

Abstract
NMR spectra of human lymphotactin (hLtn), obtained under various solution conditions, have revealed that the protein undergoes a major conformational rearrangement dependent on temperature and salt concentration. At high salt (200 mM NaCl) and low temperature (10 °C), hLtn adopts a chemokine-like fold, which consists of a three-stranded antiparallel β-sheet and a C-terminal α-helix (Kuloğlu, E. S., McCaslin, D. R., Kitabwalla, M., Pauza, C. D., Markley, J. L., and Volkman, B. F. (2001) Biochemistry 40, 12486–12496). We have used NMR spectroscopy, sedimentation equilibrium, and intrinsic fluorescence to monitor the reversible conformational change undergone by hLtn as a function of temperature and ionic strength. We have used two-, three- and four-dimensional NMR spectroscopy of isotopically enriched protein samples to determine structural properties of the conformational state stabilized at 45 °C and 0 mM NaCl. Patterns of NOEs and 1Hα and 13C chemical shifts show that hLtn rearranges under these conditions to form a four-stranded, antiparallel β-sheet with a pattern of hydrogen bonding that is completely different from that of the chemokine fold stabilized at 10 °C and 200 mM NaCl. The C-terminal α-helix observed at 10 °C and 200 mM NaCl, which is conserved in other chemokines, is absent at 45 °C and no salt, and the last 38 residues of the protein are completely disordered, as indicated by heteronuclear 15N-1H NOEs. Temperature dependence of the tryptophan fluorescence of hLtn in low and high salt confirmed that the chemokine conformation is stabilized by increased ionic strength. Sedimentation equilibrium analytical ultracentrifugation showed that hLtn at 40 °C in the presence of 100 mM NaCl exists mainly as a dimer. Under near physiological conditions of temperature, pH, and ionic strength, both the chemokine-like and non-chemokine-like conformations of hLtn are significantly populated. The functional relevance of this structural interconversion remains to be elucidated.
Chemokines are small chemoattractant proteins that facilitate leukocyte migration and activation through binding to their seven-transmembrane helix G protein-coupled receptors and play a role in homeostasis, inflammation, and disease (1–3). Chemokine gradients formed through their interaction with cell surface GAGs,1 together with chemokine oligomerization, are thought to be important in leukocyte recruitment (4). Chemokines are also involved in transendothelial migration of leukocytes, maturation of leukocytes, traffic and homing of lymphocytes, development of lymphoid tissues, and angiogenesis (5–10). Because of the broad range of their immunoregulatory roles, chemokines and their receptors are targets for drug development for control of allergic and autoimmune diseases as well as for HIV infection, because entry of the virus into cells is dependent on binding to a chemokine receptor.
Chemokines are divided into four subclasses on the basis of the number of conserved cysteine residues and their spacing. Most of the ~50 known chemokines belong to either the CC or CXC subclass. The two other subclasses of chemokine each have a single known member: fractalkine for the CX3C class and lymphotactin for the C class (11, 12). Three-dimensional structures determined for a variety of chemokines have revealed a conserved, disulfide-stabilized chemokine fold that consists of a three-stranded antiparallel β-sheet and a C-terminal α-helix. Some CXC and CC chemokines form dimers, and while the tertiary structure of the subunits is invariant, substantial differences have been observed in quaternary structure (13–23). In CXC-class chemokines, the dimer interface primarily involves joining the β1-strand of each monomer to form a single six-stranded antiparallel β-sheet; by contrast, CC chemokines typically self-associate through an additional β-strand (β0) in the N terminus of each monomer. Novel quaternary interactions have also been observed in x-ray crystal structures of fractalkine (24) and MCP-1 (25).
Lymphotactin (Ltn) is unique among chemokines in that it (i) contains only one of the two disulfide bridges that are conserved in all other chemokines and (ii) possesses a unique C-terminal extension, which is required for biological activity (26, 27). Originally identified as a T and NK cell-specific chemokine (12, 28, 29), Ltn has recently been found to chemoattract neutrophils and B cells through the XCR1 receptor (30–32). A mediator of mucosal immunity, Ltn is thought to be a factor in acute allograft rejection (33) and inflammatory bowel diseases (34, 35). We recently used NMR spectroscopy to determine the three-dimensional structure of human lymphotactin (hLtn) at 10 °C in a solution containing 200 mM NaCl (23). Under these conditions, Ltn is predominantly monomeric and adopts the canonical chemokine fold; its C-terminal extension is disordered and highly mobile.
NMR spectra of hLtn in the absence of NaCl acquired between 10 and 35 °C show chemical shift patterns dramatically dependent on temperature, which have been attributed to conformational heterogeneity (27). In the present work, we have demonstrated that hLtn undergoes a structural transition that is dependent both on temperature and salt concentration. The form stabilized at low temperature/high salt has the structure determined previously (23). We have used NMR spectroscopy, analytical ultracentrifugation, and fluorescence spectroscopy to characterize the high temperature/low salt conformation of hLtn, which is shown to have a novel, non-chemokine-like fold. At intermediate conditions, both the chemokine-like and non-chemokine-like conformations of hLtn are present in solution and interconvert reversibly.
EXPERIMENTAL PROCEDURES
Sample Preparation
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). Plasmid pET-3a, Escherichia coli strain BL21(DE3) pLysS, and Factor Xa were purchased from Novagen (Madison, WI). The source for 15NH4Cl and [13C]glucose was Isotech, Inc. (Miamisburg, OH). Construction of a plasmid pET-3a containing a synthetic hLtn gene as a fusion with staphylococcal nuclease (SNase) was described previously, as were the expression, reconstitution, purification, and isotopic labeling ([U-15N] or [13C]hLtn) of recombinant hLtn (23). A truncated version of the protein (residues 1–68) also was cloned into the pET-3a vector as a fusion with SNase, expressed, and purified from the inclusion bodies in the same manner as the full-length protein.
