

SUMMARY
Altered commensal communities are associated with human disease. IgA mediates intestinal homeostasis and regulates microbiota composition. Intestinal IgA is produced at high levels as a result of T follicular helper cell (TFH) and B cell interactions in germinal centers. However, the pathways directing host IgA responses towards the microbiota remain unknown. Here, we report that signaling through the innate adaptor MyD88 in gut T cells coordinates germinal center responses, including TFH and IgA+ B cell development. TFH development is deficient in germfree mice and can be restored by feeding TLR2 agonists that activate T cell intrinsic MyD88 signaling. Loss of this pathway diminishes high affinity IgA targeting of the microbiota and fails to control the bacterial community, leading to worsened disease. Our findings identify that T cells converge innate and adaptive immune signals to coordinate IgA against the microbiota, constraining microbial community membership to promote symbiosis.
INTRODUCTION
The development and function of the mammalian immune system is dependent upon signals conveyed by the microbiota (Belkaid and Hand, 2014; Hooper et al., 2012; Kamada et al., 2013). In particular, the abundance and type of T lymphocytes in the gut is severely reduced in germfree (GF) mice (Atarashi et al., 2011; Ivanov et al., 2008; Mazmanian et al., 2005; Round and Mazmanian, 2010). While T cell activation is governed by ligation of the T cell receptor (TCR), the quality and nature of the response is dependent on secondary signals such as the cytokine milieu. The identification that T cells express receptors associated with innate signaling such as Toll like receptors (TLRs) and the IL-1R suggests that T cells could directly utilize these signals as an additional mechanism to control responses (Caramalho et al., 2003; Kubinak and Round, 2012). This would be particularly relevant within the gut where a constant and abundant source of commensal ligands exists. Supporting this, a single commensal species utilizes TLR2 to promote its own colonization (Round et al., 2011). Recent studies have identified that MyD88 functions within splenic T cells to overcome Treg suppression during immunization (Schenten et al., 2014), identifying the relevance of this pathway to immunity. However, it remains unknown whether these signals provided by the microbiota act directly on T cells in the gut to influence mutualism.
The synthesis of IgA has been shown to promote intestinal health (Berry et al., 2012; Brandtzaeg, 2013; Fagarasan et al., 2002; Kawamoto et al., 2012; Lindner et al., 2012; Slack et al., 2009). IgA is the most abundantly produced antibody in mammals with most being secreted into the intestine. Because of this, IgA represents a key host mechanism for regulating commensal microbial communities. A recent study has shown that IgA binds colitogenic members of the microbiota (Palm et al., 2014), which highlights the role of IgA as an important mediator of microbiota-induced inflammatory disease and a potential diagnostic biomarker. T cell help is required for the generation of high affinity antibody production. In particular, TFH cells directly interact with B cells in the germinal center (GC) to induce somatic hypermutation and class switching (Crotty, 2011). Our understanding of the molecular pathways that influence GC formation in the gut and how the microbiota influences these pathways remains incomplete.
In this present study we identify that a classic innate immune molecule, MyD88, can function within the T cell compartment in the gut. Loss of MyD88 signaling in T cells leads to reductions in TFH cells and IgA producing B cells, demonstrating a key role for molecular pathways that converge on this adapter molecule leading to appropriate GC formation. Moreover, GC formation in the gut is orchestrated by signals provided by the microbiota in a T cell intrinsic MyD88 dependent manner. Loss of GC formation leads to reduced IgA production and disrupted targeting of commensal bacterial populations. Animals lacking MyD88 within the T cell compartment fail to control mucosally associated communities of bacteria resulting in dysbiosis. Finally, we demonstrate that animals lacking T cell intrinsic MyD88 develop worsened disease that can be rescued by a microbial transplant from a healthy donor. Thus, we have identified a host molecular pathway that can integrate signals from the microbiota to promote GC formation and IgA production against intestinal bacteria to control the composition of these communities to ensure a benign symbiotic interaction.
RESULTS
MyD88 Dependent Signaling in T cells Influences GC Responses in the Gut
Whether innate signaling by T cells influences the establishment of beneficial bacterial communities and host health remains to be elucidated. As MyD88 is a key molecule that governs signaling through multiple innate receptors, we crossed a MyD88-floxed animal with a T cell-specific Cre-driver to produce an animal model where MyD88 is specifically knocked out within T cells but retained in other cell types (the T-MyD88−/− mouse) (Figure S1) (Chang et al., 2013; Schenten et al., 2014). This allowed us to test whether innate and adaptive immune pathways converge to promote host-microbiota symbiosis within the gut. Recent studies have identified that T cell intrinsic MyD88 signaling influences systemic induction of TH1 and TH17 cells during immunization (Chang et al., 2013; Raetz et al., 2013; Schenten et al., 2014). Thus, we first broadly examined CD4+ T cell development during steady state conditions. We did not observe differences in TH17, TH1 or T regulatory cells (Tregs) within the spleen, mesenteric lymph nodes (MLNs), or colonic lamina propria (cLP) (Figures S2, S3). Consistent with previous reports, we observed slight defects in the abundance of TH1 and TREGS within the small intestinal lamina propria (siLP) and TH17 cells within the Peyer’s patch (PP) (Figure S3) (Reynolds et al., 2010; Schenten et al., 2014). However, the most striking difference observed was within the TFH cell compartment in both the PP and MLNs in T-MyD88−/− animals (Figure 1A and Figures S4A–K). TFH cells are one of the most abundant subsets of CD4+ helper T cells within the PPs and play a fundamental role in the generation of antibody-mediated responses against the microbiota (Crotty, 2011; Linterman et al., 2012). Therefore, we focused our analysis on GC formation in the gut. TFH cells are broadly characterized by the expression of two or more of the following surface markers; ICOS, CXCR5, and PD-1. Sub-types of TFH cells can be delineated using these markers. These include GC-resident TFH (GC-TFH) cells (CXCR5hi PD-1hi) whose function is to promote class switching and somatic hypermutation of naive B cells to produce high affinity IgA, and non-GC-resident TFH (non-GC-TFH) cells (CXCR5intPD-1int) that will ultimately migrate into the GC to promote B cell activity (Shulman et al., 2013). Any combination of these markers identifies a defect in any TFH subset in the PPs of T-MyD88−/− animals that cannot be accounted for by changes in PP cellularity (Figures 1A–C and Figure S4A–I). These data suggest that T cell intrinsic MyD88 signaling impacts multiple stages of TFH development. Importantly, these effects were gut-specific because they were not observed in the spleen (Figures S4L and S4M), indicating that T cell intrinsic MyD88 signaling influences steady state development of TFH cells in the intestine.
