

Abstract
Purpose
To use manganese-enhanced MRI (MEMRI) to detect changes in calcium handling associated with cardiac hypertrophy in a mouse model, and to determine whether the impact of creatine kinase ablation is detectable using this method.
Materials and Methods
Male C57BL/6 (C57, n=11) and male creatine kinase double knockout (CK-M/Mito−/−, DBKO, n=12) were imaged using the saturation recovery Look-Locker T1 mapping sequence before and after development of cardiac hypertrophy. Hypertrophy was induced via subcutaneous continuous 3 day infusion of isoproterenol while sham mice not subjected to cardiac hypertrophy were also imaged. During each scan the contrast agent Mn was administered and the resulting change in R1 (=1/T1) was calculated. Two anatomical regions-of-interest (ROIs) were considered: the left-ventricular free wall (LVFW) and the septum, and one ROI in a Mn-containing standard placed next to the mouse.
Results
We found statistically significant (p < 0.05) decreases in the uptake of Mn in both the LVFW and septum following induction of cardiac hypertrophy. No statistically significant decreases were detected in the standard, and no statistically significant differences were found among the sham mice.
Conclusion
Using a murine model, we successfully demonstrated that changes in manganese uptake due to cardiac hypertrophy are detectable using the functional contrast agent and calcium mimetic manganese. Our measurements showed a decrease in the relaxivity (R1) of the myocardium following cardiac hypertrophy compared to normal control mice.
Keywords: MEMRI, creatine kinase, cardiac hypertrophy, manganese, saturation recovery
Graphical abstract
In this study we investigated use of manganese-enhanced MRI (MEMRI) for the detection of alterations in calcium dynamics in normal (C57BL/6) and creatine kinase (CK) knockout mice (M-/Mito-CK −/−). We found statistically significant (p < 0.05) decreases in manganese (Mn) uptake following cardiac hypertrophy in both C57BL/6 and CK knockout mice, and found no differences in uptake between the two strains. Our results provide the first evidence that changes in calcium handling due to cardiac hypertrophy are detectable using MEMRI.
Introduction
Despite intense research, cardiovascular disease remains the leading cause of death in the United States and accounts for approximately 1 in every 3 deaths (1). Cardiomyopathies are a group of diseases of the myocardium which frequently have underlying genetic causes (2). Hypertrophic cardiomyopathy (HCM) is the most common inherited heart defect, affecting approximately 500,000 individuals in the United States (1,3,4). Among HCM patients, approximately 12% will die of HCM-related causes including sudden cardiac death (SCD), which itself makes up approximately 6% of HCM-related fatalities (5). For the those patients experiencing SCD, 71% have no or mild symptoms (5), thus illustrating the need for improved techniques to monitor disease progression and to identify at-risk patients.
Cardiac hypertrophy is a condition generally typified by increased left-ventricular wall thickness and ECG abnormalities. The etiology of cardiac hypertrophy includes, but is not limited to, hypertensive heart disease and familial HCM (3,6). Cardiac hypertrophy can also be considered an adaptive response in which the heart attempts to compensate for increased wall stress by thickening the myocardium. While this may serve to preserve cardiac function in the short-term, longstanding cardiac hypertrophy is associated with increased risk of heart failure, arrhythmia, myocardial infarction and sudden cardiac death (7,8). In the case of HCM, therapies to regress hypertrophy do not exist and implantable cardioverter-difibrillators (ICDs) are the only treatment which has been shown conclusively to prolong life (9). In other cases of cardiac hypertrophy, such as those induced by hypertension, disease regression can be attained by pharmaceutical intervention such as angiotensin-converting enzyme (ACE) inhibitors directed at reducing cardiac burden, and calcium channel blockers (e.g. diltiazem) have been shown to prevent hypertrophy and progression to heart failure in mouse models (10). Sodium-calcium exchanger (NCX) inhibitors have also been considered for the treatment of cardiac hypertrophy (11). However, the NCX inhibitors presently available (SEA-0400 and KB-R7943) have been shown to lack the specificity required to be clinically useful (11,12).
Alterations in calcium regulation have been associated with cardiac hypertrophy (13–16), but presently detection of these changes requires the use of invasive methods not conducive for in vivo experimentation or clinical use. For HCM in particular there are a wide variety of mutations that lead to the hypertrophic phenotype (17), and it has been suggested that these mutations result in increased calcium sensitivity which then causes hypertrophy (13,15). This suggestion is based on evidence demonstrating increased calcium sensitivity in a number of studies of various sarcomere mutations as measured by the log of the calcium ion concentration needed to give 50% of maximal activation (ΔpCa50). It is not presently known whether the degree of calcium perturbation has any prognostic value in predicting overall phenotype or in stratifying risk for heart failure, arrhythmia, or sudden cardiac death. Although advanced morphological changes due to hypertrophy can be easily detected using MRI and echocardiography, early detection requires sensitivity to cellular changes in gene expression and calcium homeostasis, which precede morphological changes. Unfortunately, these early cellular changes are undetectable using standard imaging techniques but can be exploited using new functional imaging techniques such as manganese-enhanced MRI (MEMRI) (18–22).
In addition to force generation, ion homeostasis requires significant energy expenditure by the cardiac myocyte, due to regulating calcium at concentrations ranging from micro to millimolar levels across the cell and within a single heartbeat. In order to maintain the large calcium gradient across the sarcoplasmic reticulum (SR) membrane in the cardiac myocyte, the SR Ca2+-ATPase requires 85–90% of ΔG from ATP (23) and requires sufficient myocardial metabolism. Myocardial energetics play a central role in maintaining normal cardiac function and when unable to maintain the PCr to ATP ratio needed to sustain Ca2+ homeostasis in excitation-contraction coupling (E–C coupling) (24–28) heart failure results.
Creatine kinase in an enzyme abundant in striated muscle which catalyzes the equilibrium reaction between adenosine triphosphate (ATP) and creatine (Cr) to ADP and phosphocreatine (PCr). Previous investigations on left- ventricular hypertrophy (LVH) and heart failure with CK-deficient mice have shown mixed results. Studies in which aortic banding was used to induce cardiac LVH have found that C57BL/6 mice do not have significant alterations in CK activity or total myocardial creatine concentrations (26). In contrast, CK-Mito−/− and CK-M/Mito−/− mice on a C57BL/6-129/SV background have shown significant increases at baseline in left ventricular mass and end-diastolic wall thickness (29). It is possible that CK-deficiency causes alterations of the calcium handling proteins and the calcium transients could explain these cardiac complications. It has been recently shown that ablations or mutations of proteins of the sarcoplasmic reticulum (SR) can alter Ca2+ handling and ultimately cardiac hypertrophy in the murine heart (30–32).