Sedimentation Equilibrium
A Beckman Optima XL-A analytical ultracentrifuge was used for sedimentation equilibrium experiments. Protein samples at 20, 124, and 220 μM in 20 mM sodium phosphate, pH 6.0, containing 100 mM NaCl were dialyzed against 2 liters of the same buffer overnight at room temperature. Double-sector charcoal-filled Epon centerpieces were used with path lengths of 3 mm (for samples at 124 and 220 μM) and 12 mm (for the 20 μM sample). The concentration gradients were recorded at 280 nm every 2–3 h until gradients became superimposable. Equilibrium data were collected at 18,000, 24,000, 29,000, 33,000, and 39,000 rpm at 40 °C. At 24,000 rpm the equilibrium gradient was monitored for over 12 h and found to be stable. To test the reversibility of the gradients after equilibrium had been reached at 39,000 rpm, the rotor speed was returned to 24,000 rpm and samples allowed to equilibrate. At the end of the run, a baseline absorbance was recorded for each cell after a high speed depletion of protein from the cell (4–5 h at 52,000 rpm). The molecular weight and partial specific volume of hLtn were calculated from the amino acid sequence to be 10,254 and 0.735 ml/g, respectively. The extinction coefficient calculated from the tryptophan, tyrosine, and cystine content was 7,115 M−1 cm−1 (36). The buffer density was measured with an Anton Paar DMA5000 density meter and found to be 0.99825 g/ml at 40 °C.
A program written for Igor Pro (Wavemetrics Inc., Lake Oswego, OR) by Darrell R. McCaslin was used for the analysis of the sedimentation equilibrium data. Various models were fit to all data simultaneously, with data from the 12-mm path length cell (20 μM protein) normalized to a 3-mm path length. Models evaluated included single species, two and three species in equilibrium, and two independent non-interacting species. All models included a fitting parameter to account for the non-sedimenting baseline absorbance measured by high-speed depletion at the end of the experiment. In evaluating the fits, models yielding baselines substantially different from the measured value were taken as incorrect. The data collected at 33,000 and 39,000 rpm did not fit well to the model (monomer-dimer equilibrium) developed for the lower speed data. Although these data suggested that higher order aggregates form at the higher concentrations developed at these speeds, the data are insufficient to clarify what these species might be. The possibility of higher order, irreversible aggregation is further suggested by the non-superimposability of the 24,000-rpm gradients before and after equilibration at higher speeds. Because of the likelihood of the presence of these higher order aggregates, equilibrium data from the 33,000, 39,000, and 24,000 reversal were not included in the final analysis.
Fluorescence Measurements
Intrinsic fluorescence emission spectra were measured in a QuantaMaster C-60/2000 spectrofluorimeter (Photon Technologies International). Temperature was varied from 2.6 to 87.7 °C by an external circulating water bath. Sample temperatures were directly measured by a Sensortek BAT-12 digital thermometer with a thermocouple probe. Excitation was at 295 nm; all slits were set at 2 nm. Spectra were recorded at 0.5-nm intervals from 310 to 450 nm with a 1-s averaging time. Samples in a 1-cm-square cuvette contained 15 μM protein in 20 mM sodium phosphate buffer, pH 6.0, with and without 200 mM NaCl.
NMR Spectroscopy
Protein samples used for NMR measurements were dissolved in 90% H2O/10% D2O. All samples contained 20 mM sodium phosphate (pH 6.0), 0.05% sodium azide, and sodium chloride ranging in concentration from 0 to 200 mM. NMR spectra were collected either at NMRFAM on Bruker DMX 600 and 750 MHz spectrometers, both equipped with triple axis gradients and triple resonance probes, or at the Medical College of Wisconsin on a Bruker DRX 600 spectrometer equipped with a z axis gradient and a triple resonance cryoprobe. Chemical shifts were referenced to the methyl signal of internal 2,2-dimethylsilapentane-5-sulfonic acid (DSS) directly for 1H and indirectly for 15N and 13C as prescribed by IUPAC recommendations (37). 15N-1H HSQC spectra were collected as a function of temperature from 10 to 45 °C, and one-dimensional 1H spectra were acquired over the range of 10 to 75 °C. NMR spectra used for obtaining sequence-specific resonance assignments of hLtn high temperature, low salt conformation were acquired at 45 °C in the absence of NaCl.
The following data sets were collected and used to determine spectral assignments and to obtain structural constraints: two-dimensional 15N-1H HSQC (38); three-dimensional SE HNCA (39–41); three-dimensional SE HN(CO)CA (41); three-dimensional SE HNCO (40, 41); three-dimensional SE C(CO)NH (42); three-dimensional 15N NOESY-HSQC (43); three-dimensional 15N TOCSY-HSQC (38); three-dimensional 13C NOESY-HSQC (44); and three-dimensional HCCH-TOCSY (45). Heteronuclear 15N-1H NOE values were determined from an interleaved pair of two-dimensional gradient sensitivity-enhanced correlation spectra of [U-15N]hLtn acquired with and without a 3-s proton saturation period (46). The values of NOEs were obtained from the ratio of the peak intensities in proton-saturated and -unsaturated spectra. A four-dimensional 13C/15N-edited HMQC-NOESY-HSQC spectrum (47) was acquired on a sample composed of [U-15N] and [U-13C]hLtn mixed in a 1:1 ratio in order to probe a potential dimer interface. Felix95 (Molecular Simulations) or NMRPipe (48) software was used to process NMR data. The XEASY software package was used for NMR data analysis and resonance assignments (49). The CSI program was used for calculating the consensus chemical shift index from experimental 1Hα, 13Cα, 13Cβ, and 13C′ chemical shifts (50, 51).