Figure 1. T cells utilize MyD88 signaling to coordinate germinal center responses.
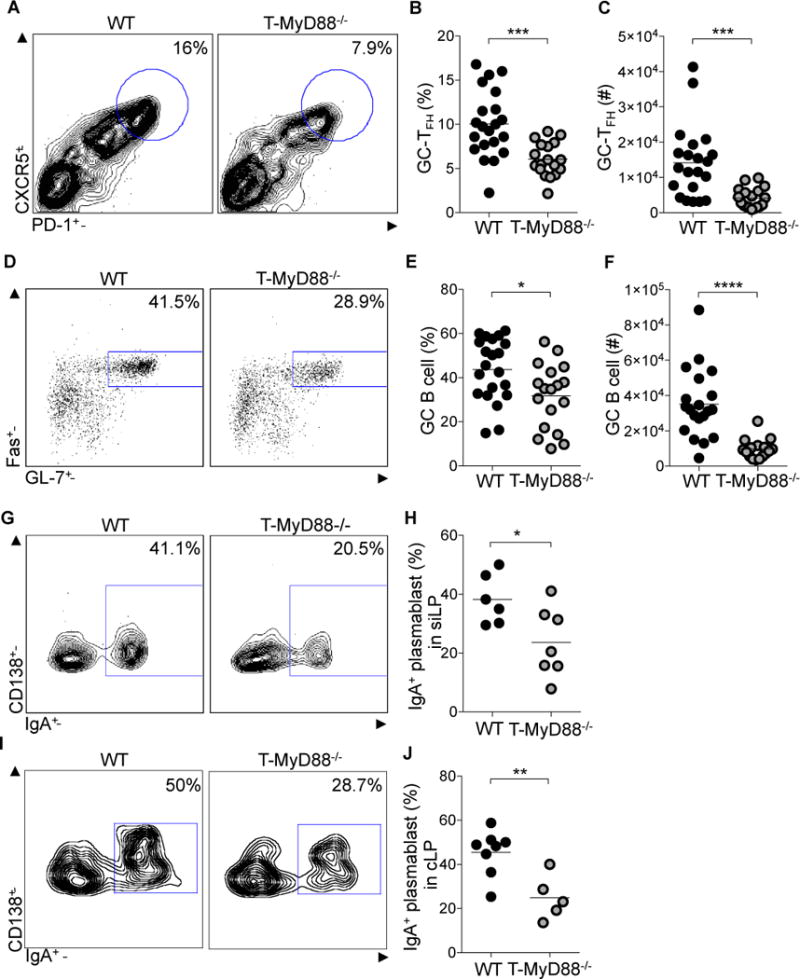
(A–C) GC-TFH cells from PPs, defined by CD3+CD4+B220−CXCR5hiPD-1hi, were measured by flow cytometry. (A) Representative plots were previously gated on CD3+CD4+B220− cells. (B) Frequency and (C) absolute numbers of GC-TFH cells (n= 21 WT, n=18 T-MyD88−/−).
(D–F) GC B cells, defined by B220+IgDlowFas+GL-7+, from PPs were measured by flow cytometry. (D) Representative plots were previously gated on B220+IgDlow cells. (E) Frequency and (F) absolute numbers of GC B cells (n=21 WT, n=18 T-MyD88−/−).
(G–J) IgA+ plasmablasts, defined as B220−CD138+IgA+, within the siLP and cLP were measured by flow cytometry (G), Representative plots were previously gated on CD138+ B220− cells from siLP. (H) Frequencies of IgA+ plasmablasts in siLP (n=6 WT, n=7 T-MyD88−/−). (I) Representative plots were previously gated on CD138+ B220− cells from cLP. (J) Frequencies of IgA+ plasmablasts in cLP (n=8 WT, n=5 T-MyD88−/−). Unpaired two-tailed Student’s t-tests were used for all comparisons. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***). Lines in scatterplots represent means. See also Figures S1–4.
TFH-dependent instruction of GC B cells induces IgA+ plasmablast migration to the lamina propria of the small and large intestine leading to secretion of high-affinity IgA into the intestinal lumen. Based on the observed deficiencies in TFH development, intestinal B cell function was examined. Percentages and absolute numbers of PP GC B cells were significantly reduced in T-MyD88−/− mice (Figures 1D–F) indicating a defect in the GC response. Consequently, IgA+ plasmablasts in the siLP and cLP were also diminished (Figures 1G–J). These results demonstrate that MyD88 signaling within T cells regulates appropriate GC formation within the gut.
Microbiota derived signals influence the Presence of TFH Cells in the Gut
B cell responses within the gut are sensitive to the presence of the microbiota. Indeed, GF mice have fewer B cells and lower IgA levels. MyD88 functions downstream of Toll-like receptors (TLRs) which are involved in responses to bacterial products. Thus, our observations suggest that TFH development within the gut is influenced by signals from the commensal microbiota. To test this we isolated PPs from either GF or specific pathogen free (SPF) mice from two different backgrounds of animals (Balb/c and C57Bl/6) and examined the TFH population. Both C57Bl/6 and Balb/c GF mice had significantly reduced numbers of GC-TFH cells within the PPs when compared to SPF mice (Figures 2A–D). Moreover, GC-TFH cell populations were severely reduced when C57Bl/6 SPF animals were treated with an antibiotic cocktail that depletes the microbiota (Figures 2E–G). This demonstrates that development and maintenance of GC-TFH cells within the gut is promoted by the presence of commensal organisms.