Manganese-enhanced MRI (MEMRI) uses the contrast agent and functional calcium analog manganese (Mn). Mn has an ionic radius similar to that of calcium, and is handled similarly in many biological systems (33). Divalent manganese ions (Mn2+) can enter cells through voltage-gated calcium channels (33) and are known to do so by this mechanism. Like Ca2+, Mn2+ can become sequestered in mitochondria and secretory granules (34) and has been used as a tool to better understand Ca2+ pathways. Mn is also an excellent NMR contrast agent due to its T1 relaxation properties (35–38) and was used as the first MRI contrast agent by Lauterbur and coworkers (39). Early work in MRI contrast agent targeting utilized liposomes to deliver Mn to the liver (40), as well as detecting active regions of the brain(41) and heart (42). Preclinical experiments by Hu et al. have shown that changes in calcium influx in cardiac myocytes due to altered cardiac inotropy are detectable using MEMRI (42), however MEMRI has not previously been applied to investigate calcium changes associated with cardiac hypertrophy. In this study, we demonstrate that manganese-enhanced MRI (MEMRI) can be used to detect changes in calcium influx in the murine heart associated with cardiac hypertrophy and creatine kinase ablation. The techniques described in this paper can be immediately applied to in vivo monitoring of cardiac hypertrophy in animal models. In the United States, the lack of FDA approval of MnCl2 for use in human patients currently limits the clinical applicability of the method. However, recent work illustrates that MnCl2 can be safely administered to humans (43), and Nordhøy et al. have suggested based on theoretical models that low micromolar concentrations will likely provide effective contrast enhancement in humans without compromising cardiac function (44).
Methods and Materials
In Vivo Studies
Male C57BL/6 (n=11, age range 11 – 12 wk, mean age 11.0 ± 1.7 wk) and male creatine kinase (M-/Mito-CK−/−) double knockout mice (DBKO, n=12, age range 8 – 12 wk, mean age 9.8 ± 1.3 wk) were imaged using the following protocol. Mice were anesthetized with 1–2% isoflurane and 100% oxygen, placed supine in a 35 mm quadrature volume coil, and imaged in a 9.4 T animal scanner (Bruker Biospec). Body temperature, ECG and respiration were monitored (SA Instruments, Billerica, MA), and ECG data were exported and saved following the experiment. Animals were infused for 30 minutes with 125 mM of MnCl2 using an intraperitoneal (IP) line. The infusion rate was determined by the mass of the mouse and the equation , where r is the infusion rate in μL/min and m is the mass of the mouse in g. This infusion protocol insures each mouse receives a Mn2+ dose of approximately 500 nmol/g body weight (BW). For contrast and signal reference purposes, a standard containing 100μM Mn2+ was placed adjacent to the mouse. Ten days following the initial imaging isoproterenol (ISO) was administered via subcutaneously implanted osmotic pumps (Alzet model 1003D) at a dose of 47 mg/kg/day for 3 days, at which time the pumps were removed and the mice re-imaged (45).
To verify that changes in Mn uptake were due to hypertrophy, a small number of sham mice of both strains were imaged using the above protocol. However, sham mice were not subjected to ISO treatment before re-imaging. For the C57 sham mice, a sample size of n=5 was used for pre-hypertrophic data and n=2 for post-hypertrophic data; for the DBKO mice, a sample size of n=4 was used for pre-hypertrophic data and n=3 for post-hypertrophic data. All procedures were carried out in compliance with our institution’s Institutional Animal Care and Use Committee.
Baseline myocardial T1 measurements were obtained using a single mid-ventricular slice (short-axis view) in the left-ventricular free wall (LVFW) and in the intraventricular septum using a saturation recovery Look-Locker sequence (SRLL) (46) with the following parameters: FOV=2.56cm2, TR=4000 ms, TE=2.4 ms, flip angle=10°, slice thickness=1mm, matrix size=64x64, 50 samples. Equilibrium magnetization (M0) was measured using a FLASH sequence with the following parameters: FOV=2.56cm2, TR=3000 ms, TE=2.4 ms, flip angle=10°, slice thickness=1 mm, matrix size=64x64. Acquisitions were triggered on each heartbeat 5 ms after the R-wave with values of sampling interval τ ranging from ~80 – 150 ms, depending on the R-R interval. To ensure accurate curve fitting, the ECG data were retrieved after the experiment in order to calculate the mean R-R interval during the window of SRLL data acquisition. This mean R-R interval gave the sampling interval τ for curve fitting. For each mouse, the mean heart rate during the time of the infusion was calculated using ECG data from our physiological monitoring station (Table 2).
Table 2.
Mean heart rate (HR) in beats per minute (bpm) for each experimental group. Results are given as mean ± SEM. No statistically significant differences were found between any of the experimental groups (Wilcoxon Rank Sum and Signed Rank tests with Benjamini-Hochberg correction for multiple comparisons).
| Strain | C57 | DBKO |
|---|---|---|
| Cardiac State | ||
| Normal | 487 ± 17 | 526 ± 8 |
| Hypertrophic | 485 ± 15 | 510 ± 8 |
For each mouse scan, the reconstructed image that best visualized the left-ventricle and septum was chosen as the image on which regions-of-interest (ROIs) were manually drawn. Single-slice ROIs were drawn in the LVFW (mean size ≈ 27 pixels), septum (mean size ≈ 24 pixels), and the standard (mean size ≈ 62 pixels). For each ROI location, the mean voxel intensity within the ROI was calculated for each for the 50 sample points to produce a T1* recovery curve; Levenberg-Marquardt three-parameter fitting was then performed on the intensity versus time curve as described in Li et al. (46). T1 values were calculated using customized Matlab software (The MathWorks, Inc., Natick, MA) by using the values from the three-parameter fit and the mean voxel intensity within the corresponding ROI in the equilibrium magnetization image (M0). Example T1 maps are illustrated in Figure 1. An example T1 curve fit is shown in Figure 2 along with several images representative of raw saturation recovery Look-Locker data acquisitions.