RESULTS
Salt- and Temperature-dependence of hLtn Chemical Shifts
We previously reported the NMR solution structure of hLtn determined at 10 °C in a buffer containing 20 mM sodium phosphate, pH 6.0, and 200 mM NaCl (23). Under these conditions, hLtn adopted the conserved chemokine fold consisting of three antiparallel β-strands and a C-terminal α-helix with unstructured N- (1–9) and C-terminal (69–93) residues. In identifying the optimal solution conditions for the study of this structure, we observed heterogeneity in the 15N-1H HSQC spectrum of hLtn that could be modulated by temperature and salt concentration (23), a phenomenon also described by Handel and co-workers (27). To characterize this effect, we acquired a series of NMR spectra under various salt concentrations (ranging from 0 to 200 mM NaCl) and temperatures (ranging from 10 to 45 °C). Under some solution conditions, two-dimensional 15N-1H HSQC spectra of hLtn displayed two sets of peaks corresponding to two distinct conformations. The relative populations of these peaks were highly dependent on the temperature and the ionic strength of the solution. One set of signals corresponding to the chemokine-like structure dominated at low temperature and high salt concentrations, and the other dominated at high temperature and low salt concentrations. The exchange between the two conformations was slow on the NMR timescale, as evidenced by the appearance of distinct resonances. To assess the role of the unique C-terminal extension of hLtn in promoting this conformational exchange, a truncated version including only residues 1–68 was produced, and its 15N-1H HSQC spectrum was compared with that of the full-length protein under a variety of solution conditions and temperatures. Removal of the 25 C-terminal residues had no effect on the temperature- and ionic strength-dependent conformational exchange, indicating that the unstructured C terminus of hLtn is not responsible for its unusual structural heterogeneity.
An overlay of 15N-1H HSQC spectra of hLtn collected at 10 °C with 200 mM NaCl and at 45 °C with 0 mM NaCl is shown in Fig. 1 with sequence-specific resonance assignments indicated for the 45 °C spectrum. The patterns of signals for the low temperature/high salt species (Fig. 1, red peaks) and the high temperature/low salt species (Fig. 1, black peaks) are strikingly different, consistent with a major structural change. The stability of this novel high temperature/low salt conformation against thermal denaturation was demonstrated by a series of one-dimensional 1H spectra acquired from 45 to 75 °C that displayed the same pattern of amide 1HN signals over the entire range (data not shown).
Fig. 1. Overlay of 15N-1H HSQC spectra of hLtn collected at 45°C with no NaCl (black) and at 10 °C with 200 mM NaCl (red).

Both samples contained 20 mM sodium phosphate, pH 6.0, and 0.05% sodium azide. Backbone NH assignments for the high temperature spectrum are indicated by the one-letter amino acid code and residue number.
Fig. 2 shows the changes in the high frequency region of the two-dimensional 15N-1H HSQC spectrum of hLtn as a result of varying salt concentrations and temperatures. At 10 °C in the presence of 200 mM NaCl, hLtn adopted one conformation corresponding to the chemokine-like fold (panel A). As the temperature was increased from 10 to 25 °C, a new set of signals appeared in the spectrum (panel B). Comparison of the spectra in panels B and C, both collected at 25 °C with and without 200 mM NaCl, clearly shows the effect of ionic strength on hLtn conformation. At 25 °C in the presence of 200 mM NaCl, signals from the chemokine-like conformation are stronger, whereas in the absence of salt, the new set of peaks is predominant. Under low salt conditions, a temperature increase from 25 to 45 °C shifted the equilibrium farther, as illustrated by the complete lack of low temperature signals in Fig. 2D. For convenience, the two human lymphotactin species will be referred to henceforth as hLtn10 (chemokine-like conformation, 10 °C, 20 mM sodium phosphate, pH 6.0, 200 mM NaCl) and hLtn45 (45 °C, 20 mM sodium phosphate, pH 6.0, 0 mM NaCl).
Fig. 2. Changes in the high frequency region of 1H-15N HSQC spectra of hLtn as a function of temperature and salt concentration.
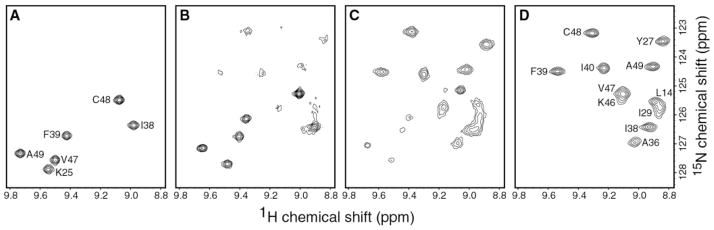
A, 10 °C, 200 mM NaCl. B, 25 °C, 200 mM NaCl. C,25 °C, 0 mM NaCl. D, 45 °C, 0 mM NaCl.
In order to estimate the relative populations of each hLtn conformation near physiologically relevant conditions, a two-dimensional 15N-1H HSQC spectrum was collected at 37 °C in the presence of 200 mM NaCl. Quantitative analysis of peak volumes for 12 residues displaying well resolved signals for both hLtn10 and hLtn45 conformations indicated that the two species are present in nearly equal amounts (46% Ltn10, 54% Ltn45).
Sequence-specific Resonance Assignments
To investigate the temperature- and salt-dependent structural changes in more detail, chemical shift assignments were obtained for the hLtn45 species. Complete 1H, 15N, and 13C resonance assignments and the three-dimensional structure of hLtn10 have been reported previously (BioMagResBank (BMRB) accession code 5240; PDB entries 1J9O and 1J8I). Backbone resonance assignments for hLtn45 were made on the basis of the following three-dimensional triple resonance experiments: HNCA, HN-(CO)CA, HNCO, C(CO)NH, 15N TOCSY-HSQC, and 15N NOESY-HSQC. The two-dimensional 15N-1H HSQC spectrum was used as a reference to correlate each cross-peak to its corresponding amide 1HN and 15N in each three-dimensional spectrum. The sequential connections obtained mainly from HNCA and HN(CO)CA experiments were confirmed by the analysis of NOE patterns from 15N NOESY-HSQC spectra. The C(CO)NH experiment provided assignments for aliphatic 13C resonances. Aliphatic side chain 1H assignments were obtained from 15N TOCSY-HSQC and HCCH-TOCSY experiments. Complete backbone and partial side chain 1H, 15N, and 13C assignments for hLtn45 have been deposited in the BMRB data bank (BMRB accession code 5251).
Chemical Shift Mapping
Perturbations to the backbone 1HN and 15N chemical shifts for each residue resulting from the conformational rearrangement of hLtn are plotted in Fig. 3. Comparisons of the resonance assignments from the two conformations of hLtn revealed substantial chemical shift perturbations for residues throughout the domain but with no obvious pattern. Backbone amide shift differences are relatively small for residues of the unstructured termini (Fig. 3), but residues 54–66 of the α-helix display consistently large 13Cα chemical shift differences between hLtn10 and hLtn45 (data not shown). Ring current effects from the aromatic side chain of Trp-55 contribute to the unusually upfield 1HN chemical shifts of Ile-19 (5.33 ppm) and Val-56 (5.91 ppm) in the hLtn10 structure (23); the transition to hLtn45 produces particularly large 1HN shift perturbations for these residues.
