

Abstract
Purpose
The identification of patients with metastatic triple-negative breast cancer (mTNBC) who are expected to benefit from platinum-based chemotherapy is of interest. We conducted a single-arm phase II clinical trial of single-agent platinum for mTNBC with biomarker correlates.
Patients and Methods
Patients with mTNBC received first- or second-line cisplatin (75 mg/m2) or carboplatin (area under the concentration-time curve 6) by physician's choice once every 3 weeks. Coprimary end points were objective response rate (RR) and response prediction by p63/p73 gene expression. Secondary and exploratory end points included toxicity assessment, RR in cisplatin versus carboplatin, and RR in molecularly defined subgroups, including BRCA1/2 mutation carriers.
Results
Patients (N = 86; 69 as first-line therapy) received cisplatin (n = 43) or carboplatin (n = 43). RR was 25.6% (95% CI, 16.8% to 36%) and was numerically higher with cisplatin (32.6%) than with carboplatin (18.7%). RR was 54.5% in patients with germline BRCA1/2 mutations (n = 11). In patients without BRCA1/2 mutations (n = 66), exploratory analyses showed that a BRCA-like genomic instability signature (n = 32) discriminated responding and nonresponding tumors (mean homologous recombination deficiency–loss of heterozygosity/homologous recombination deficiency–large-scale state transitions [HRD-LOH/HRD-LST] scores were 12.68 and 5.11, respectively), whereas predefined analysis by p63/p73 expression status (n = 61), p53 and PIK3CA mutation status (n = 53), or PAM50 gene expression subtype (n = 55) did not. Five of the six long-term responders alive at a median of 4.5 years lacked germline BRCA1/2 mutations, and two of them had increased tumor HRD-LOH/HRD-LST scores.
Conclusion
Platinum agents are active in mTNBC, especially in patients with germline BRCA1/2 mutations. A measure of tumor DNA repair function may identify patients without mutations who could benefit from platinum therapy agents. Prospective controlled confirmatory trials are warranted.
INTRODUCTION
Triple-negative breast cancer (TNBC), defined clinically as lacking estrogen receptor (ER) and progesterone receptor (PgR) expression and human epidermal growth factor receptor 2 (HER2) gene amplification, represents up to 20% of all breast cancers and is associated with a more aggressive clinical course in the metastatic setting compared with other breast cancer subtypes.1,2 Most TNBCs share common histologic and molecular features, including high grade, frequent TP53 mutation, and frequent expression of a so-called basal-like gene expression signature on hierarchical clustering analysis.3 Nonetheless, TNBC is a heterogeneous disease entity and is likely to represent multiple clinically and biologically distinct subgroups that are not yet clearly defined.4,5
Approximately 80% of breast cancers arising in BRCA1 mutation carriers are TNBCs, and these tumors are characterized by a defect in DNA double-strand break repair which is thought to render them particularly sensitive to interstrand cross-linking agents, including platinum analogs.6 Accordingly, high pathologic complete response (pCR) rates have been observed among BRCA1 carriers treated with cisplatin in the preoperative setting,7,8 and high response rates (RRs) were also observed among one small cohort of BRCA1 carriers treated with cisplatin in the metastatic setting.9 Evidence suggests that a group of sporadic TNBCs may also exhibit platinum sensitivity, possibly associated with a BRCA-like phenotype in a subset of patients.10,11 Collectively, these findings have suggested that platinum-based chemotherapy may be particularly effective for TNBC.
Enthusiasm for platinum treatment of TNBC is tempered by both retrospective analyses and small prospective studies that have yielded a mixed picture of whether patients with TNBC in general derive benefit from platinum.12 In the metastatic setting, reported RRs vary from 10% to more than 40% in prospective studies that included single-agent cisplatin or carboplatin.12,13 Such differences may reflect subtle differences in patient selection in these trials, but they also speak to the need to identify subsets of patients with TNBCs who are likely to benefit from platinum-based therapy. To address this need, we conducted a multicenter phase II clinical trial (TBCRC009: Platinum for Triple-Negative Metastatic Breast Cancer and Evaluation of p63/p73 as a Biomarker of Response) of cisplatin or carboplatin treatment in the first- or second-line setting for metastatic TNBC (mTNBC). We interrogated the BRCA1/2 pathway through germline BRCA1/2 sequencing and tumor somatic genomic analysis, and we evaluated established TNBC subsets defined by gene expression and p53 and PIK3CA mutation.