Figure 2. Microbiota dependent TFH development relies on T cell intrinsic MyD88 signaling.
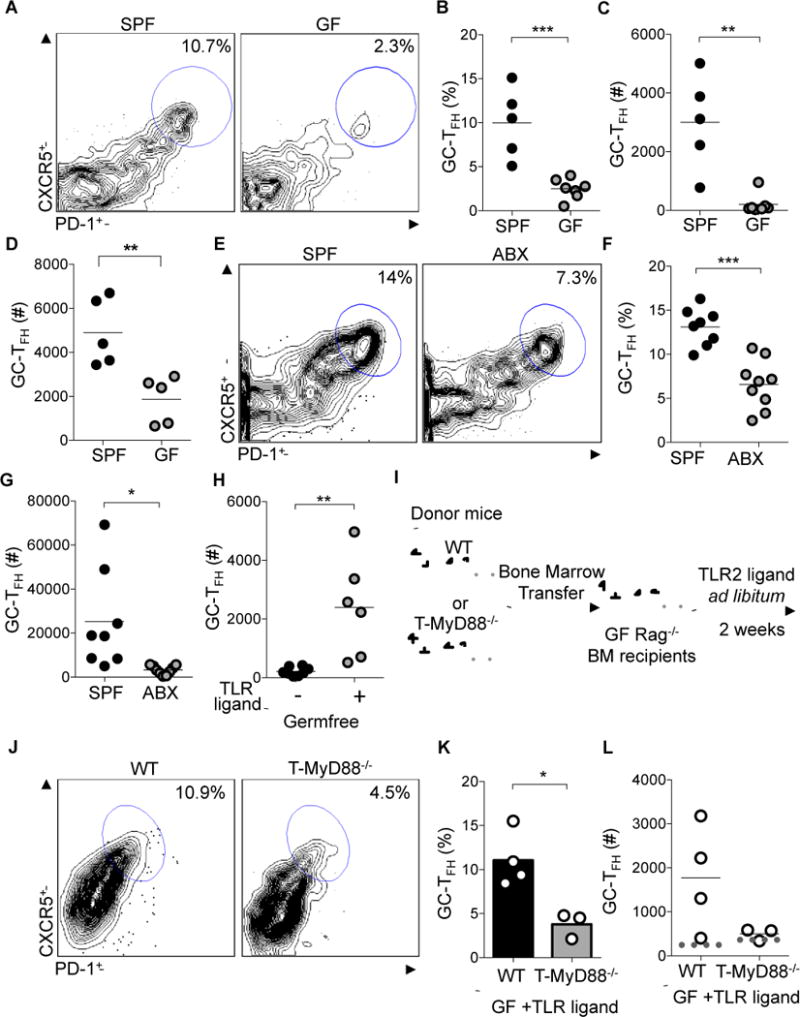
(A–C) GC-TFH cells from PPs, defined by CD3+CD4+B220−CXCR5hiPD-1hi, were compared between SPF and GF Balb/c mice with flow cytometry. (A) Representative plots were previously gated on CD3+CD4+B220− cells. (B) Frequency and (C) absolute numbers of GC-TFH cells (n= 5 SPF, n=7 GF).
(D) GC-TFH cells from PPs were compared between SPF and GF C57Bl/6 mice with flow cytometry (representative plots not shown) to obtain absolute numbers of GC-TFH cells (n=5 for each group).
(E–G) GC-TFH cells from PPs werecompared between SPF mice treated with oral antibiotics (ABX) and non-treated SPF animals with flow cytometry. (E) Representative plots were previously gated on CD3+CD4+B220− cells. (F) Frequency and (G), absolute numbers of TFH cells (n= 8 SPF, n=9 ABX).
(H) Pam3CSK was provided in the drinking water of GF animals for two weeks to calculate absolute numbers of TFH cells in animals given TLR ligand (+)(n=6) or not (−)(n=8).
(I) Experimental schematic showing GF Rag−/− mice reconstituted with either WT or T-MyD88−/− bone marrow and subsequently treated with TLR2 ligand, Pam3CSK, in the drinking water.
(J–L) GC-TFH cells from MLNs were compared between GF Rag−/− mice reconstituted with WT or T-MyD88−/− bone marrow and fed the TLR2 ligand Pam3CSK. (J) Representative plots were previously gated on CD3+CD4+B220− cells. (K) Frequency and (L) absolute numbers of GC-TFH cells (dotted line represents the number of cells within these populations when GF-Rag mice are given bone marrow for either genotype and not stimulated) (n=4 WT and n=3 T-MyD88−/−). P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***). Lines in scatterplots and bar graphs represent means.
To determine whether live bacterial colonization is required for induction of GC responses or whether bacterial products alone are sufficient, we placed the TLR2 ligand, Pam3CysK, in the drinking water of germfree mice. Oral treatment of GF mice with a purified TLR2 agonist alone was capable of significantly increasing GC-TFH abundance within PPs (Figure 2H). To definitively test the relevance of T cell intrinsic MyD88 signaling during microbiota dependent TFH development in vivo, we reconstituted GF Rag−/− mice with bone marrow from either WT or T-MyD88−/− animals (Figure 2I). As GF Rag−/− animals do not develop PPs and a bone marrow reconstitution does not correct this, we analyzed the defect in TFH development within the MLNs of these mice. Eight weeks after transfer, animals were orally treated with a purified TLR2 agonist. Strikingly, TFH development occurred in GF WT animals in response to purified TLR2 ligands, while TFH development was not induced in TLR2 treated GF animals reconstituted with T-MyD88−/− bone marrow (Figures 2J–L). This was not a result of differences in reconstitution of GF Rag−/− animals, as baseline TFH abundance is the same in Rag−/− animals receiving WT or T-MyD88−/− bone marrow in the absence of TLR2 treatment (Figure 2L). Collectively, these data argue strongly that cues from the microbiota can act through MyD88 within T cells to coordinate TFH biology. Thus, identifying a pathway by which commensal bacteria can directly influence GC responses within the intestine.