Fig. 3. Chemical shift changes for each residue of hLtn accompanying the structural rearrangement dependent on temperature and salt conditions.

The chemical shift changes are for conversion of hLtn10 (10 °C in the presence of 200 mM NaCl) to hLtn45 (45 °C in the absence of NaCl). The values shown are weighted averages (Δav) calculated using the equation Δav = [((Δ1HN)2 + (Δ15N/4.6)2)/2]1/2 (69, 70). The scaling factor of 0.22 for 15N was derived from the ratio of the chemical shift ranges for 1HN (9.76–5.31 = 4.45 ppm) and 15N (127.8–107.3 = 20.5 ppm).
Effects of Salt and Temperature on the Aggregation State of hLtn
Although structural analysis of hLtn at 10 °C in the presence of 200 mM NaCl provided no specific evidence of aggregation, our sedimentation equilibrium studies and NMR pulsed-field gradient diffusion measurements have shown that the protein exists as a monomer-dimer equilibrium with an equilibrium association constant of 850 ± 10 M−1 (23). Broader lines were observed in the two-dimensional 15N-1H HSQC spectrum of hLtn45 than in hLtn10 (Fig. 1), suggesting that a change in aggregation state might accompany the transition. Many chemokines form dimers (14–16, 52–55), and the aggregation of MIP-1α has been shown to be inhibited by elevated salt concentrations (56, 57). In order to determine the aggregation state of the high temperature/low salt hLtn species, a sedimentation equilibrium study was performed at 40 °C in a buffer containing 20 mM sodium phosphate, pH 6.0, 100 mM NaCl. The conditions used for ultracentrifugation represented the closest approximation to the hLtn45 NMR conditions that could be accommodated experimentally. Global analysis of the sedimentation equilibrium behavior of hLtn under these conditions indicated the presence of monomer-dimer equilibrium with an association constant of 26,000 ± 2,000 M−1. In contrast, our previous study of hLtn at 10 °C and 200 mM NaCl revealed a much weaker association (Ka = 850 ± 10 M−1). These results clearly show that the high temperature/low salt form of hLtn has a greater tendency to self-associate. Fig. 4 illustrates sedimentation equilibrium under each of the conditions. For the theoretical curves, the molecular weight of the monomer and the baseline absorbance were fixed. The experimental data were then fit while also holding the association constant to the values indicated.
Fig. 4. Equilibrium analytical ultracentrifugation of ~0.2 mM Ltn at 10 °C with 200 mM NaCl and 40 °C with 100 mM NaCl.

Data at 10 °C (black symbols) and 40 °C (gray symbols) were acquired at 26,000 and 24,000 rpm, respectively. Theoretical curves corresponding to Ka values of 850 M−1 (black) and 27,000 M−1 (gray) for each velocity are shown to illustrate the change in monomer-dimer equilibrium constant. Data acquired at 10 °C were shifted up by 0.2 absorbance units for clarity.
Fluorescence Measurements
The fluorescence emission from tryptophan residues is very sensitive to the environment of the side chain and is often used as a probe of structural perturbations. Red shifts in the emission maximum and decreases in fluorescence intensity generally suggest an increased exposure of tryptophan residues to solvent (58). hLtn has a single tryptophan residue (Trp-55) located near the start of the C-terminal α-helix in the chemokine-like hLtn10 conformation that participates in hydrophobic core interactions. Therefore, changes in the emission spectra of hLtn directly reflect alterations in the Trp-55 environment. Fig. 5A shows the emission spectra at 10.6 and 53.9 °C in the absence of salt. Going from low temperature to high, we see both a loss in intensity and a red shift in the emission maximum, indicating the tryptophan has become more solvent exposed at the higher temperature. Addition of salt to the 53.9 °C sample results in both a shift back to lower wavelengths and enhanced fluorescence yield. These results show that the exposure of the single tryptophan of hLtn to solvent is dependent on both temperature and salt concentration. As further characterization of the structural interconversion, emission spectra were recorded as a function of temperature in the presence and absence of salt. The assignment of precise peak positions in intrinsic fluorescence spectra is difficult, but the use of the intensity-weighted average emission wavelength has proven effective in quantitating shifts in emission maximum positions (58). Fig. 5B shows a plot of the average emission position versus temperature, and clearly the presence of salt has shifted the exposure of the tryptophan to higher temperatures.
Fig. 5. Tryptophan fluorescence as a function of salt concentration and temperature.
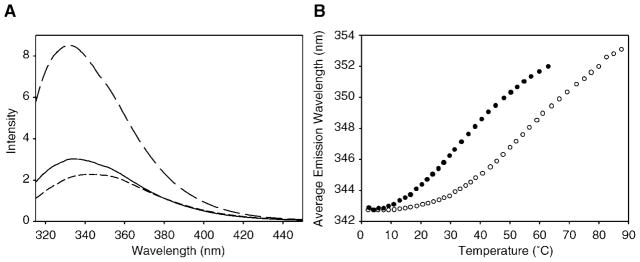
A, fluorescence emission spectra of hLtn at 10.6 (long dashes) and 53.9 °C (short dashes) in the absence of NaCl and at 53.9 °C in the presence of 200 mM NaCl (solid line). B, intrinsic fluorescence of Trp-55 of hLtn as a function of temperature at 0 and 200 mM NaCl. Average emission wavelengths are plotted for hLtn in 0 mM (solid circles) and 200 mM NaCl (open circles).