PATIENTS AND METHODS
Patients
Eligible patients had metastatic or locally recurrent unresectable TNBC. ER, PgR, and HER2 status were determined locally and were not centrally reviewed. Paraffin blocks or unstained slides of tumor were required from all patients for correlative studies. Additional eligibility criteria included measurable disease according to RECIST (Response Evaluation Criteria in Solid Tumors) 1.0, Eastern Cooperative Oncology Group performance status ≤ 2, life expectancy more than 12 weeks, normal organ and bone marrow function, no more than one prior cytotoxic chemotherapy for metastatic disease, no prior cisplatin or carboplatin therapy, and no active brain metastases. All patients provided written informed consent, and the study was approved by the institutional review boards at each participating site.
Study Design and Treatment Plan
This open-label, single-arm, two-stage phase II clinical trial was conducted at eight Translational Breast Cancer Research Consortium centers in the United States. Eligible patients were assigned at the discretion of their treating physician to receive either cisplatin 75 mg/m2 (cohort 1) or carboplatin at area under the time-concentration curve 6 (cohort 2) once every 3 weeks. Accrual to each cohort was limited to ensure an equal number of patients in each cohort. At the discretion of the treating physician after cycle 1, growth factors were permitted, and treatment cycles could be extended to 4 weeks. Oral magnesium and potassium supplementation was encouraged. Tumor response was assessed locally by computed tomography according to RECIST 1.0 every two cycles for the first four cycles and every three cycles thereafter. Patients were treated until disease progression, unacceptable toxicity, or withdrawal of consent. Treatment delay up to 3 weeks and two dose reductions were permitted. Blood was collected from all patients for germline BRCA1/2 analysis. Patients were observed annually until death or the October 2013 data cutoff.
Further details of all correlative studies are described in the Data Supplement. All correlative studies were performed blinded to clinical end points. The study was designed in accordance with REMARK biomarker guidelines, recognizing that homologous recombination deficiency (HRD) analysis was developed subsequent to study initiation.14
RESULTS
Patient Characteristics
Patient demographics and baseline characteristics are provided in Table 1. Between June 2007 and October 2010, 86 patients were accrued. The original study design called for 41 patients to be treated with first-line cisplatin. After a total of 15 patients were accrued to first-line cisplatin therapy, a protocol amendment was activated on July 28, 2008, that increased the planned accrual to 82 patients and expanded protocol therapy to include carboplatin or cisplatin according to physician's choice as first- or second-line therapy. A total of 43 patients were accrued to each treatment cohort. The majority of patients (86%) had received prior adjuvant or neoadjuvant therapy, most with an anthracycline (74%) and taxane (78%). Patients had a median of two sites of disease, and 45% of patients had at least three sites. Lymph nodes were the most common metastatic site (63%), followed by lungs (51%), liver (29%), bone (29%), and skin (19%).
Table 1.
Patient Demographics and Baseline Characteristics (N = 86)
| Characteristic | No. | % |
|---|---|---|
| Age, years | ||
| Median | 52 | |
| Range | 30-78 | |
| < 40 | 13 | 15 |
| 40-65 | 63 | 73 |
| > 65 | 10 | 12 |
| Race/ethnicity | ||
| White | 69 | 80 |
| African American | 7 | 8 |
| Asian | 4 | 5 |
| > One or other | 6 | 7 |
| ECOG PS (median, 0) | ||
| 0 | 55 | 64 |
| 1 | 20 | 23 |
| 2 | 5 | 6 |
| Not reported | 6 | 7 |
| Sites of metastases | ||
| Lymph nodes | 54 | 63 |
| Lung | 44 | 51 |
| Bone | 25 | 29 |
| Liver | 25 | 29 |
| Skin | 16 | 19 |
| Brain | 4 | 5 |
| Stage at initial diagnosis | ||
| 1 | 10 | 12 |
| 2 | 32 | 37 |
| 3 | 37 | 43 |
| 4 | 7 | 8 |
| Prior chemotherapy | ||
| Adjuvant/neoadjuvant | 74 | 86 |
| Anthracycline | 65 | 74 |
| Taxane | 67 | 78 |
| Metastatic | 17 | 20 |
| Treatment cohort | ||
| Cisplatin (cohort 1) | 43 | 50 |
| Carboplatin (cohort 2) | 43 | 50 |
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Efficacy
The overall RR was 25.6%, including three complete responses (CRs) and 19 partial responses (Table 2). An additional four patients (4.7%) had stable disease for more than 6 months. The median number of cycles delivered was four (range, one to 23 cycles), and 12 patients (14%) received more than 10 cycles of therapy. Preplanned exploratory subgroup analysis included determination of RR by line of therapy and by chemotherapy agent. The RR was 29% with first-line therapy and 11.8% with second-line therapy (P = .22). Cisplatin therapy had an RR of 32.6% compared with 18.6% for carboplatin (P = .22). At a median follow-up of 49.9 months, 75 patients had died; the median progression-free survival (PFS) was 2.9 months and overall survival (OS) was 11 months (Figs 1A and 1C). There was, however, considerable clinical heterogeneity; by the time of the first tumor assessment at 6 weeks, 33% of patients had progressed, but 25% of patients had a PFS beyond 6 months.