Figure 1.

Representative short-axis T1 maps from C57 mouse during diastole before and after manganese contrast. (a) Normal heart, pre-contrast (b) Normal heart, post-contrast (c) Hypertrophic heart, pre-contrast (d) Hypertrophic heart, post contrast.
Figure 2.
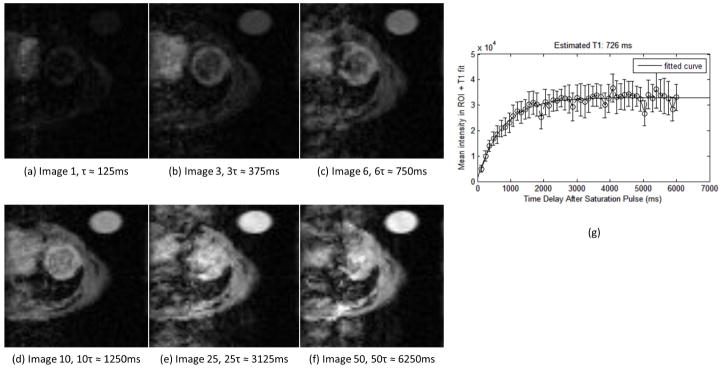
Example of raw data from saturation recovery Look-Locker (SRLL) sequence. A series of 50 images (single mid-ventricular slice, short-axis view) was acquired after an initial saturation pulse. Time delay between sequential images was τ and was computed as the average R-R interval during the acquisition. The images shown here were acquired at (a) 1τ ≈ 125ms (b) 3τ ≈ 375ms (c) 6τ ≈ 750ms (d) 10τ ≈ 1250ms (e) 25τ ≈ 3125ms (f) 50τ ≈ 6250ms. Frame (g) shows an example T1* recovery curve fitted from these data from a region-of-interest (ROI) taken in the left-ventricular free wall. Error bars indicate the standard deviation of the intensity within the ROI.
To estimate the time scale for detectable changes in calcium flux we performed a preliminary experiment using a single C57 mouse given an ≈18 hour exposure of isoproterenol at a dose of 47 mg/kg/day via a subcutaneously implanted osmotic pump.
The ParaVision software package (Bruker Biospec) was used to measure the interior diameter of the left ventricular for measurement of left-ventricular end diastolic diameter to body weight ratio (LVEDd/BW). The diastolic diameter was measured using a mid-ventricular, short-axis view. This interior diameter result was then divided by the body weight, with values given in Table 1.
Table 1.
Left ventricular end-diastolic diameter (LVEDd) normalized to body weight in C57BL/6 and DBKO (M-/Mito-CK −/−) mice, as measured in short-axis MR images. Results are given as mean ± standard deviation in units of mm/g. These results indicate DBKO mice have greater LV dilatation post-hypertrophy vs. C57 mice.
Significant vs. C57 Control (p=0.0023),
Significant vs. DBKO Control (p=0.0013),
Significant vs. C57 Hypertrophic (p=0.0136). Significance is determined by p < 0.05 by Student t- test (C57 Control vs DBKO control, C57 Hypertrophic vs. DBKO Hypertrophic) or paired Student t-test (C57 Control vs. C57 Hypertrophic, DBKO Control vs. DBKO Hypertrophic).
Statistical Analysis
The change in relaxivity, ΔR1 = 1/T1,post-contrast − 1/T1,pre-contrast, was used as the measure of contrast agent uptake. For non-sham mice, statistical analysis on ΔR1 values was performed using a Wilcoxon Signed Rank test to test significance (p < 0.05) within each strain after hypertrophy, and a Wilcoxon Rank Sum test was used to test for differences between the two strains. Rather than pre-hypertrophic and post-hypertrophic data sets, data from sham mice were separated into control and re-imaged data sets since sham mice were not exposed to isoproterenol and thus were not made hypertrophic. Tests for differences between the control and re-imaged data sets were performed using the Wilcoxon Rank Sum test. All p-values were corrected for multiple comparisons using the Benjamini-Hochberg post-hoc test. Mean heart rate data were analyzed using Wilcoxon Rank Sum and Signed Rank tests with Benjamini-Hochberg correction for multiple comparisons.
Linear regression analysis was conducted on ΔR1 vs. heart weight to body weight ratio (HW/BW) measurements. For left-ventricular end diastolic diameter to body weight ratio (LVEDd/BW), a paired Student’s t-test was used to test for changes within each strain after hypertrophy, and an unpaired Student’s t-test was used to test for differences between the two strains. All statistical analyses were performed using the R programming environment (http://www.r-project.org/).
Results
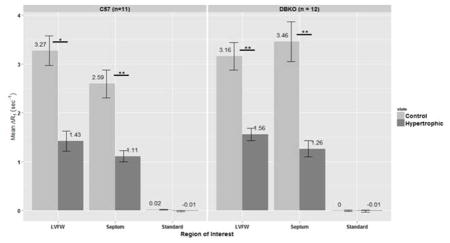
Significant differences in mean ΔR1 values were found between control and hypertrophic mice in the left-ventricular free wall (LVFW) and septum of both the C57 and DBKO strains (p < 0.05, Figure 3). No significant differences in ΔR1 values were found between the two strains (i.e. C57 vs DBKO) in either the LVFW or the septum (also in Figure 3). Figure 4 shows data from our sham mice experiments. We again tested the change in relaxivity, ΔR1, for changes before and after hypertrophy in the LVFW and septum and did not find any statistically significant differences. As an internal control the standard was also examined for changes in ΔR1, yielding no statistically significant results and demonstrating the consistency of the data across all acquisitions.
Figure 3.

Mean ΔR1 by region-of-interest (ROI) for C57 and DBKO mice. ROIs used were: left-ventricular free wall (LVFW), septum and a 100μM MnCl2 standard placed next to the mouse. Error bars represent SEM. Significant differences in ΔR1 are found in both C57 and DBKO mice for the LVFW and septum. No significant differences were found between the two strains in any of the ROIs. Asterisks indicate significance by Wilcoxon Signed Rank test with Benjamini-Hochberg correction for multiple comparisons: * p < 0.01, ** p < 0.001
Figure 4.