Secondary Structure
Secondary structural elements of hLtn45 were determined from chemical shift index (CSI) and NOE patterns and compared with those of the hLtn10 conformation. Fig. 6 shows the CSI and heteronuclear 15N-1H NOE values for hLtn10 and hLtn45. A clear difference in the CSI predictions for hLtn10 and hLtn45 is observed for residues 53–68, which normally form an α-helix in chemokines. In hLtn10, this stretch is predicted by chemical shift and NOE patterns to be α-helical and confirmed to be α-helical in the NMR structure (23). In hLtn45, the same stretch is predicted to be non-helical. Diminished heteronuclear NOE values obtained at 45 °C for residues 55–70 suggest that these residues are considerably more dynamic in hLtn45, consistent with a loss of the helical structure found in hLtn10. The absence of characteristic helical (i, i+3) and (i, i+4) NOEs for these residues in the three-dimensional 15N NOESY-HSQC spectrum provides additional evidence for this conformational change.
Fig. 6.

Comparison of secondary structure elements from the chemical shift index and relative mobility of the protein backbone from 15N-1H heteronuclear NOE values for hLtn10 (A) and hLtn45 (B).
A total of four β-strands is predicted by the CSI for both hLtn conformations. However, the strand predicted for residues 10–16 did not participate in a β-sheet in the NMR structure of hLtn10. In contrast, NOESY spectra of hLtn45 contain a series of strong cross-strand NOEs between the β0- (residues 11–14) and β3- (residues 44–48) strands, which participate in a four-stranded antiparallel β-sheet structure. Further analysis of the NOE data showed that, apart from the presence of the new β-strand in hLtn45, the sheet structures of hLtn45 and hLtn10 are entirely different. Fig. 7 shows the NOE and hydrogen bonding patterns defining the two β-sheets. Compared with the pairing of strands in the hLtn10 sheet, β1 and β3 in the hLtn45 sheet are each shifted in opposite directions by one residue relative to the central β2-strand. The hLtn45 sheet is slightly longer: strand β1 starts at R23 instead of K25 and extends to P51 at the end of β3, whereas the same strand in hLtn10 ends at D50. In the hLtn10 sheet, both β-turns are irregular 3-residue turns, but in the hLtn45 sheet, the β-turns are regular and each contains 4 residues. In addition to changing the position and types of turns at the β1-β2 and β2-β3 junctions, the rearrangement completely changes the pattern of cross-strand hydrogen bonding partners within the sheet.
Fig. 7. β-sheet diagrams for the two hLtn conformations.
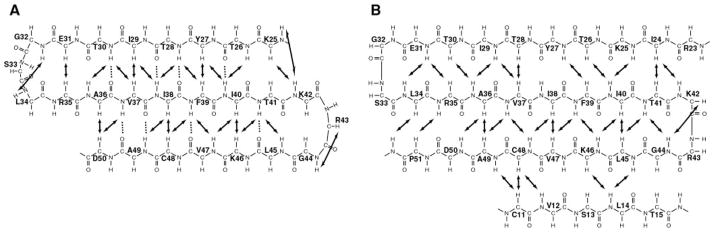
A, the chemokine-like conformation of hLtn10 (solution conditions: 10 °C, 200 mM NaCl). B, the non-chemokine-like conformation of hLtn45 (solution conditions: 45 °C, no NaCl). Solid arrows indicate interstrand NOEs. Dashed lines indicate the hydrogen bonds observed in the hLtn10 NMR structure.
An N-terminal β0-strand is formed in a number of CC chemokines, including RANTES and MCP-1, but in those structures the β0 strands from two monomers pair up to form part of the dimer interface (14, 15). On this basis we considered the possibility that β0 in hLtn45 could form part of a dimer interface. While no NOEs were observed that would indicate dimeric pairing of the β0, the altered contacts between β0 and β3 in hLtn45 (Fig. 7B) might arise from the interaction of two separate Ltn molecules. To test the hypothesis that β0-β3 inter-strand NOEs are intermolecular, we acquired a four-dimensional 13C/15N-edited HMQC-NOESY-HSQC experiment on a sample containing 50% 13C-labeled Ltn and 50% 15N-labeled hLtn. Because neither protein component was labeled with both 15N and 13C, the combination of 13C and 15N editing should have detected only NOEs arising from the association of 13C- and 15N-labeled monomers. No intermolecular NOEs were detected in the 13C/15N-edited spectrum, and we have therefore concluded that the β0-β3 cross-strand NOEs result from intramolecular β-sheet contacts.
DISCUSSION
Chemokines and their receptors, which play important roles in inflammation and disease, are attractive candidates for drug development. Characterization of the biologically active structure and aggregation state for a chemokine is key to understanding its mode of receptor binding and activation. We had shown previously that human lymphotactin adopts the conserved chemokine fold and is predominantly monomeric under specific conditions of low temperature and elevated salt concentration (23). In the present study we have demonstrated that at physiologically relevant temperatures hLtn exists in a conformational equilibrium between the chemokine domain fold and a fold with a substantially different tertiary and quaternary structure.
Low Salt and High Temperature Conformation of hLtn Is Dimeric
Analytical ultracentrifugation measurements showed that hLtn associates to a greater extent at high temperature and low salt concentration than at 10 °C and 200 mM NaCl. The association constant of 850 M−1 (Kd = ~1.18 mM) determined for hLtn10 indicates that 0.5 mM hLtn10 is predominantly monomeric (23), whereas the association constant of 26,000 ± 2,000 M−1 (Kd = ~39 μM) determined for hLtn at 40 °C in the presence of 100 mM NaCl indicates that it is essentially all in the dimeric form. Although sedimentation studies indicated that hLtn45 is dimeric, we failed to observe any intermolecular NOEs in the 13C/15N-edited HMQC-NOESY-HSQC spectrum of 50% 13C/50% 15N-labeled hLtn. One possible reason for this might be that the dimer interface in hLtn45 is composed entirely of side chain-side chain interactions and that no backbone 1HN protons are within the 5 Å NOE range of 13C-bound protons of the other monomer. Alternatively, the rate of monomer-dimer exchange may be on an intermediate chemical shift timescale, resulting in the broadening of resonances at the interface to the point that intermolecular NOEs are too weak to be observed.