Table 2.
Treatment Response and Exploratory Subgroup Analysis
| RECIST Response | No. of Patients | % | 95% CI | Response |
|---|---|---|---|---|
| Overall RR (N = 86) | 22 | 25.6 | 16.8 to 36.1 | |
| CR | 3 | 3.5 | ||
| PR | 19 | 22.1 | ||
| SD > 6 months | 4 | 4.7 | ||
| PD | 57 | 66.3 | ||
| NE | 3 | 3.5 | ||
| RR by subgroup | ||||
| BRCA1/2 status | ||||
| Mutated (n = 11) | 6 | 54.5 | 23.4 to 83.3 | 6 PR |
| Wild type (n = 66) | 13 | 19.7 | 10.9 to 31.3 | 10 PR, 3 CR |
| Unknown (n = 9) | 3 | 33.3 | 7.5 to 70.1 | 3 PR |
| Cisplatin (n = 43) | 14 | 32.6 | 19.1 to 48.5 | |
| First line (n = 34) | 12 | 35.3 | 10 PR, 2 CR | |
| Second line (n = 9) | 2 | 22.2 | 2 PR | |
| Carboplatin (n = 43) | 8 | 18.6 | 8.4 to 33.4 | |
| First line (n = 35) | 8 | 22.9 | 7 PR, 1 CR | |
| Second line (n = 8) | 0 | 0 | ||
| First line (n = 69) | 20 | 29.0 | ||
| Second line (n = 17) | 2 | 11.8 |
Abbreviations: CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; RR, response rate; SD, stable disease.
Fig 1.

(A) Progression-free survival and (C) overall survival estimates for all 86 enrolled patients. (B) Progression-free survival and (D) overall survival of BRCA1/2 mutation (MUT) carriers versus BRCA1/2 wild type (WT). Hazard ratios (HRs) and 95% CIs estimated by the Kaplan-Meier method. Patients were censored at last follow-up or at time of change in therapy without progression.
Toxicity
Single-agent cisplatin or carboplatin had generally mild toxicity, with fatigue, nausea, electrolyte abnormalities, and hematologic toxicity among the most common (Data Supplement). Most events were similar between the two cohorts. However, thrombocytopenia was more common with carboplatin (47% v 19%). Anemia (81% v 65%), neutropenia (49% v 35%), hypomagnesemia (42% v 23%), tinnitus (37% v 0%), and anorexia (26% v 9%) were more common with cisplatin. Grade 3 and 4 adverse events were rare and generally similar between cisplatin and carboplatin. Grade 3 and 4 adverse events occurring in at least 5% of all patients included fatigue, neutropenia, dyspnea, anemia, hyperglycemia, and hyponatremia (Data Supplement). No treatment-related deaths were reported. Ten patients discontinued study treatment for treatment-related toxicity, including neuropathy (5), fatigue (4), allergic reaction (3; all with carboplatin), renal dysfunction (2), transaminitis (1), and ototoxicity (1).
p63/p73 Ratio and Response
A coprimary end point of this study was prediction of treatment response through analysis of messenger RNA expression for the two p53-related genes p63 (TP63) and p73 (TP73). Preclinical data suggests that the ΔNp63 isoform functions as a survival factor in TNBC and other cancers, in part by inhibiting the proapoptotic p73 isoform TAp73.15 Applying a prespecified ΔNp63/TAp73 ratio cutoff more than 2 and by using the same methodology as in a prior study,8 we did not observe a significant association of this ratio with RR or clinical benefit among 61 evaluable tumors (Data Supplement, and data not shown). However, we did note the association of this ratio with clinical benefit among the small group of patients (n = 7) who presented with de novo metastatic disease and therefore had not received prior therapy (Data Supplement).
BRCA1/2 Status and Response
The status of germline BRCA1 and BRCA2 was assessable in 77 of 86 patients. Eleven patients with germline BRCA1/2 deleterious mutations were identified (Table 2) and, as anticipated, most harbored BRCA1 mutations (nine of 11). These patients roughly matched the cohort as a whole in terms of prior adjuvant therapy (10 of 11) and first-line therapy (eight of 11), although a higher fraction received cisplatin than carboplatin (seven of 11). Individuals with BRCA1/2 mutations were more likely to achieve a response than those without mutations (54.5% v 19.7%; 95% CI, 23.4% to 83.3% v 10.9% to 31.3%; P = .022; Table 2). Notably, however, responses in these BRCA1/2 carriers were not durable, none achieved a CR, and PFS was not significantly different between carriers and noncarriers (median 3.3 v 2.8 months; P = .92; Fig 1B). No significant difference in OS was observed between carriers and noncarriers (median 13.7 v 10.9 months; P = .58; Fig 1D). Among all BRCA1/2 carriers, the two BRCA2 carriers demonstrated the longest PFS and the longest OS (data not shown).