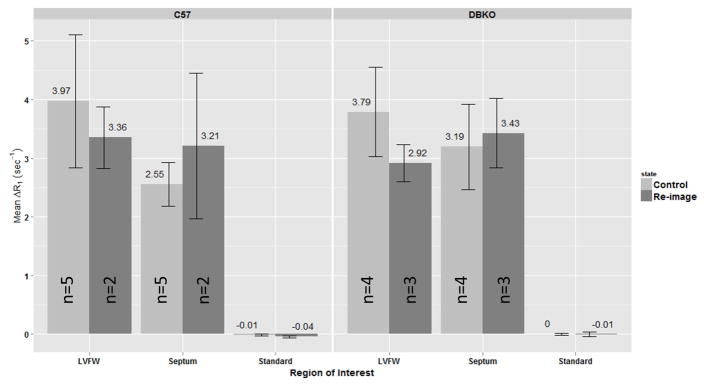
Mean ΔR1 by region-of-interest (ROI) for sham C57 and DBKO mice. ROIs used were: left-ventricular free wall (LVFW), septum and a 100μM MnCl2 standard placed next to the mouse. Error bars represent SEM. No significant differences were found in any of the ROIs considered for either strain of mice (Wilcoxon Rank Sum test with Benjamini-Hochberg correction for multiple comparisons). These data are consistent with our interpretation that the induction of hypertrophy is responsible for the observed changes in calcium handling.
For each strain of mouse, Figure 5 shows the relationship between the degree of hypertrophy, measured in terms of the heart weight to body weight ratio (HW/BW), and ΔR1 for both the LVFW and septum. The grey shaded areas indicate the 95% of confidence interval of the fitted linear regression line. No statistically significant relationship between ΔR1 and HW/BW was found for either strain in either the LVFW or the septum.
Figure 5.
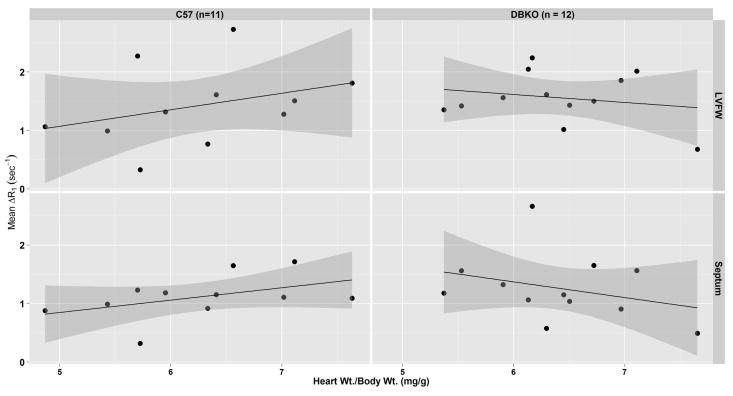
ΔR1 vs. heart weight/body weight ratio for C57 and DBKO mice in the left-ventricular free wall (LVFW) and septum. For each strain and region-of-interest (ROI) a linear model is used to test for correlation between the two parameters. Shaded gray areas represent 95% confidence intervals on the linear models. No significant differences were found between either strain or ROI.
Measurements of left-ventricular end-diastolic diameter to body weight ratio (LVEDd/BW) are given in Table 1. Significant differences were found in hypertrophic C57 versus control C57, for hypertrophic DBKO versus control DBKO, and for hypertrophic DBKO versus hypertrophic C57 (p < 0.05). Table 2 provides data regarding the mean heart rate of each group of mice during the Mn infusion. No statistically significant differences in heart rate were found among the four groups of mice.
For the experiment utilizing a single C57 mouse exposed to approximately 18 hours of isoproterenol treatment a LVFW ΔR1 = 1.05 ± 0.70 sec−1 was measured, demonstrating a marked reduction from the mean ΔR1=3.27 ± 1.0 sec−1 for C57 control mice. In this same experiment we found that the HW/BW = 5.26 mg/g BW, which was less than the mean 6.25 ± 0.81 mg/g BW for C57 mice treated with isoproterenol for 3 days; however, we found that LVEDd/BW=1.86 ± 0.21 mm/g BW, indicating dilatation of the heart and the development of hypertrophy with a much shorter window of stimulation.
To support our results we also attempted to measure left-ventricular blood R1 signal from our 11 C57 mice. Using manual segmentation, we found that blood R1 signal in hypertrophic mice is approximately 63% ± 15% of that in non-hypertrophic controls following Mn administration. However, the R1 measurements acquired using the SRLL pulse sequence depend on data sampled over a time period of several minutes. During this time period variations in heart rate can cause substantial differences in the amount of blood observed in the left-ventricular lumen which can cause large variations in apparent R1. Furthermore, cardiac motion itself results in partial volume and averaging effects within the lumen which contribute additional uncertainty and also act to lower the apparent R1 of blood. For these reasons blood R1 signal within the left-ventricular lumen cannot be accurately measured using our present data.
Discussion
Significant differences in mean ΔR1 were detected between control (i.e., pre-hypertrophic) and hypertrophic mice of both strains (Figure 3). Although changes in calcium handling have been previously associated with cardiac hypertrophy (13,15,17,47,48), we believe we are the first to show the relationship of hypertrophy to changes in ΔR1 using MEMRI.
Although expected, there were no significant differences in ΔR1 between mouse strains, i.e., C57 and DBKO mice, either before or after hypertrophy. There are several possible explanations for this. One possible scenario is that DBKO mice have adapted to the absence of CK by up- or down-regulating other genes involved in calcium handling in order to achieve normal calcium homeostasis. Jameel and Zhang demonstrated that although CK flux decreased in a canine model of left-ventricular hypertrophy, total CK activity remained at normal levels (27). It is also possible that the perturbation to calcium homeostasis caused by the ablation of CK is too small or too fast to be detected using the MEMRI technique. In previous studies in murine skeletal muscle with CK-M ablation, differences in CK function was only observed at high temporal resolution following brief muscle contraction (49). A similar scenario would be difficult to perform in the heart in vivo. It is likely that an exercise stress test performed in the hypertrophic or infarcted heart may reveal a functional difference in the DBKO mice. The data from our sham mice experiments showed no significant changes in ΔR1. This is consistent with the literature that calcium regulation is altered due to the induction of hypertrophy rather than exposure to the contrast agent itself. We showed only very negligible changes in ΔR1 of the Mn-containing standard, again demonstrating the consistency of data acquisition and the quality of our data.