Other chemokines display similar aggregation behavior: MCP-1 is dimeric above 100 μM (15) and RANTES is dimeric above 35 μM (14). The functional relevance of CC and CXC chemokine dimerization has been studied extensively. A monomeric variant of interleukin-8 has been shown to be fully functional in activating neutrophils (59), but a cysteine cross-linked interleukin-8 dimer is also similarly active (60). Monomeric CC chemokine variants P8A-MIP-1β (61) and P8A-MCP-1 have been shown to bind and activate their receptors as effectively as the wild type proteins (62). Whereas chemokines are presumed to be predominantly monomeric at physiological concentrations, the MCP-1 dimer has been shown to exist at physiological concentrations, and covalently cross-linked MCP-1 dimers have been shown to be fully active (63). Furthermore, physiological solution conditions have been shown to enhance the formation of dimeric MIP-1β (64), and naturally produced heterodimers of MIP-1α and MIP-1β have been identified (65). Fully functional disaggregated mutants of MIP-1α and RANTES have also been reported (66). Some chemokines can inhibit HIV infection by preventing coreceptor-mediated cell entry. Although disaggregated RANTES variants retain their HIV-inhibitory activities, high concentrations of wild type RANTES, but not the disaggregated variants, have been found to enhance HIV infection (66). These findings suggest that chemokine oligomerization, and in turn chemokine function, may be altered depending on the local environment, the solution conditions, and chemokine concentration.
hLtn45 Has a Novel Secondary Structure
The conformational equilibrium for hLtn is affected dramatically by changes in temperature and ionic strength, and the conserved chemokine fold is stabilized by elevated salt concentrations and low temperature. The structural rearrangement involves the disruption of all hydrogen bonds in the three-stranded antiparallel β-sheet of hLtn10 and the formation of a new four-stranded β-sheet, as well as the loss of the C-terminal α-helix. Although quaternary structural rearrangements have been described for other chemokines (24, 25), this is the first example of a major conformational rearrangement for a chemokine at the level of secondary or tertiary structure.
Rearrangement of the β-sheet in hLtn10 requires the shift of the β1- and β3-strand by one residue in opposite directions relative to the β2-strand. As a result of this shift, residues 25 of β1 and 45 of β3, which are bulged out in the hLtn10 structure, become part of the sheet in hLtn45. In the course of this rearrangement, all hydrogen bonds in the β2-strand of hLtn10 are broken and replaced by new bonds. This rearrangement not only changes the entire hydrogen bonding network of the β-sheet but also results in rearrangement of the side chains of residues in the β1- and β3-strand to opposite sides of the sheet. Side chains of residues in β1 and β3 that point toward the α-helix in hLtn10 move to the other face of the sheet in hLtn45. This facet of the β-sheet rearrangement may help explain the loss of the C-terminal α-helix in hLtn45. Conserved hydrophobic core interactions involving β-sheet residues Tyr-27, Ile-29, Val-37, and Phe-39 and α-helix residues Trp-55, Val-60, and Met-63 in hLtn10 are broken in hLtn45 because the hydrophobic side chains of Tyr-27 and Ile-29 are replaced by side chains of Thr-26 and Thr-28. The β3-strand in hLtn45 extends to residue 51 as opposed to residue 50 in hLtn10, which might also contribute to the unfolding of the α-helix by shortening the connection between the sheet and the helix, which normally starts at residue 54. The temperature dependence of the fluorescence emission maximum of hLtn suggests that the single tryptophan in hLtn is more solvent-exposed in the high temperature/low salt form. This is consistent with changes in the structure observed by NMR, in which this residue at the start of the helix in hLtn10 leaves the hydrophobic core for a more dynamic and solvent-exposed environment upon conversion to hLtn45.
Role of Unique hLtn Sequences in Structural Rearrangement
All known chemokines, except Ltn, contain at least two disulfide bonds. Ltn lacks the first and the third cysteine residues present in other chemokines. The second disulfide bond in the other chemokines tethers the N-terminal region to the 30’s loop. This additional covalent cross-link probably would preclude a β-sheet rearrangement of the kind observed for hLtn. The additional β0-strand present in hLtn45 positions Cys-11 and Cys-48 directly across from each other on adjacent strands of the sheet, thereby accommodating the single disulfide linkage. A C-terminal extension is the other unique feature of the Ltn sequence, and previous structural studies showed that these residues are dynamically disordered (23). NMR studies reported here of a version of hLtn truncated after residue 68 showed that the unstructured 25 residues of the C terminus do not play a role in the temperature- and ionic strength-dependent conformational changes.
Biological Consequences of hLtn Conformational Changes
Both the hLtn10 and hLtn45 conformations of hLtn are present at physiological temperature and salt concentration. This structural heterogeneity may have functional consequences. For example, others have reported difficulties in obtaining reproducible functional assays with recombinant commercial hLtn (67). If the two conformational states reported here have different activities, the variability in activity could be explained by differences in solution conditions that would alter the relative concentrations of the two species. For elucidating the functional roles of each conformation, it will be of interest to develop variants of hLtn that preferentially adopt either the chemokine (hLtn10) or non-chemokine (hLtn45) fold. Changes in aggregation behavior accompanying the tertiary structural transition undergone by lymphotactin may also be relevant to its role as a ligand for either the XCR1 receptor or cell surface GAGs. A recent study of the GAG binding activity of interleukin-8 showed that both heparin and heparan sulfate oligosaccharides display substantially higher affinity for monomeric interleukin-8 than for the dimer (68). Moreover, soluble interleukin-8·GAG complexes were more stabilized against thermal unfolding than the free protein. The potential role for GAGs in regulating the tertiary and quaternary structural features of lymphotactin will be the subject of future investigations.
Acknowledgments
This study made use of the National Magnetic Resonance Facility at Madison (NMRFAM), using equipment purchased with funds from the University of Wisconsin, the National Science Foundation Biological Instrumentation Program (DMB-8415048), the National Institutes of Health Biomedical Research Technology Program (RR02301), National Science Foundation Academic Research Instrumentation Program (BIR-9214394), National Institutes of Health Shared Instrumentation Program (RR02781 and RR08438), and the United States Department of Agriculture. Sedimentation equilibrium data were obtained at the University of Wisconsin-Madison Biophysics Instrumentation Facility, which is supported by the University of Wisconsin-Madison and National Science Foundation Grant BIR-9512577 and National Institutes of Health Grant S10 RR13790.