T cell Intrinsic MyD88 Signaling Coordinates IgA Responses Against the Microbiota
GC responses function within the intestine to produce high affinity IgA against food, self and microbial antigens (Bemark et al., 2012). As animals that lack MyD88 within the T cell compartment have reduced TFH and B cell responses within the gut, we wished to test whether these deficiencies resulted in impaired IgA production. Intestinal contents were isolated from WT and T-MyD88−/− and analyzed for the production of secretory IgA (SIgA) by ELISA. We found that total SIgA was reduced in T-MyD88−/− mice (Figure 3A), illustrating the importance of this pathway in T cells for the generation of productive GC responses. To determine whether this pathway regulates the generation of antigen specific IgA against the microbiota, we mono-colonized GF Rag−/− mice previously reconstituted with WT or T-MyD88−/− bone marrow with a commensal bacteria strain that was engineered to express the model antigen ovalbumin (Bacteroides fragilis-OVA). This allowed us to track a commensal specific antibody response (Figure 3B). GF Rag−/− animals have no detectable OVA-specific IgA within the intestine (data not shown). While B. fragilis-OVA colonization in the lumen was similar between cohorts (Figure 3C), there was a complete absence of OVA-specific SIgA production in mono-associated T-MyD88−/− animals (Figure 3D), despite similar levels of total SIgA being produced (Figure 3E). These data indicate that MyD88 signaling within T cells directs antigen-specific SIgA against the microbiota.
Figure 3. IgA production against commensal antigens is perturbed in the absence of MyD88 signaling in T cells.
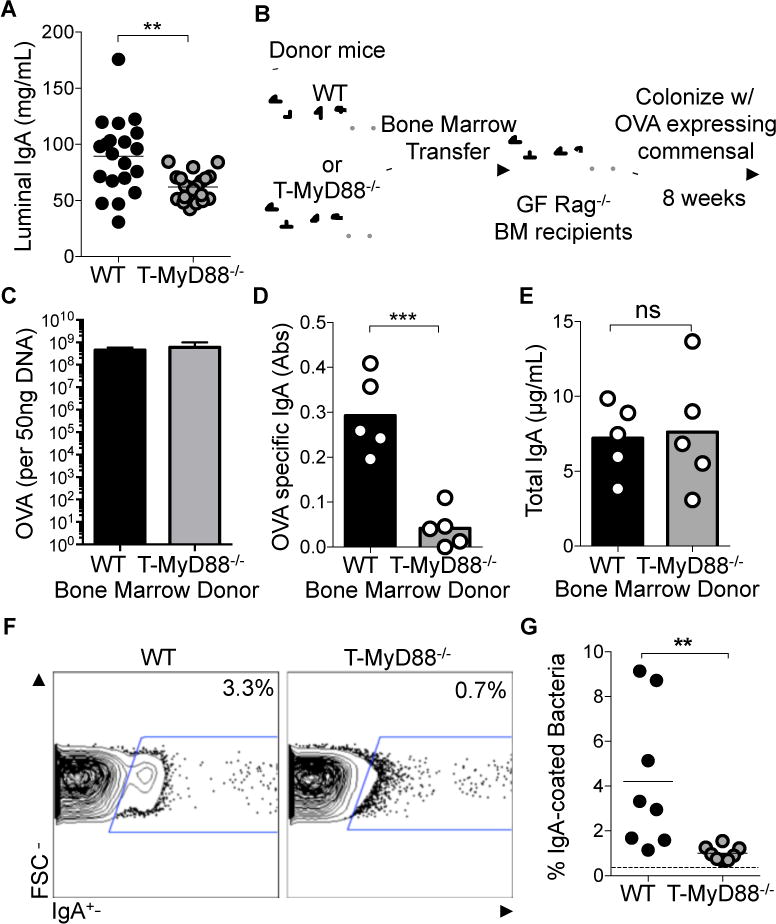
(A) Concentrations of soluble IgA within gut luminal contents measured by ELISA and normalized to fecal weight (n=20 per group).
(B) Experimental schematic showing GF Rag−/− mice reconstituted with either WT or T-MyD88−/− bone marrow and subsequently mono-colonized with B. fragilis-OVA.
(C) Raw abundances of DNA specific to the OVA gene was detected via q-RTPCR to quantify the loads of B. fragilis-OVA per 50ng of fecal DNA (n=5 per group).
(D) ELISA was used to measure the concentration of IgA within the lumen of these animals, normalized to fecal weight (n=5 per group).
(E) Abundance of OVA-specific IgA within gut luminal contents measured by ELISA and normalized to fecal weight. Data shown as absorbance at 450nm (n=5 per group).
(F) IgA-bound bacteria in intestinal lumen was measured by flow cytometry. Representative plots were previously gated on SYBR green+ events (=bacteria).
(G) Frequencies of IgA-bound bacteria (n=8 per group) with dashed line showing mean non-specific binding in RAG−/− controls. Unpaired two-tailed Student’s t-tests were used for all comparisons. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***). Lines in scatterplots represent means. Bar graphs represent means ±SD.