Our mean left-ventricular T1 and R1 measurements were similar to other values in the literature for mice (50,51) and rats (52), although a wide range of values have been reported. In particular, our results differed from Li et al. (46) who used the saturation recovery Look-Locker (SRLL) sequence with similar concentrations of Mn. Coolen et al. have suggested that the phrase “apparent T1” be used to describe myocardial T1 measurements due to dependence of the measurement on the inflow of magnetized blood (50). Wansapura and co-authors have shown that the average drop in T1 from end-systole to end-diastole due to the presence of blood in the LV lateral wall in human patients at 3 T is 43±13% (53). Our technique differs from Li et al. in that rather than attempting to hold the mouse’s heart rate constant by varying the temperature, we have chosen to hold the temperature constant and allow the heart rate of the mouse to vary as described in the Methods section. Moreover, we acquired 50 data points and sampled more of the recovery curve than did Li et al. In addition to these factors, with the relatively low matrix size used, we cannot rule out partial volume effects where blood signal has been included in the ROI.
Two recent papers have examined the issues surrounding the accuracy of T1 mapping sequences. Stikov et al. evaluated 3 common methods for T1 mapping, including Look-Locker, using Bloch equation simulations, phantoms, and human subjects (54). They found that T1 measurements performed in vivo on white matter brain tissue did not agree. They concluded that inaccurate B1 field mapping and incomplete spoiling were the likely causes. Kellman and Hansen examined the question of accuracy and precision of cardiac T1 measurements specifically and found a number of factors which affect the outcome of measurements and can lead to inter-observer discrepancies, including choice of parameters, flip angle, shimming, flow, and magnetization transfer effects (55). Measurements on our standard have proved consistent throughout data acquisition, and because relative changes in relaxivity are considered rather than absolute, the differences in relaxivity measurements among different observers should not affect the results reported here.
As the severity of hypertrophy increases, one might expect larger decreases in ΔR1. We tested this hypothesis using a linear regression model (Figure 3) and no significant relationship was found between the severity of isoproterenol-induced hypertrophy, as measured by the heart weight to body weight ratio (HW/BW), and ΔR1. Experimental results from a single C57 mouse exposed to approximately 18 hours of isoproterenol treatment showed that the development of hypertrophy and decreases in ΔR1 are manifested within a relatively short period of time following the initiation of beta adrenergic stimulation. This suggests that further studies should examine the effects of different amounts of isoproterenol stimulation and correlate them with measured changes in ΔR1 in order to obtain a better understanding of the relationship between morphological change and changes in ionic regulation.
Work by Nahrendorf et al. has shown that CK knockout mice develop left ventricular hypertrophy and dilatation, even without external stimulation (29). Our measurements of LVEDd/BW showed larger ventricular dilatation for DBKO mice, consistent with the results of Nahrendorf et al. However, our results showed no detectable difference in calcium uptake between CK deficient DBKO mice and C57 mice either before or after hypertrophy as measured by Mn uptake. It should also be noted that the Nahrendorf study used 41 wk old CK deficient mice on a mixed C57BL/6 - 129/SVJ background, whereas our study used 8–12 wk old CK deficient mice backcrossed to a pure C57BL/6 background; it is therefore possible that improved ability to detect changes in Mn uptake would be observed by using older mice. Data from Shah and collaborators showed that 129SV/J mice differ from C57 mice in sarcoplasmic reticulum calcium content, left-ventricular ejection fraction, fractional shortening, and cellular volume (56). Prior work from our laboratory demonstrated increased sensitivity to isoproterenol-induced cardiac hypertrophy in 129SV/J mice, as measured by the heart weight to body weight ratio (57). Although no differences between the strains were observed under baseline conditions, it is possible that increased cardiac stress due to exercise or to positive inotropic stimulation (e.g., dobutamine) could reveal differences not appreciated here.
A recent paper by Coelho-Filho et al. investigated the detection of cardiac myocyte hypertrophy directly by measuring the intracellular lifetime (τic) of water molecules. This was done by infusing mice with gadolinium diethylenetriaminepentacetic acid (Gd-DTPA) (58). Using a two-compartment exchange model between the extracellular volume (ECV) and cardiac myocytes, they showed a significantly increased τic in hypertrophic mice versus control mice. The relationship between blood R1 and myocardial R1 determines τic, which can then be related to myocyte volume. Our results showed increased R1 following hypertrophy, as indicated by the observed reduction in ΔR1. These results are consistent with Coelho-Filho et al., whose observations show reduced R1 in tissue compared to blood in hypertrophic mice. The changes in R1 observed in Coelho-Filho et al., however, are attributed to changes in myocyte volume that affect the amount of water exchange between myocytes and the ECV.
A recent paper examining the effects of isoproterenol on myocardial perfusion in the rat heart demonstrated significantly higher myocardial blood flow following isoproterenol treatment (59) and suggests consideration of the delivery pathway of Mn. Intraperitoneal (IP) rather than intravenous (IV) infusion of Mn performed in these experiments means that Mn is available for direct absorption by multiple organs including the liver, pancreas, and kidneys. Potential increases in the amount of perfusion or Mn uptake in these other organ systems following isoproterenol treatment represents an alternative explanation for the decrease in R1 observed in this experiment If changes in perfusion were the dominant effect in determining the amount of Mn uptake by the heart then the perfusion of other organ systems must increase in kind and exceed that of the heart. Isoproterenol treatment is not known to significantly increase perfusion to these other organ systems, and our results showed no statistically significant differences in heart rate before and after hypertrophy in both the experimental and sham groups. Furthermore, the high concentration (125 mM) and dose (500 nmol/kg) of Mn ensure Mn availability is not limiting. Additionally, these results are consistent with other investigations demonstrating decreased calcium uptake in the heart following isoproterenol treatment (57,60). Thus, the conclusion that the measured decrease in Mn uptake is due to isoproterenol-induced hypertrophy and not to changes in the relative changes in perfusion or Mn uptake to other organ systems remains the most parsimonious interpretation of these data. The analysis presented here can be expanded and improved upon in several different ways. Prolonged exposure to the anesthetic isoflurane is known to affect myocardial blood flow (61) and calcium channels (62), however how isoflurane affects the magnitude of the results reported here remains unknown. The experiments should be repeated with lower concentrations of manganese to see if the dynamic range of the technique can be improved. Dobutamine has been shown to increase image contrast in T1-weighted MEMRI experiments and, due to the increased energy demands of the myocardium under stress, would be useful for probing possible differences between C57 and DBKO mice (42). Our preliminary results using only one day of isoproterenol treatment suggest measurable changes in ΔR1 occur in as few as ≈18 hours, suggesting that lower doses and/or shorter durations of isoproterenol could be used to detect altered calcium homeostasis in pre-hypertrophic mice. Isoproterenol-induced hypertrophy is a limited model in which chronic β-adrenergic signaling is used as the initiating stimulus. Absolute quantification of Mn concentrations in the heart and other organ systems by means of mass spectrometry was not investigated as part of this experiment but would provide useful information on absolute changes in Mn in the heart following isoproterenol treatment. This may also help to verify that the observed changes in this experiment are not due to increased Mn uptake in other organ systems. Investigation of other models of cardiac hypertrophy, such as aortic banding and angiotensin II, would serve to widen the scope of this investigation and make its results more generalizable to the human population.