To extend the characterization of the BRCA1/2 pathway, we interrogated available tumors from these patients (n = 32) for somatic BRCA1/2 mutations, and in addition, we used two assays that measured discrete patterns of BRCA-associated genomic instability, originally derived through comparison of BRCA1/2 versus nonmutant tumors.10,11 The HRD large-scale state transition (HRD-LST) assay identifies chromosome breaks (translocations, inversions, or deletions) resulting in adjacent segments of at least 10 Mb,10 whereas the HRD loss of heterozygosity (HRD-LOH) assay identifies regions showing LOH of intermediate size.11 As predicted, values for these two assays among all the tumors were significantly correlated (Data Supplement). Also as expected, significantly higher values for HRD-LST and for the mean of HRD-LST plus HRD-LOH were observed in tumors from patients with BRCA1/2 mutations (including one identified somatic BRCA1 mutation) than in tumors from noncarriers (carrier v noncarrier mean HRD-LST plus HRD-LOH, 13.81 v 6.52; P = .0089; Fig 2A). Most notably, tumors from patients who responded to platinum but lacked germline mutations exhibited higher values for both of the assays and a statistically significant difference for the combined values (response v no response mean HRD-LST plus HRD-LOH, 12.68 v 5.11; P = .0318; Fig 2B). Greater correlation was observed when patients with BRCA1 methylation were excluded from the BRCA1/2 wild-type analysis (Data Supplement) and when response was analyzed as a continuous variable (Data Supplement). Thus, responses of mTNBC to platinum therapy in this trial are associated with either BRCA1/2 mutation or with a tumor-specific genomic instability pattern characteristic of BRCA1/2 deficiency.
Fig 2.

High homologous recombination deficiency–loss of heterozygosity (HRD-LOH) and homologous recombination deficiency–large-scale state transition (HRD-LST) scores are associated with BRCA1/2 mutation and with platinum sensitivity. Primary tumor DNA was analyzed for two distinct genomic aberration patterns (HRD-LST, HRD-LOH) as described in Results. Higher scores indicate increased aberrations. Horizontal lines show mean values ± SEM. (A) BRCA1/2-mutant (MUT) versus wild-type (WT) tumors. (B) Tumors of responding patients (complete response [CR] plus partial response [PR]) versus nonresponding patients (stable disease [SD] plus progressive disease [PD]), excluding BRCA1/2-mutant tumors. Unpaired t test (Welch's correction) was used to calculate P values. Gray circle indicates germline BRCA1 mutation; blue circle indicates patient without germline or tumor somatic BRCA1/2 mutation, BRCA1 methylation, or long-term response; gold diamond indicates somatic BRCA1 mutation; red diamond indicates germline BRCA2 mutation; gold triangle indicates BRCA1 promoter methylation; gold star indicates long-term responder.
Intrinsic Subtyping and Other Gene Expression Signatures
We performed global gene expression profiling on available tumors (n = 55) to determine whether predefined molecular subsets of TNBC benefited selectively from platinum. We first used the PAM50 gene set to determine intrinsic subtypes among these TNBCs.16 In line with previous studies that used similar methodology,3 62% (34 of 55) of these were basal-like tumors. We observed a nonsignificant trend toward increased RR in basal versus nonbasal TNBC (Fig 3 and Data Supplement), and no differences in PFS or OS after platinum therapy (Data Supplement). We next analyzed other previously described gene expression signatures reported by Lehmann et al17 to subset TNBC (Fig 3).19 Although most were not statistically associated with response to platinum in this cohort, the luminal androgen receptor signature showed a significant negative association with RR (Fig 3 and Data Supplement) but no association with PFS or OS (data not shown).
Fig 3.