Reports have indicated that IgA targeting of the microbiota is modulated during intestinal disease in humans (van der Waaij et al., 2004) and may target colitogenic members of the microbiota (Palm et al., 2014). SIgA can be produced in the intestine as a result of either T cell dependent (TD) or independent (TiD) mechanisms (Mantis et al., 2011; Stephens and Round, 2014). TiD often results in low affinity, cross-reactive antibody. TD responses, mediated by TFH cells, result in commensal targeting via specific, high affinity IgA binding (Lycke and Bemark, 2012). To better characterize the TD antibody defect in T-MyD88−/− animals we utilized a flow cytometry based assay to look at host-generated SIgA against the microbiota. T cell deficient animals have undetectable IgA-bound bacteria with this method, so this assay quantifies high affinity TD antibody production (Slack et al., 2012; Slack et al., 2009). The microbiota was isolated from animals and immunostained with antibody against IgA and treated with the DNA dye SYBR green. Consistent with other reports (Palm et al., 2014), 1% to 10% of SYBR+ microbiota was bound by IgA in WT mice, while a ten-fold reduction in IgA targeting of the microbiota was observed in T-MyD88−/− animals (Figure 3F and 3G). These results reveal that innate recognition of commensal products by T cells influences the abundance and quality of the TD SIgA response towards commensal bacteria.
MyD88 within T cells Prevents Dysbiosis of Tissue-Associated Microbial Communities
Defects in commensal directed SIgA might have important effects on the microbial ecology of the gut (Cerutti et al., 2011; Cullender et al., 2013; Fagarasan et al., 2002; Peterson et al., 2007). Therefore, we sought to determine via high-throughput 16S rRNA sequencing whether the loss of T cell intrinsic MyD88 signaling could influence microbial composition. To control for differences in bacterial communities based on housing conditions, we performed analyses on separately housed animals whose breeding lines were originally derived from heterozygote crosses as well as co-housed animals. Regardless of housing conditions, fecal and mucosal communities were distinct in both genotypes (Figure 4A and Table S1). Interestingly, greater variation exists between fecal communities among individuals when compared to mucosa-associated communities (Fig. 4B). Since mucosal communities reside in closer proximity to host tissue, this result suggests that the immune system exerts stronger selection on community membership at this site. Consistent with this, the differences in community composition between genotypes are only maintained in mucosa-associated communities (Figure 4C), but not fecal communities (Table S1), during cohousing. Among these differences, we observed increases in multiple taxa containing mucolytic members in T-MyD88−/− animals including Desulfovibrionaceae and Mucispirillum (Figures S5A and S5B), as well as significant increases in Ruminococcus (Figures S5A and S5B). Collectively, these data indicate that loss of T cell intrinsic TLR signaling results in significant shifts in microbial composition in the gut.
Figure 4. Innate recognition by T cells influences the composition of mucosally associated microbial communities.
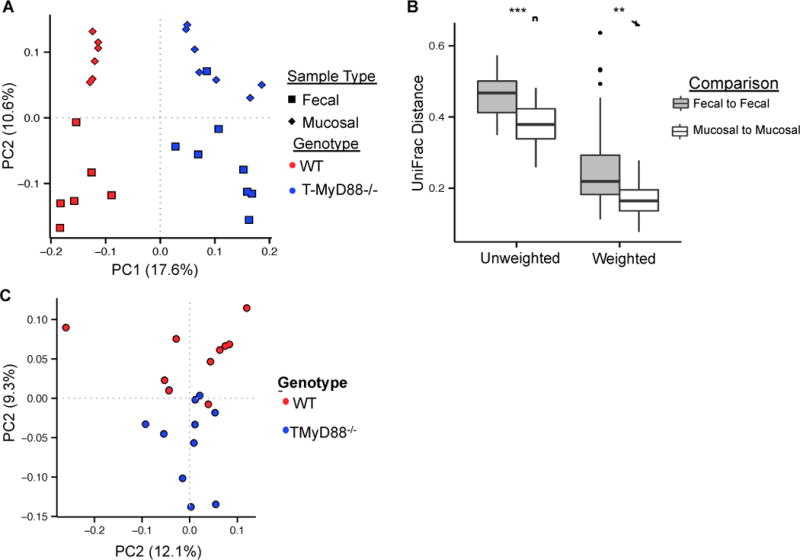
(A) Principal coordinates plot of unweighted UniFrac distances shows separation of microbial communities by genotype and sample type within separately housed animals. See also Table S1.
(B) Mucosal communities are more similar than fecal communities (Non-parametric t-test; ** P-value<0.001, *** P-value=0.0001). Boxplot whiskers=interquartile range.
(C) Differences in mucosal communities between genotypes are maintained in cohoused animals (PERMANOVA; P-value = 0.0111). See also Table S1.
(D) UniFrac distances between fecal and mucosal communities are not significantly different between genotypes (Non-parametric t-test). Boxplot whiskers=interquartile range.
T cell intrinsic MyD88 Signaling Governs IgA Targeting of Mucosa Associated Microbiota
We next investigated how innate signaling in T cells would influence SIgA targeting of microbial communities. To do this, we developed an assay to purify IgA-bound bacteria with 94% purity (Supplementary Data) and characterized SIgA-targeted bacterial communities via high-throughput sequencing. IgA binds a distinct group of bacteria when compared to either the total fecal or mucosal community in either genotype, indicative of specific targeting of commensal species by SIgA (Figure 5A and Table S1). Additionally, the assemblage of bacterial species targeted by the host via SIgA is altered in T-MyD88−/− mice compared to WT animals (Figure 5A). The SIgA-bound fraction is more similar to the mucosa-associated community than the fecal community in WT animals, indicating that SIgA tends to target mucosa associated organisms more so than fecal community members (Figure 5B). Remarkably, this IgA bias toward mucosal communities is lost in T-MyD88−/− animals as IgA-bound species are equally representative of mucosal and fecal communities (Figures 5B). This is not just a result of blending of the mucosa-associated and fecal communities because these communities are as dissimilar in T-MyD88−/− animals as they are in WT animals (Fig. 5C). These results indicate that the host IgA response is primarily directed toward specific bacteria within the mucosa and that this bias is disrupted in T-MyD88−/− animals.