Footnotes
This study was supported by National Institutes of Health Grant R01 AI45843.
The abbreviations used are: GAG, glycosaminoglycan; Ltn, lymphotactin; h, human; CSI, chemical shift index; HIV, human immunodeficiency virus; NOE, nuclear Overhauser effect; RANTES, regulated on activation normal T cell expressed and secreted; SE, sensitivity-enhanced.
The NMR spectrosocopy data reported in this paper have been deposited in the BioMagResBank under BMRB accession number 5251.
References
- 1.Baggiolini M, Dewald B, Moser B. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 2.Rossi D, Zlotnik A. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 3.Mackay CR. Nat Immunol. 2001;2:95–101. doi: 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- 4.Proudfoot AEI. Eur J Immunol. 1998;8:147–157. [Google Scholar]
- 5.Worthylake RA, Burridge K. Curr Opin Cell Biol. 2001;13:569–577. doi: 10.1016/s0955-0674(00)00253-2. [DOI] [PubMed] [Google Scholar]
- 6.Luther SA, Cyster JG. Nature Immunol. 2001;2:102–107. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 7.Campbell JJ, Hedrick JA, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 8.Gale LM, McColl SR. Bioessays. 1999;21:17–28. doi: 10.1002/(SICI)1521-1878(199901)21:1<17::AID-BIES3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Baggiolini M. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 10.Sallusto F, Mackay CR, Lanzavecchia A. Annu Rev Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- 11.Bazan JF, Bacon KB, Hardiman G, Wang W, Rossi D, Greaves DR, Zlotnik A, Schall TJ. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 12.Kelner G, Kennedy J, Bacon K, Kleyensteuber S, Largaespada D, Jenkins N, Copeland N, Bazan J, Moore K, Schall T, Zlotnik A. Science. 1994;266:1395–1399. doi: 10.1126/science.7973732. [DOI] [PubMed] [Google Scholar]
- 13.Clore GM, Gronenborn AM. FASEB J. 1995;9:57–62. doi: 10.1096/fasebj.9.1.7821760. [DOI] [PubMed] [Google Scholar]
- 14.Skelton NJ, Aspiras F, Ogez J, Schall TJ. Biochemistry. 1995;34:5329–5342. doi: 10.1021/bi00016a004. [DOI] [PubMed] [Google Scholar]
- 15.Handel TM, Domaille PJ. Biochemistry. 1996;35:6569–6584. doi: 10.1021/bi9602270. [DOI] [PubMed] [Google Scholar]
- 16.Shao W, Jerva LF, West J, Lolis E, Schweitzer BI. Biochemistry. 1998;37:8303–8313. doi: 10.1021/bi980112r. [DOI] [PubMed] [Google Scholar]
- 17.LiWang AC, Wang ZX, Sun Y, Peiper SC, LiWang PJ. Protein Sci. 1999;8:2270–2280. doi: 10.1110/ps.8.11.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizoue LS, Bazan JF, Johnson EC, Handel TM. Biochemistry. 1999;38:1402–1414. doi: 10.1021/bi9820614. [DOI] [PubMed] [Google Scholar]
- 19.Young H, Roongta V, Daly TJ, Mayo KH. Biochem J. 1999;338:591–598. [PMC free article] [PubMed] [Google Scholar]
- 20.Sticht H, Escher SE, Schweimer K, Forssmann WG, Rosch P, Aderman K. Biochemistry. 1999;38:5995–6002. doi: 10.1021/bi990065i. [DOI] [PubMed] [Google Scholar]
- 21.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer KL, Stone MJ. Biochemistry. 2000;39:8382–8395. doi: 10.1021/bi000523j. [DOI] [PubMed] [Google Scholar]
- 23.Kuloğlu ES, McCaslin DR, Kitabwalla M, Pauza CD, Markley JL, Volkman BF. Biochemistry. 2001;40:12486–12496. doi: 10.1021/bi011106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoover DM, Mizoue LS, Handel TM, Lubkowski J. J Biol Chem. 2000;275:23187–23193. doi: 10.1074/jbc.M002584200. [DOI] [PubMed] [Google Scholar]
- 25.Lubkowski J, Bujacz G, Boque L, Domaille PJ, Handel TM, Wlodawer A. Nat Struct Biol. 1997;4:64–69. doi: 10.1038/nsb0197-64. [DOI] [PubMed] [Google Scholar]
- 26.Hedrick JA, Zlotnik A. Methods Enzymol. 1997;287:206–215. doi: 10.1016/s0076-6879(97)87016-4. [DOI] [PubMed] [Google Scholar]
- 27.Marcaurelle LA, Mizoue LS, Wilken J, Oldham L, Kent SBH, Handel TM, Bertozzi CR. Chem Eur J. 2001;7:1129–1132. doi: 10.1002/1521-3765(20010302)7:5<1129::aid-chem1129>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy J, Kelner G, Kleyensteuber S, Schall T, Weiss M, Yssel H, Schneider P, Cocks B, Bacon K, Zlotnik A. J Immunol. 1995;155:203–209. [PubMed] [Google Scholar]
- 29.Hedrick JA, Saylor V, Figueroa D, Mizoue L, Xu Y, Menon S, Abrams J, Handel T, Zlotnik A. J Immunol. 1997;158:1533–1540. [PubMed] [Google Scholar]
- 30.Yoshida T, Imai T, Kakizaki M, Nishimura M, Takagi S, Yoshie O. J Biol Chem. 1998;273:16551–16554. doi: 10.1074/jbc.273.26.16551. [DOI] [PubMed] [Google Scholar]
- 31.Huang H, Li F, Cairns CM, Gordon JR, Xiang J. Biochem Biophys Res Commun. 2001;281:378–382. doi: 10.1006/bbrc.2001.4363. [DOI] [PubMed] [Google Scholar]
- 32.Cairns CM, Gordon JR, Li F, Baca-Estrada ME, Moyana T, Xiang J. J Immunol. 2001;167:57–65. doi: 10.4049/jimmunol.167.1.57. [DOI] [PubMed] [Google Scholar]
- 33.Wang JD, Nonomura N, Takahara S, Li BS, Azuma H, Ichimaru N, Kokado Y, Matsumiya K, Miki T, Suzuki S, Okuyama A. Immunology. 1998;95:56–61. doi: 10.1046/j.1365-2567.1998.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boismenu R, Feng L, Xia YY, Chang JC, Havran WL. J Immunol. 1996;157:985–992. [PubMed] [Google Scholar]