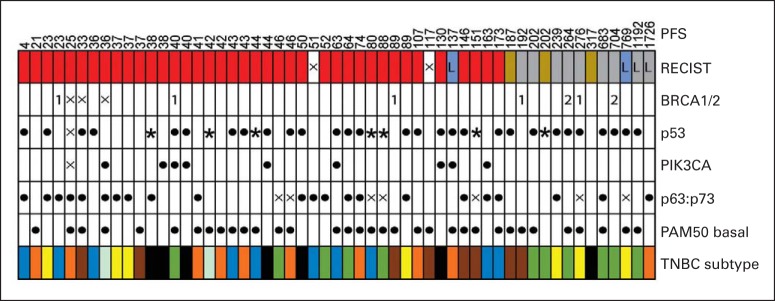
Summary of gene expression and mutational analysis. Each column represents one patient. Row 1: Progression-free survival (PFS) in days. Row 2: The four RECIST response categories shown here are complete response (light blue), partial response (gray), stable disease (gold), and progressive disease (red). Row 3: Germline BRCA1 (1) and BRCA2 (2); no mutation (blank). Row 4: Tumor p53 mutation analyses; no mutation (blank); missense mutation ([solid dot]); nonsense/splice-site mutation ([solid star]). Row 5: Tumor PIK3CA mutation analyses; no mutation (blank); missense mutation ([solid dot]). Row 6: ΔNp63:TAp73 messenger RNA expression ratio (ie, p63:p73) > 2 ([solid dot]); < 2 (blank).8 Row 7: PAM50 basal signature according to Nielson et al18; basal ([solid dot]); nonbasal (blank). Row 8: Gene expression signatures derived from Lehmann et al17: unstable (dark blue), immunomodulatory (orange), mesenchymal stem-like (yellow), basal-like 1 (brown), basal-like 2 (light blue), luminal androgen receptor (black), and mesenchymal (green). (L) Long-term responders; (X) not available.
Tumor Mutational and Genomic Analyses
The two genes most commonly subject to somatic mutation in TNBC are the p53 tumor suppressor and the phosphatidylinositol 3-kinase (PI3K) catalytic subunit gene PIK3CA.3 Accordingly, we sequenced these genes in available tumors (n = 53). As expected, p53 mutation was observed in approximately 60% of patients (36 of 52), including the majority of BRCA1/2-associated and basal-like tumors (Fig 3 and data not shown). Although a somewhat larger proportion of patients with p53-mutant tumors responded to platinum (Data Supplement), there was a consistent trend toward inferior OS for those with p53-mutant versus wild-type tumors (Data Supplement).20 As expected, relatively few tumors (nine of 53) harbored PIK3CA mutations, and all but one of these patients experienced progressive disease as best response (Fig 3 and Data Supplement).
Patients With Durable Responses and No Active Disease
A small but notable subset of 6 long-term responding (LTR) patients remain alive, progression free, and not receiving any therapy at a median of more than 4 years after platinum treatment (Table 3). Each of these patients achieved a partial response or CR to protocol therapy; two of six eventually discontinued treatment because of toxicity and received no additional therapy, although the others went on to consolidative local or systemic therapy. The majority (four of six) had received prior standard adjuvant chemotherapy before systemic relapse, and all received platinum as first-line therapy. Sites of disease varied (Table 3), but no patients had liver or bone involved. None of the tested (five of six) LTR patients harbored germline BRCA1/2 mutations or rearrangements. However, the two tumors from LTR patients available for HRD-LST/HRD-LOH analysis did show values in line with those of other responding patients (Fig 2). None of the other molecular analyses carried out in this cohort identified these patients (Table 3).
Table 3.
Clinical, Genetic, and Tumor Molecular Features of Long-Term Responders
| Patient | BRCA | Subtype | PIK3CA | p53 | p63/p73 | OS (months)* | Adjuvant Therapy | Therapy | Best Response | Site of Disease | Therapy After Platinum Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | WT | B | Missense mutation | Missense mutation | < 2 | 69 | None | Cisplatin | CR | Breast, lymph nodes | Surgery, chemotherapy, and radiation |
| 28 | WT | N | WT | WT | > 2 | 58 | Anthracycline-taxane | Cisplatin | PR | Lymph nodes | None |
| 45 | WT | X | WT | WT | X | 48 | None | Cisplatin | CR | Lung, breast, lymph nodes | Surgery, chemotherapy, and radiation |
| 53 | WT | B | WT | Missense mutation | < 2 | 40 | Anthracycline-taxane | Cisplatin | PR | Lung | None |
| 69 | X | X | X | X | X | 41 | Anthracycline-taxane | Carboplatin | PR | Lung, lymph nodes | Radiation |
| 77 | WT | N | WT | Missense mutation | X | 34 | Anthracycline-taxane | Carboplatin | CR | Lymph nodes | Stereotactic radiosurgery to brain metastasis, chemotherapy |
Abbreviations: B, basal (PAM50); CR, complete response; N, nonbasal; OS, overall survival; PR, partial response; WT, wild type; X, not available/not assessed.
To data lock in October 2013.
DISCUSSION
This study was designed to test the efficacy of single-agent platinum-based chemotherapy for mTNBC and to determine whether defined genetic and molecular disease subsets exhibited significantly different RRs. The observed RR of 25.6% was higher than the 10% RR seen by using the same dose of cisplatin in a randomized phase II trial of cisplatin plus cetuximab for mTNBC.21 Similarly, in the TBCRC (Translational Breast Cancer Research Consortium) 001 trial for mTNBC, an RR of 16% was observed with the combination of carboplatin and cetuximab after progression on cetuximab alone.22 Although we observed a higher RR for cisplatin versus carboplatin in our study, this difference must be interpreted with caution, given that our study was a nonrandomized trial. Most likely, the explanation for the differences in RR among these trials is multifactorial and reflects diversity in patient populations or molecular subsets. For example, given the significant difference we report in RRs between BRCA1/2 wild-type and mutation carriers, small differences in the number of carriers (not reported in the other two studies) could have an impact on the apparent overall RRs.