Figure 5. T cell intrinsic MyD88 signaling regulates IgA selection of the microbiota.
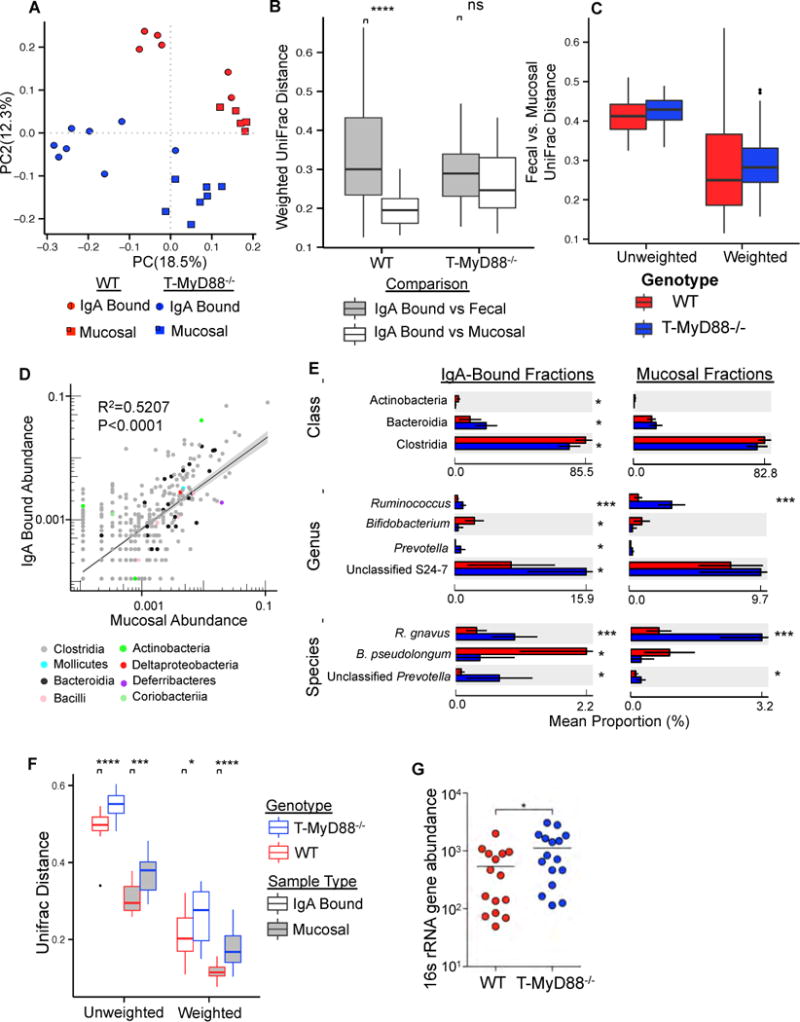
(A) PCoA plot of unweighted UniFrac distances comparing IgA-bound and mucosa-associated bacterial communities between genotypes. (PERMANOVA; P-value<0.01 for all pairwise comparisons) See also Table S1.
(B) Community dissimilarity (weighted UniFrac distance) between IgA-bound vs. fecal and IgA-bound vs. mucosal communities for each genotype. Non-parametric t-test. P-value=0.0001 (****). Increasing values along the y-axis represents increasing community dissimilarity among individual samples within a given cohort. Boxplot whiskers=interquartile range.
(C) Plot of the mean relative abundance of individual OTUs in all mucosal and IgA-bound samples colored by class of the OTU. A linear model fit to the data are shown, and is plotted on log scale.
(D) Class, genus and species abundances detected as significantly different in the IgA-bound fraction are shown along with their abundances in mucosa-associated communities. Welch’s t-test. P-value<0.05 (*); P-value<0.001(***). Bar graphs represent mean taxa abundance ±SD. See also Table S2.
(E) Comparison of community dissimilarity between genotypes for IgA bound and mucosa-associated communities. Non-parametric t-test, P-value<0.05 (*); P-value<0.001 (***); P-value=0.0001 (****). Boxplot whiskers=interquartile range.
(F) Total bacterial load in mucosa of WT and T-MyD88−/− animals as measured by qPCR. Student’s t-test. P-value<0.05 (*). Bars represent means.
Given these results, we focused on how a loss of IgA mucosal bias, as seen in T-MyD88−/− animals, would influence the microbial community. There is a significant positive correlation between bacterial species (OTU) abundance in the total mucosal community and in the IgA-bound fraction, which implies that in general IgA binds the most abundant organisms (Fig. 5D). Additionally, several taxonomic groups were targeted differently between WT and T-MyD88−/− animals (Figure 5E). These include reduced binding of the classes Clostridia and Actinobacteria, and more robust targeting of many OTUs in the Bacteroidia (primarily within the uncultured S24–7 family) in T-MyD88−/− animals (Figure 5E). Resolution of microbial communities at finer taxonomic depth reveals a reduction of IgA targeting of Bifidobacterium pseudolongum and increased IgA binding of Ruminococcus gnavus and unclassified Prevotella species in T-MyD88−/− animals (Figure 5E and Figure S5A). Additionally, OTUs of the genus Lachnospiraceae were the most differentially targeted by IgA (including cohoused animals)(Table S2), as has been observed in a mouse model of colitis (Berry et al., 2012). Differences in IgA targeting in T-MyD88−/− animals did not simply reflect significant differences in OTU abundance in the mucosa-associated community (Figure 5E), suggesting that abundance alone does not dictate the specificity of IgA targeting. Importantly, we observed significantly increased dissimilarity among individual T-MyD88−/− animals in both their IgA-bound and mucosa-associated communities compared to WT animals (Figure 5F), suggesting that there is greater variability in which species persist in the gut of a T-MyD88−/− animal. We also observed an increased total bacterial load at the mucosa in T-MyD88−/− animals (Figure 5G). Collectively, these data demonstrate that innate signaling by T cells dictates IgA specificity to constrain the composition of the microbial community, while also limiting mucosal association of commensal microbes.