In this paper, we have shown that MEMRI can be applied to study alterations in calcium handling in preclinical models of cardiac hypertrophy. Our results have demonstrated significant reduction in ΔR1 in hypertrophic mice versus non-hypertrophic controls. Moreover, the detection of hypertrophy by changes in R1 alone is completely non-invasive in that it does not require the use of blood samples to measure hematocrit (58). We investigated whether the ablation of CK produces measurable differences in ΔR1 by studying both C57BL/6 and M-/Mito-CK−/− double knockout (DBKO) mice and found no difference between the two strains. Finally, we have found no measurable correlation between heart weight to body weight ratio and ΔR1. Because changes to calcium handling typically precede changes in gross morphology, this study represents an important initial step toward the early detection of cardiac hypertrophy. Moreover, it opens the possibility of detecting different signal changes in ΔR1 for different pathways of hypertrophy. This may allow more targeted and effective treatment or improved identification of patients at risk for sudden cardiac death. Although the administration of free manganese to human patients lacks FDA approval, mangafodipir trisodium (Teslascan®), a chelated, hepatocyte-specific manganese-based contrast agent, has been approved. Recent human trials have shown free manganese can be administered without adverse side-effects (43), leaving open the possibility for future human studies using the techniques presented in this paper.
Acknowledgments
Grant Support: Research reported in this publication was supported by the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000430 and NIH 1R01CA133490. This research was also supported, in part, by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under grant number T32 EB002103, and The Florsheim Foundation.
We would to thank the Xin Yu, Ph.D., Wen Li, Ph.D., and Kai Jiang of the Yu laboratory at Case Western Reserve University for providing us with the SRLL pulse sequence, and for all their support in helping us to implement it on our equipment.
Thanks to Gregory S. Karczmar, Ph.D. for many productive conversations relating to our experiments. Thanks also to Jim Vosciky, Erica Markiewicz, and Marta Zamora for the assistance with the animal setup.
List of Abbreviations
- ACE
Angiotensin-converting enzyme
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- BW
Body weight
- C57
C57BL/6 mice
- CK
Creatine kinase
- DBKO
CK-M/Mito −/− double knockout mice
- ECG
Electrocardiogram
- ECV
Extracellular volume
- FDA
Food and Drug Administration (United States)
- FLASH
Fast, low-angle shot
- FOV
Field of view
- Gd-DTPA
Gadolinium diethylenetriaminepentacetic acid
- HCM
Hypertrophic cardiomyopathy
- HW
Heart weight
- HW/BW
Heart weight to body weight ratio
- ICD
Implantable cardioverter-difibrillator
- IP
Intraperitoneal
- LVEDd
Left-ventricular end-diastolic diameter
- LVEDd/BW
LVEDd to body weight ratio
- LVFW
Left-ventricular free wall
- LVH
Left-ventricular Hypertrophy
- MEMRI
Manganese-enhanced magnetic resonance imaging
- Mn
Manganese
- NCX
Sodium-calcium exchanger
- PCr
Phosphocreatine
- ROI
Region-of-interest
- SCD
Sudden cardiac death
- SEM
Standard error of the mean
- SR
Sarcoplasmic reticulum
- SRLL
Saturation recovery Look-Locker
Contributor Information
Martin Andrews, Email: mmandrews@uchicago.edu, University of Chicago, 5812 S. Ellis Ave, Chicago, Il, 60637.
Maryellen L. Giger, Email: m-giger@uchicago.edu, University of Chicago, 5812 S. Ellis Ave, Chicago, Il, 60637.