Compared with BRCA1/2 noncarriers, mutation carriers in our study exhibited a significantly higher RR but no difference in PFS, likely explained by relatively rapid subsequent disease progression in carriers and the presence of a subset of noncarriers who exhibited durable responses. Both the RR and PFS among carriers were substantially lower than those observed in a small trial of single-agent cisplatin in Poland that enrolled 20 BRCA1 carriers with metastatic breast cancer.9 Possible explanations for these differences include unidentified patient-specific factors and the inclusion of patients with ER-positive and/or PgR-positive tumors in the prior study (two of which were among four patients who exhibited prolonged responses). Nonetheless, both studies support a selective sensitivity of BRCA1/2-associated tumors to platinum-based therapy, even in the advanced setting.
Underscoring the functional link between the BRCA1/2 pathway and platinum response in TNBC, we found that responses among those who did not carry mutations were associated with the presence of tumor genomic instability patterns characteristic of BRCA1/2-mutant tumors. This finding is in line with other recent work reporting the ability of a genomic signature derived from BRCA1/2-mutant tumors to predict clinical benefit from platinum-containing therapy for breast cancer.23 We used two independent assays (HRD-LST and HRD-LOH) that were both defined through analysis of BRCA1/2-mutant (including BRCA1-methylated) versus wild-type tumors.10,11 Accordingly, we found that the results of the two assays were highly correlated, and they identified the tumors of BRCA1/2 mutation carriers and those of responding patients without germline BRCA1/2 mutations.
This is among the first studies to evaluate the HRD-LST and HRD-LOH biomarkers in a metastatic population. Given the lack of any predictive markers for chemotherapy responses in noncarriers with TNBC, such assays could have major clinical impact. Our data suggest that HRD-LOH and HRD-LST assays might be useful in identifying a subset of patients who are unlikely to benefit from platinum-based therapy; high HRD scores identified responders (high sensitivity) but also some nonresponders (low specificity), whereas low HRD scores were strongly associated with lack of response. Taken together, our findings suggest that platinum sensitivity may be largely or wholly a result of a defect in a BRCA1/2-related pathway for abnormal DNA repair. Future randomized studies incorporating HRD analysis will be needed to determine whether such biomarkers are limited to predicting benefit specifically from platinum or more generally to other cytotoxic therapy.
A coprimary end point of the study was the ability of a gene expression biomarker, the ΔNp63:TAp73 ratio, to predict responses to platinum-based therapy. In a prior study of preoperative cisplatin treatment for TNBC, a prespecified cutoff ratio of ΔNp63:TAp73 more than 2 was observed in 75% of tumors (3 of 4) with pathologic CR (pCR) compared with 27% of tumors (three of 11) with poor response.8 Although this end point was not met in this study, it is relevant to note that patients with early-stage TNBC who obtain a pCR (and thus for whom this biomarker might be predictive) are unlikely to develop metastatic disease and therefore would be largely excluded from our cohort. Collectively, these observations may serve as a cautionary note for the application of biomarkers developed in the early-stage setting to the metastatic population.
Established molecular biologic tumor features were of limited value in predicting response to therapy in our small study. Most notably, none of these established markers, including p53 and PIK3CA mutations, basal subtype, or p63:p73 ratio identified the subset of patients who exhibited prolonged responses and remain alive and progression free to date. Five of these six patients were tested, and all lacked germline BRCA1/2 mutations, although the two tumors from this group tested did have HRD-LST and HRD-LOH scores in the range of other responding patients (Fig 2). Notably, the ability of patients who have relapsed after standard adjuvant chemotherapy (four of the six LTRs) to achieve LTRs with platinum agents supports the notion that platinum sensitivity may be a distinct form of chemotherapy sensitivity.
In summary, this work demonstrates the meaningful activity of platinum-based chemotherapy for mTNBC. Our exploratory findings in tumors lacking BRCA1/2 mutations suggest a genomic instability pattern associated with defects in the BRCA1/2 pathway that may predict response to therapy (platinum, in this study). Properly designed prospective controlled trials that use analytically validated assays and predefined cutoffs are warranted to test the clinical utility of measures of DNA repair status to predict responsiveness to platinum and other DNA-damaging agents.