We next sought to determine whether any of the observed defects in the host humoral response were associated with changes in microbial community composition. Consistent with previous reports, we found a significant positive correlation between the relative abundance of GC B cells and mucosal community diversity (Fig. 6A). This implies that a stronger GC response promotes diversity, perhaps by opening niches for rare species by limiting the densities of abundant members. Similarly, we utilized a Mantel’s test to demonstrate that the magnitude of difference in immune phenotype among individuals was positively associated with dissimilarity between microbial communities (Figure 6B). This effect was observed for multiple immune parameters, and was driven by differences between host genotypes (Figure 6B). Thus, differences in microbial composition between WT and T-MyD88−/− animals are directly associated with differences in their immune response. Collectively, these data demonstrate that host humoral responses sculpt microbial communities in the gut through IgA-mediated selection and that innate sensing of microbial products by T cells regulates this process.
Figure 6. Phenotypic Variation in Immune response Correlates with Microbial Community Similarity.
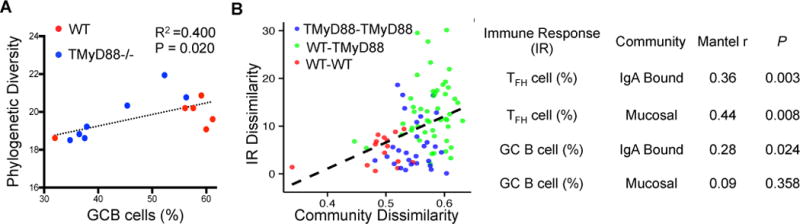
(A) Phylogenetic diversity in mucosal communities is correlated with GC B cell abundance (%).
(B) (left panel) a representative plot defines the axes and pairwise comparisons used to derive results of a Mantel’s test. (right panel) Correlations and p-values for the association between the differences in immune response (IR) and community dissimilarity among individuals. IR dissimilarity and community dissimilarity are estimates calculated from distance matrices built from immune phenotype and microbial community phylogenetic (UniFrac) data, respectively.
Alterations to the Microbiota in T-MyD88−/− Animals Results in Increased Intestinal Disease
Changes to microbial communities have been shown to promote inflammatory disease in the gut. Our observed shifts in Lachnospiraceae are similar to those seen in colitogenic animals, and R. gnavus has been associated with intestinal disease in humans (Hansen et al., 2013; Png et al., 2010). Moreover, defects in antibody targeting of bacteria have been seen in patients with IBD (Harmsen et al., 2012; van der Waaij et al., 2004), suggesting that a reduced ability to control the microbiota within T-MyD88−/− animals could lead to harmful consequences to the host. To determine whether changes in microbial community composition observed in T-MyD88−/− mice led to worsened disease, we examined colitis susceptibility in this model. Typical of the TNBS colitis model in C57Bl/6 animals, WT animals lost approximately 10% of their body weight and quickly regained their original mass. However, T-MyD88−/− animals lost more weight and did not recover to WT levels by the end of the experiment (Figure 7A). Histological analysis of these colons revealed that T-MyD88−/− mice had increased inflammatory infiltrate and greater crypt loss than WT animals (Figures 7B and 7C). Increased bacterial loads at the mucosa or differences in the assemblage of species living at this site could explain this. To determine whether microbial composition in the gut influenced disease progression, we tested whether susceptible T-MyD88−/− animals could be rescued with a WT microbiota. To this end, cohorts of T-MyD88−/− animals were treated with antibiotics to clear endogenous commensals and subsequently given a transplant with either WT or T-MyD88−/− microbiota. Animals that received a microbial transplant from a T-MyD88−/− animal developed severe crypt loss (Figures 7B and 7C). Remarkably, the extent of disease observed in T-MyD88−/− animals provided a microbiota from WT mice was significantly reduced and indistinguishable from that observed in WT animals (Figure 7C). Notably, WT GF mice given a T-MyD88−/− microbiota had no difference in disease severity (data not shown), indicating that the microbial composition formed in T-MyD88−/− animals is not sufficient to cause disease in an immune-competent host. Thus, this model closely mirrors the complexity seen in human IBD whereby genetic and environmental factors interact to promote disease. These data also highlight the potential curative value of fecal transplantations from healthy donors.
Figure 7. Increased intestinal disease observed in T-MyD88−/− animals is dependent on the composition of the microbiota.
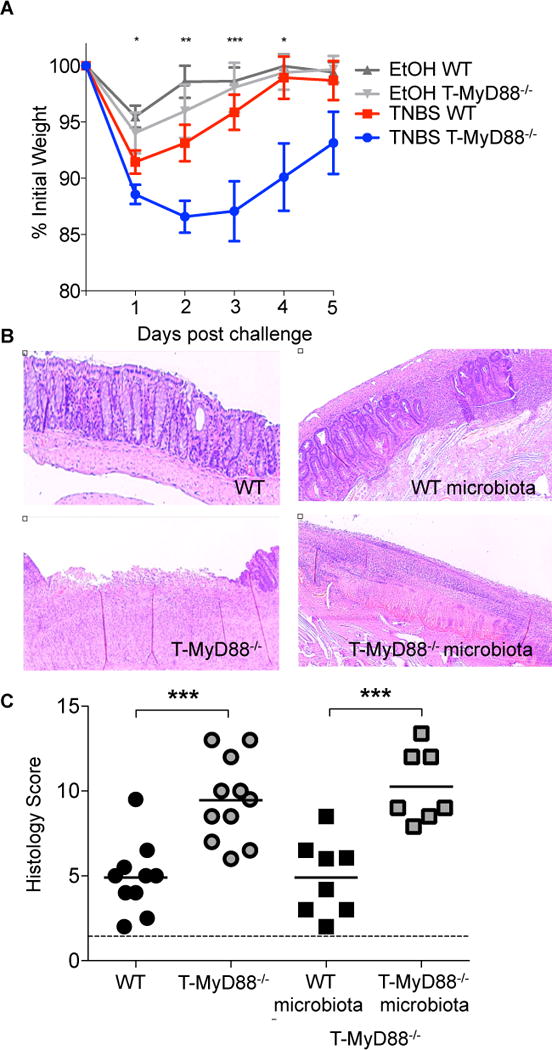
(A) TNBS-induced weight loss (represented as the percentage of initial weight) was compared among WT and T-MyD88−/− animals. EtOH vehicle controls are shown for comparison. Points represent means ±S.E. (representative of two independent experiments).