- 35.Middel P, Thelen P, Blaschke S, Polzien F, Reich K, Blaschke V, Wrede A, Hummel KM, Gunawan B, Radzun HJ. Am J Pathol. 2001;159:1751–1761. doi: 10.1016/S0002-9440(10)63022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pace N, Schmid FX. In: Protein Structure: A Practical Approach. Creighton TE, editor. IRL Press; Oxford: 1997. pp. 253–259. [Google Scholar]
- 37.Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K. Eur J Biochem. 1998;256:1–15. doi: 10.1046/j.1432-1327.1998.2560001.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhang O, Kay LE, Olivier JP, Forman-Kay JD. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 39.Kay LE, Xu GY, Yamazaki T. J Magn Reson Ser A. 1994;109:129–133. [Google Scholar]
- 40.Muhandiram DR, Kay LE. J Magn Reson Ser B. 1994;103:203–216. [Google Scholar]
- 41.Grzesiek S, Bax A. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 42.Grzesiek S, Anglister J, Bax A. J Magn Reson Ser B. 1993;101:114–119. [Google Scholar]
- 43.Talluri S, Wagner G. J Magn Reson Ser B. 1996;112:200–205. doi: 10.1006/jmrb.1996.0132. [DOI] [PubMed] [Google Scholar]
- 44.Muhandiram DR, Farrow NA, Xu GY, Smallcombe SH, Kay LE. J Magn Reson Ser B. 1993;102:317–321. [Google Scholar]
- 45.Kay LE, Xu GY, Singer AU, Muhandiram DR, Forman-Kay JD. J Magn Reson Ser B. 1993;101:333–337. [Google Scholar]
- 46.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 47.Morshauser RC, Zuiderweg ERP. J Magn Reson. 1999;139:232–239. doi: 10.1006/jmre.1999.1802. [DOI] [PubMed] [Google Scholar]
- 48.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 49.Bartels CH, Xia TH, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 50.Wishart DS, Sykes BD, Richards FM. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 51.Wishart DS, Sykes BD. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 52.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 53.Chung CW, Cooke RM, Proudfoot AEI, Wells TNC. Biochemistry. 1995;34:9307–9314. doi: 10.1021/bi00029a005. [DOI] [PubMed] [Google Scholar]
- 54.Lodi PJ, Garrett DS, Kuszewski J, Tsang MLS, Weatherbee JA, Leonard WJ, Gronenborn AM, Clore GM. Science. 1994;263:1762–1767. doi: 10.1126/science.8134838. [DOI] [PubMed] [Google Scholar]
- 55.Meunier S, Bernassau JM, Guillemot JC, Ferrara P, Darbon H. Biochemistry. 1997;36:4412–4422. doi: 10.1021/bi9627929. [DOI] [PubMed] [Google Scholar]
- 56.Patel SR, Evans S, KD, Knight GC, Morgan PJ, Varley PG, Craig S. Biochemistry. 1993;32:5466–5471. doi: 10.1021/bi00071a024. [DOI] [PubMed] [Google Scholar]
- 57.Varley PG, Brown AJ, Dawkes HC, Burns NR. Eur Biophys J. 1997;25:437–443. doi: 10.1007/s002490050058. [DOI] [PubMed] [Google Scholar]
- 58.Royer CA. Meth Mol Biol. 1995;40:65–89. doi: 10.1385/0-89603-301-5:65. [DOI] [PubMed] [Google Scholar]
- 59.Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, Clark-Lewis I. Science. 1994;264:90–92. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 60.Leong S, Lowman H, Liu J, Shire S, Deforge L, Gillece-Castro B, McDowell R, Hebert C. Protein Sci. 1997;6:609–617. doi: 10.1002/pro.5560060310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laurence JS, Blanpain C, Burgner JW, Parmentier M, LiWang PJ. Biochemistry. 2000;39:3401–3409. doi: 10.1021/bi9923196. [DOI] [PubMed] [Google Scholar]
- 62.Paavola CD, Hemmerich S, Grunberger D, Polsky I, Bloom A, Freedman R, Mulkins M, Bhakta S, McCarley D, Wiesent L, Wong B, Jarnagin K, Handel TM. J Biol Chem. 1998;273:33157–33165. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Rollins B. Mol Cell Biol. 1995;15:4851–4855. doi: 10.1128/mcb.15.9.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laurence JS, LiWang AC, LiWang PJ. Biochemistry. 1998;37:9346–9354. doi: 10.1021/bi980329l. [DOI] [PubMed] [Google Scholar]
- 65.Guan E, Wang J, Norcross MA. J Biol Chem. 2001;276:12404–12409. doi: 10.1074/jbc.M006327200. [DOI] [PubMed] [Google Scholar]
- 66.Czaplewski LG, McKeating J, Craven CJ, Higgins LD, Appay V, Brown A, Dudgeon T, Howard LA, Meyers T, Owen J, Palan SR, Tan P, Wilson G, Woods NR, Heyworth CM, Lord BI, Brotherton D, Gilbert R, Morgan P, Randle E, Schofield N, Varley PG, Fisher J, Waltho JP, Hunter MG. J Biol Chem. 1999;274:16077–16084. doi: 10.1074/jbc.274.23.16077. [DOI] [PubMed] [Google Scholar]
- 67.Shan L, Qiao X, Oldham E, Catron D, Kaminski H, Lundell D, Zlotnik A, Gustafson E, Hedrick JA. Biochem Biophys Res Commun. 2000;268:938–941. doi: 10.1006/bbrc.2000.2235. [DOI] [PubMed] [Google Scholar]
- 68.Goger B, Halden Y, Rek A, Mosl R, Pye D, Gallagher J, Kungl AJ. Biochemistry. 2002;41:1640–1646. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- 69.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 70.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]