Supplementary Material
Acknowledgment
We thank Charles Perou and Chris Fan for conducting PAM50 gene expression subtype analysis, Carrie Quadrino for regulatory assistance and trial management, Ranjit Shetty, Susan Boivert, and Hector Lopez for help with correlative studies, Nora Horick for help with statistical analyses, and the patients who participated in this trial. We also thank the Translational Breast Cancer Research Consortium and its three founding partners (the Avon Foundation, the Breast Cancer Research Foundation, and Susan G. Komen for the Cure), Robert Howe, Lee Kennedy, and the Boston Chapter of Golfers Against Cancer for research support.
Glossary Terms
- basal-like:
a gene expression pattern displayed by a subset of breast cancers (approximately 15%-20%), usually in the setting of a triple-negative phenotype, with increased expression of a variety of cytokeratins and other markers such as epidermal growth factor receptor typically found in the basilar epithelium of the breast.
- BRCA1:
a tumor suppressor gene known to play a role in repairing DNA breaks. Mutations in this gene are associated with increased risks of developing breast or ovarian cancer.
- BRCA2:
a tumor suppressor gene whose protein product is involved in repairing chromosomal damage. Although structurally different from BRCA1, BRCA2 has cellular functions similar to BRCA1. BRCA2 binds to RAD51 to fix DNA breaks caused by irradiation and other environmental agents. Also known as the breast cancer 2 early onset gene.
- cisplatin:
an inorganic platinum agent (cis-diamminedichloroplatinum) with antineoplastic activity. Cisplatin forms highly reactive, charged, platinum complexes, which bind to nucleophilic groups such as GC-rich sites in DNA, inducing intrastrand and interstrand DNA cross-links as well as DNA-protein cross-links. These cross-links result in apoptosis and cell growth inhibition. Carboplatin and oxaliplatin are other members of this class.
- homologous recombination:
genetic recombination whereby nucleotide sequences are exchanged between two similar or identical strands of DNA to facilitate accurate repair of DNA double-strand breaks.
- RECIST (Response Evaluation Criteria in Solid Tumors):
a model proposed by the Response Evaluation Criteria Group by which a combined assessment of all existing lesions, characterized by target lesions (to be measured) and nontarget lesions, is used to extrapolate an overall response to treatment.
- triple-negative breast cancer (TNBC):
breast tumors that are negative for estrogen and progesterone receptor expression and that also underexpress HER-neu.
Footnotes
Supported by The Translational Breast Cancer Research Consortium and its three foundation partners (The AVON Foundation, The Breast Cancer Research Foundation, and Susan G. Komen for the Cure); National Cancer Institute (NCI)-AVON Partners for Progress Award 3P30CA06516-45S2 (S.J.I.); Golfers Against Cancer, Boston Chapter; the Tracey Davis Breast Cancer Research Fund; the Clinical Investigator Training Program of Harvard-Massachusetts Institute of Technology Health Sciences and Technology; and the Dana-Farber/Harvard Cancer Center Tumor Imaging Metrics Core.
Presented in part at the 47th Annual Meeting of the American Society of Clinical Oncology (ASCO), Chicago, IL, June 3-7, 2011; the 50th Annual Meeting of the ASCO, Chicago, IL, May 30-June 3, 2014; and the Clinical and Translational Research Center-American Association for Cancer Research 35th Annual San Antonio Breast Cancer Symposium, San Antonio, TX, December 4-8, 2012.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00483223.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Steven J. Isakoff, Paula D. Ryan, Eric P. Winer, Paul E. Goss, Leif W. Ellisen
Provision of study materials or patients: Steven J. Isakoff, Erica L. Mayer, Tiffany A. Traina, Lisa A. Carey, Karen J. Krag, Hope S. Rugo, Minetta C. Liu, Vered Stearns, Steven E. Come, Judy E. Garber, Paula D. Ryan, Eric P. Winer
Collection and assembly of data: Steven J. Isakoff, Lei He, Darrell R. Borger, Leif W. Ellisen
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Steven J. Isakoff
Consulting or Advisory Role: Myriad Genetics, BIND Biosciences
Research Funding: Genentech, PharmaMar, AbbVie
Travel, Accommodations, Expenses: Genentech, Myriad Genetics
Erica L. Mayer
Research Funding: Myriad Genetics, Pfizer, Eisai
Lei He
Employment: AbbVie
Tiffany A. Traina
Consulting or Advisory Role: Eisai, Genentech, ProStrakan, Halozyme Therapeutics, Celgene, Medivation
Research Funding: Medivation, AstraZeneca, Eisai, Janssen Biotech, Genentech, Novartis
Lisa A. Carey
Research Funding: gsk, Genentech/Roche
Karen J. Krag
No relationship to disclose
Hope S. Rugo
Honoraria: Genomic Health
Speakers' Bureau: Genomic Health