Brian B. Roman, Email: broman@uchicago.edu, University of Chicago, 5812 S. Ellis Ave, Chicago, Il, 60637.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functio. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 3.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–831. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 4.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults: Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Circulation. 1995;92:785–9. doi: 10.1161/01.CIR.92.4.785. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of Hypertrophic Cardiomyopathy-Related Death: Revisited in a Large Non-Referral-Based Patient Population. Circulation. 2000;102:858–64. doi: 10.1161/01.CIR.102.8.858. [DOI] [PubMed] [Google Scholar]
- 6.Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–39. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–6. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 8.Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–41. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Braunwald E. Evolution of hypertrophic cardiomyopathy to a contemporary treatable disease. Circulation. 2012;126:1640–4. doi: 10.1161/CIRCULATIONAHA.112.123174. [DOI] [PubMed] [Google Scholar]
- 10.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–20. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hobai IA, O’Rourke B. The potential of Na+/Ca2+ exchange blockers in the treatment of cardiac disease. Expert Opin Investig Drugs. 2004;13:653–64. doi: 10.1517/13543784.13.6.653. [DOI] [PubMed] [Google Scholar]
- 12.Reuter H, Henderson SA, Han T, Matsuda T, Baba A, Ross RS, Goldhaber JI, Philipson KD. Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ Res. 2002;91:90–2. doi: 10.1161/01.res.0000027529.37429.38. [DOI] [PubMed] [Google Scholar]
- 13.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–9. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacques A, Hoskins AC, Kentish JC, Marston SB. From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J Muscle Res Cell Motil. 2008;29:239–46. doi: 10.1007/s10974-009-9174-0. [DOI] [PubMed] [Google Scholar]
- 15.Marston SB. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J Cardiovasc Transl Res. 2011;4:245–55. doi: 10.1007/s12265-011-9266-2. [DOI] [PubMed] [Google Scholar]
- 16.Van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJM, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–83. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 17.Teekakirikul P, Padera RF, Seidman JG, Seidman CE. Hypertrophic cardiomyopathy: Translating cellular cross talk into therapeutics. J Cell Biol. 2012;199:417–21. doi: 10.1083/jcb.201207033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendland MF. Applications of manganese-enhanced magnetic resonance imaging (MEMRI) to imaging of the heart. NMR Biomed. 2004;17:581–94. doi: 10.1002/nbm.943. [DOI] [PubMed] [Google Scholar]
- 19.Massaad CA, Pautler RG. Manganese-enhanced magnetic resonance imaging (MEMRI) Methods Mol Biol. 2011;711:145–74. doi: 10.1007/978-1-61737-992-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva AC, Lee JH, Aoki I, Koretsky AP. Manganese-enhanced magnetic resonance imaging (MEMRI): methodological and practical considerations. NMR Biomed. 2004;17:532–43. doi: 10.1002/nbm.945. [DOI] [PubMed] [Google Scholar]
- 21.Boretius S, Frahm J. Manganese-enhanced magnetic resonance imaging. Methods Mol Biol. 2011;771:531–68. doi: 10.1007/978-1-61779-219-9_28. [DOI] [PubMed] [Google Scholar]
- 22.Delattre BMA, Braunersreuther V, Hyacinthe J-N, Crowe LA, Mach F, Vallée J-P. Myocardial infarction quantification with Manganese-Enhanced MRI (MEMRI) in mice using a 3T clinical scanner. NMR Biomed. 2010;23:503–13. doi: 10.1002/nbm.1489. [DOI] [PubMed] [Google Scholar]
- 23.Tian R. Thermodynamic limitation for the sarcoplasmic reticulum Ca(2+)-ATPase contributes to impaired contractile reserve in hearts. Ann N Y Acad Sci. 1998;853:322–4. doi: 10.1111/j.1749-6632.1998.tb08290.x. [DOI] [PubMed] [Google Scholar]
- 24.Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 26.Lygate CA, Fischer A, Sebag-Montefiore L, Wallis J, ten Hove M, Neubauer S. The creatine kinase energy transport system in the failing mouse heart. J Mol Cell Cardiol. 2007;42:1129–36. doi: 10.1016/j.yjmcc.2007.03.899. [DOI] [PubMed] [Google Scholar]
- 27.Jameel MN, Zhang J. Myocardial energetics in left ventricular hypertrophy. Curr Cardiol Rev. 2009;5:243–50. doi: 10.2174/157340309788970379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 29.Nahrendorf M, Spindler M, Hu K, Bauer L, Ritter O, Nordbeck P, Quaschning T, Hiller K-H, Wallis J, Ertl G, Bauer WR, Neubauer S. Creatine kinase knockout mice show left ventricular hypertrophy and dilatation, but unaltered remodeling post-myocardial infarction. Cardiovasc Res. 2005;65:419–27. doi: 10.1016/j.cardiores.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Guo W, Jorgensen AO, Jones LR, Campbell KP. Biochemical characterization and molecular cloning of cardiac triadin. J Biol Chem. 1996;271:458–65. doi: 10.1074/jbc.271.1.458. [DOI] [PubMed] [Google Scholar]
- 31.Kirchhefer U, Jones LR, Begrow F, Boknik P, Hein L, Lohse MJ, Riemann B, Schmitz W, Stypmann J, Neumann J. Transgenic triadin 1 overexpression alters SR Ca2+ handling and leads to a blunted contractile response to beta-adrenergic agonists. Cardiovasc Res. 2004;62:122–34. doi: 10.1016/j.cardiores.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Kirchhefer U, Baba HA, Kobayashi YM, Jones LR, Schmitz W, Neumann J. Altered function in atrium of transgenic mice overexpressing triadin 1. Am J Physiol Heart Circ Physiol. 2002;283:H1334–43. doi: 10.1152/ajpheart.00937.2001. [DOI] [PubMed] [Google Scholar]
- 33.Dryselius S, Grapengiesser E, Hellman B, Gylfe E. Voltage-dependent entry and generation of slow Ca2+ oscillations in glucose-stimulated pancreatic beta-cells. Am J Physiol. 1999;276:E512–8. doi: 10.1152/ajpendo.1999.276.3.E512. [DOI] [PubMed] [Google Scholar]
- 34.Rorsman P, Hellman B. The interaction between manganese and calcium fluxes in pancreatic beta-cells. Biochem J. 1983;210:307–14. doi: 10.1042/bj2100307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angtuaco TL, Mattison DR, Thomford PJ, Jordan J. Effect of manganese on human placental spin-lattice (T1) and spin-spin (T2) relaxation times. Physiol Chem Phys Med NMR. 1986;18:41–8. [PubMed] [Google Scholar]
