the lesion, suggesting a role for this glycoprotein in tissue remodeling. Consistent with this theory, Ptx3−/− macrophages were less efficient at degrading fibrin matrix in vitro. Fibrin is a provisional extracellular matrix protein that is transiently deposited after tissue injury, but must be degraded for proper tissue remodeling and scar formation to occur.
To understand the link between PTX3 and fibrin matrix degradation, the authors conducted an elegant series of experiments involving tissue acidification showing that PTX3 serves as a molecular bridge between fibrin and plasminogen and promotes fibrinolysis. So, PTX3 appears to have an important role in fibrin breakdown, and tissue acidification serves as the switch that promotes this wound-healing activity following injury.
These data demonstrate that PRMs such as PTX3 can have different functions that depend on the tissue milieu and the manner by which they are activated. In fact, another recently published study from this same group in Cell showed that PTX3 also has anti-oncogenic properties linked to its ability to quench complement activation and reduce tumor infiltration by macrophages. We now know that induction of this protein has the potential to accelerate wound healing, ward off microbes, and suppress tumor growth. Given its ability to coordinate such diverse immune activities, PTX3 is a relevant and interesting target for therapeutic manipulation in damaged tissue as well as in a variety of diseases. Future avenues of research may uncover other unexpected roles for this fascinating molecule.
References
- Bonavita, E., et al. 2015. Cell. 160:700–714. [DOI] [PubMed] [Google Scholar]
- Doni, A., et al. 2015. J. Exp. Med. 10.1084/jem.20141268 [DOI] [Google Scholar]