Research Funding: Plexxikon (Inst), Macrogenics (Inst), Ontology for Biomedical Investigations (OBI) (Inst), Eisai (Inst), Pfizer (Inst), Novartis (Inst), Lilly (Inst), GlaxoSmithKline (Inst), Genentech (Inst), Celsion (Inst), Nektar (Inst), Merck (Inst)
Travel, Accommodations, Expenses: Novartis, Nektar, Roche/Genentech, OBI, Mylan
Minetta C. Liu
Research Funding: Eisai (Inst), Seattle Genetics (Inst), Celgene (Inst), Veridex (Inst), Novartis (Inst), Roche/Genentech (Inst)
Vered Stearns
Research Funding: AbbVie, Celgene, Merck, MedImmune, Novartis, Pfizer, Puma Biotechnology
Steven E. Come
Consulting or Advisory Role: Genentech/Roche
Expert Testimony: Pfizer
Kirsten M. Timms
Employment: Myriad Genetics
Stock or Other Ownership: Myriad Genetics
Research Funding: Myriad Genetics
Patents, Royalties, Other Intellectual Property: Myriad Genetics
Anne-Renee Hartman
Employment: Myriad Genetic Laboratories
Stock or Other Ownership: Myriad Genetics
Darrell R. Borger
Consulting or Advisory Role: Bioreference Laboratories
Travel, Accommodations, Expenses: Cambridge Healthtech Institute
Dianne M. Finkelstein
No relationship to disclose
Judy E. Garber
Consulting or Advisory Role: Pfizer (I), Novartis (I), Sequenom
Research Funding: Myriad Genetics, Novartis (I), Pfizer (I)
Paula D. Ryan
Employment: McKesson (I)
Leadership: McKesson (I)
Stock or Other Ownership: McKesson (I)
Travel, Accommodations, Expenses: McKesson (I)
Eric P. Winer
Research Funding: Genentech/Roche (Inst), Novartis (Inst)
Other Relationship: Genentech, Novartis
Paul E. Goss
Honoraria: GlaxoSmithKline
Leif W. Ellisen
Consulting or Advisory Role: BioReference Laboratories
Patents, Royalties, Other Intellectual Property: I hold a patent on use of p63/p73 expression as a biomarker to predict response to platinum chemotherapy
REFERENCES
- 1.Kassam F, Enright K, Dent R, et al. Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clin Breast Cancer. 2009;9:29–33. doi: 10.3816/CBC.2009.n.005. [DOI] [PubMed] [Google Scholar]
- 2.Hudis CA, Gianni L. Triple-negative breast cancer: An unmet medical need. Oncologist. 2011;16:1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 3.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Criscitiello C, Azim HA, Jr, Schouten PC, et al. Understanding the biology of triple-negative breast cancer. Ann Oncol. 2012;23:vi13–vi18. doi: 10.1093/annonc/mds188. [DOI] [PubMed] [Google Scholar]
- 5.Turner NC, Reis-Filho JS. Tackling the diversity of triple-negative breast cancer. Clin Cancer Res. 2013;19:6380–6388. doi: 10.1158/1078-0432.CCR-13-0915. [DOI] [PubMed] [Google Scholar]
- 6.Turner NC, Tutt AN. Platinum chemotherapy for BRCA1-related breast cancer: Do we need more evidence? Breast Cancer Res. 2012;14:115. doi: 10.1186/bcr3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrski T, Gronwald J, Huzarski T, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 8.Silver DP, Richardson AL, Eklund AC, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrski T, Dent R, Blecharz P, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012;14:R110. doi: 10.1186/bcr3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Popova T, Manié E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 11.Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M, Mo QG, Wei CY, et al. Platinum-based chemotherapy in triple-negative breast cancer: A meta-analysis. Oncol Lett. 2013;5:983–991. doi: 10.3892/ol.2012.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isakoff SJ. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010;16:53–61. doi: 10.1097/PPO.0b013e3181d24ff7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies. J Clin Oncol. 2005;23:9067–9072. doi: 10.1200/JCO.2004.01.0454. [DOI] [PubMed] [Google Scholar]
- 15.Leong CO, Vidnovic N, DeYoung MP, et al. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest. 2007;117:1370–1380. doi: 10.1172/JCI30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res. 2010;16:5222–5232. doi: 10.1158/1078-0432.CCR-10-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakha EA, Elsheikh SE, Aleskandarany MA, et al. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Clin Cancer Res. 2009;15:2302–2310. doi: 10.1158/1078-0432.CCR-08-2132. [DOI] [PubMed] [Google Scholar]
- 21.Baselga J, Gómez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013;31:2586–2592. doi: 10.1200/JCO.2012.46.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carey LA, Rugo HS, Marcom PK, et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30:2615–2623. doi: 10.1200/JCO.2010.34.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vollebergh MA, Lips EH, Nederlof PM, et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014;16:R47. doi: 10.1186/bcr3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.