- 36.Kang YS, Gore JC. Studies of tissue NMR relaxation enhancement by manganese. Dose and time dependences. Invest Radiol. 19:399–407. doi: 10.1097/00004424-198409000-00012. [DOI] [PubMed] [Google Scholar]
- 37.Mamourian AC, Burnett KR, Goldstein EJ, Wolf GL, Kressel HY, Baum S. Proton relaxation enhancement in tissue due to ingested manganese chloride: time course and dose response in the rat. Physiol Chem Phys Med NMR. 1984;16:123–8. [PubMed] [Google Scholar]
- 38.Wydrzynski TJ, Marks SB, Schmidt PG, Govindjee, Gutowsky HS. Nuclear magnetic relaxation by the manganese in aqueous suspensions of chloroplasts. Biochemistry. 1978;17:2155–62. doi: 10.1021/bi00604a020. [DOI] [PubMed] [Google Scholar]
- 39.Lauterbur P, MM-D, Rudin A. Augmentation of tissue water proton spin-lattice relaxation rates by in-vivo addition of paramagnetic ions. Front Biol Energy. 1978;1 [Google Scholar]
- 40.Bacic G, Niesman MR, Magin RL, Swartz HM. NMR and ESR study of liposome delivery of Mn2+ to murine liver. Magn Reson Med. 1990;13:44–61. doi: 10.1002/mrm.1910130107. [DOI] [PubMed] [Google Scholar]
- 41.Lin YJ, Koretsky AP. Manganese ion enhances T1-weighted MRI during brain activation: an approach to direct imaging of brain function. Magn Reson Med. 1997;38:378–88. doi: 10.1002/mrm.1910380305. [DOI] [PubMed] [Google Scholar]
- 42.Hu TC, Pautler RG, MacGowan GA, Koretsky AP. Manganese-enhanced MRI of mouse heart during changes in inotropy. Magn Reson Med. 2001;46:884–90. doi: 10.1002/mrm.1273. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes JL, Storey P, da Silva JA, de Figueiredo GS, Kalaf JM, Coelho OR. Preliminary assessment of cardiac short term safety and efficacy of manganese chloride for cardiovascular magnetic resonance in humans. J Cardiovasc Magn Reson. 2011;13:6. doi: 10.1186/1532-429X-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nordhøy W, Anthonsen HW, Bruvold M, Jynge P, Krane J, Brurok H. Manganese ions as intracellular contrast agents: proton relaxation and calcium interactions in rat myocardium. NMR Biomed. 2003;16:82–95. doi: 10.1002/nbm.817. [DOI] [PubMed] [Google Scholar]
- 45.Slawson SE, Roman BB, Williams DS, Koretsky AP. Cardiac MRI of the normal and hypertrophied mouse heart. Magn Reson Med. 1998;39:980–7. doi: 10.1002/mrm.1910390616. [DOI] [PubMed] [Google Scholar]
- 46.Li W, Griswold M, Yu X. Rapid T1 mapping of mouse myocardium with saturation recovery Look-Locker method. Magn Reson Med. 2010;64:1296–303. doi: 10.1002/mrm.22544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J Biol Chem. 1994;269:1603–5. [PubMed] [Google Scholar]
- 48.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–83. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roman BB, Meyer RA, Wiseman RW. Phosphocreatine kinetics at the onset of contractions in skeletal muscle of MM creatine kinase knockout mice. AJP Cell Physiol. 2002;283:C1776–C1783. doi: 10.1152/ajpcell.00210.2002. [DOI] [PubMed] [Google Scholar]
- 50.Coolen BF, Geelen T, Paulis LEM, Nauerth A, Nicolay K, Strijkers GJ. Three-dimensional T1 mapping of the mouse heart using variable flip angle steady-state MR imaging. NMR Biomed. 2011;24:154–62. doi: 10.1002/nbm.1566. [DOI] [PubMed] [Google Scholar]
- 51.Naresh NK, Xu Y, Klibanov AL, Vandsburger MH, Meyer CH, Leor J, Kramer CM, French BA, Epstein FH. Monocyte and/or macrophage infiltration of heart after myocardial infarction: MR imaging by using T1-shortening liposomes. Radiology. 2012;264:428–35. doi: 10.1148/radiol.12111863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Ye Q, Zheng J, Schelbert EB, Hitchens TK, Ho C. Improve myocardial T1 measurement in rats with a new regression model: Application to myocardial infarction and beyond. Magn Reson Med. 2013 doi: 10.1002/mrm.24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wansapura J, Gottliebson W, Crotty E, Fleck R. Cyclic variation of T1 in the myocardium at 3 T. Magn Reson Imaging. 2006;24:889–93. doi: 10.1016/j.mri.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 54.Stikov N, Boudreau M, Levesque IR, Tardif CL, Barral JK, Pike GB. On the accuracy of T 1 mapping: Searching for common ground. Magn Reson Med. 2014 doi: 10.1002/mrm.25135. [DOI] [PubMed] [Google Scholar]
- 55.Kellman P, Hansen MS. T1-mapping in the heart: accuracy and precision. J Cardiovasc Magn Reson. 2014;16:2. doi: 10.1186/1532-429X-16-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah AP, Siedlecka U, Gandhi A, Navaratnarajah M, Al-Saud SA, Yacoub MH, Terracciano CM. Genetic background affects function and intracellular calcium regulation of mouse hearts. Cardiovasc Res. 2010;87:683–93. doi: 10.1093/cvr/cvq111. [DOI] [PubMed] [Google Scholar]
- 57.Waters SB, Diak DM, Zuckermann M, Goldspink PH, Leoni L, Roman BB. Genetic background influences adaptation to cardiac hypertrophy and Ca(2+) handling gene expression. Front Physiol. 2013;4:11. doi: 10.3389/fphys.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coelho-Filho OR, Shah RV, Mitchell R, Neilan TG, Moreno H, Simonson B, Kwong R, Rosenzweig A, Das S, Jerosch-Herold M. Quantification of cardiomyocyte hypertrophy by cardiac magnetic resonance: implications for early cardiac remodeling. Circulation. 2013;128:1225–33. doi: 10.1161/CIRCULATIONAHA.112.000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desrois M, Kober F, Lan C, Dalmasso C, Cole M, Clarke K, Cozzone PJ, Bernard M. Effect of isoproterenol on myocardial perfusion, function, energy metabolism and nitric oxide pathway in the rat heart - a longitudinal MR study. NMR Biomed. 2014;27:529–38. doi: 10.1002/nbm.3088. [DOI] [PubMed] [Google Scholar]
- 60.Gøtzsche O. Decreased myocardial calcium uptake after isoproterenol in streptozotocin-induced diabetic rats. Studies in the in vitro perfused heart. Lab Invest. 1983;48:156–61. [PubMed] [Google Scholar]
- 61.Kober F, Iltis I, Cozzone PJ, Bernard M. Myocardial blood flow mapping in mice using high-resolution spin labeling magnetic resonance imaging: influence of ketamine/xylazine and isoflurane anesthesia. Magn Reson Med. 2005;53:601–6. doi: 10.1002/mrm.20373. [DOI] [PubMed] [Google Scholar]
- 62.Camara AK, Begic Z, Kwok WM, Bosnjak ZJ. Differential modulation of the cardiac L- and T-type calcium channel currents by isoflurane. Anesthesiology. 2001;95:515–24. doi: 10.1097/00000542-200108000-00038. [DOI] [PubMed] [Google Scholar]