(B) Representative H&E stained colon sections from WT and T-MyD88−/− mice following TNBS-induced colitis.
(C) Histology scores reflecting disease severity for TNBS-treated animals (dotted line represents score for mice treated with EtOH only) WT animals (n=10); T-MyD88−/− animals (n=11); T-MyD88−/− given WT (n=8) or T-MyD88−/− (n=7) microbiota transplants. A student’s t-test was used for all comparisons. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***).
DISCUSSION
While originally thought to play a role only within the innate immune compartment, emerging data supports that MyD88 functions also in cells of the adaptive immune system such as T lymphocytes (Chang et al., 2013; Schenten et al., 2014). How this pathway could function to influence resident commensal communities has remained unexplored. MyD88 functions downstream of multiple receptors including the cytokine receptors, IL-1, IL-18, and IL-33 as well as all the TLRs with the exception of TLR3. The microbiota is an abundant source of TLR ligands and can induce the basal expression of these cytokines, making MyD88 an attractive molecule to integrate multiple signals induced by the microbiota to directly control host T cell function. Indeed, here we identify that MyD88 signaling in T cells governs the development of functional GCs within the gut. It is unclear at this stage what exact signals are triggering activation of MyD88 within T cells to control GC responses. However, it is likely that multiple signals act directly on T cells and converge on MyD88 as T cells express several of these TLRs and cytokine receptors (Reynolds et al., 2010; Kabelitz, 2007). The use and development of new conditional animal models to study these individual signals will be required to fully understand how innate signals elicited by the microbiota can directly influence T cell development and function within the gut.
One of the primary mechanisms to promote homeostasis within the gut is through the production of antigen specific IgA. Understanding the mechanisms by which this pathway governs antigen specific TFH responses will be an important future endeavor. By employing a newly developed technology we were able to determine that antigen specific IgA toward the microbiota is influenced by MyD88 in T cells (Cullender et al., 2013; Kawamoto et al., 2012; Palm et al., 2014). There is emerging evidence that there are niche-specific microbial communities within the gut, including organisms that are in close proximity to the host (tissue or mucosa associated communities) and those that are found to reside away from the host within the intestinal lumen. We identify that host IgA tends to target tissue-associated microbial communities in a healthy animal. However, in the absence of MyD88 signaling within the T cells, the mucosal bias of IgA is lost, indicating that this pathway serves to dictate IgA specificity within the gut. More importantly, loss of this signaling pathway in T cells leads to defective control over microbial community composition resulting in worsened intestinal disease.
Immune systems evolved within a microbe dominated world under strong selection to protect host tissues from pathogenic invasion. However, the dependence of host health on the microbiota strongly suggests that the immune system evolved to serve an additional purpose; to promote colonization by microbial species whose presence can be exploited to benefit host health (McFall-Ngai et al., 2013). Thus, co-evolutionary forces between resident microbes and hosts have resulted in mechanisms to respond to one another, creating a flexible dialog to ensure stability of symbiosis. IgA represents one host tool to directly influence bacterial communities, however it has been unclear whether IgA binds to specific members of the community and more importantly what host molecular machinery governs this specificity. Our data supports a model whereby signals from commensal microbes are perceived directly by host T cells to promote the production of TFH and GC B cell responses leading to the generation of high-affinity IgA toward commensals. This represents an important mechanism through which hosts can maintain a benign assemblage of microbial species in the gut.
Segregation of innate and adaptive arms of the immune system would be appropriate in sterile environments (like the systemic compartment) as this would provide a binary switch to respond to microbial invasion. However, in tissues with persistent bacterial exposure, cellular convergence of innate and adaptive immune pathways could provide more flexibility to fine tune antigen-specific signaling and respond to environmental changes properly. Thus, modulation of T cell responses through microbiota derived signals might provide a therapeutic target to prevent disease associated with human autoimmunity.
EXPERIMENTAL PROCEDURES
Animal models
MyD88fl/fl (Jackson Labs) mice were crossed to CD4-cre+ mice (Taconic) (both on C57/B6 background) to produce the T-MyD88−/− (MyD88fl/fl-CD4cre+) mouse model. MyD88+/+-CD4-cre+ mice were used as wildtype (WT) controls for all comparisons. Germfree animals were on a C57Bl/6 background except when noted in the figure legend. Animal use adhered strictly to Federal guidelines and those set forth by the University of Utah’s Institutional Animal Care and Use Committee.
In vitro activation experiments
Purified cells were plated in 250 μL of supplemented RPMI containing 20ng/mL IL-2 in 96 well plates (2×105 cells per plate) that had been previously coated with 5μg/mL purified anti-CD3. For TFH skewing conditions, 1μg/mL of the co-stimulatory antibody anti-CD28, 5μg/mL anti-CD3e, 10μg/mL anti-IFNγ, 50ng/mL IL-21, and 50ng/mL IL-6 were added. For TLR ligand stimulation, 1μg/mL of either Pam2CSK (Invivogen), Pam3CSK (Invivogen), Flagellin from S. typhimurium (Invivogen), or LPS (Invivogen) was added. Cells were cultured for four days at 37°C in these conditions before being analyzed by flow cytometry.
RNA-Isolation and RNA-Sequencing of purified TFH cells
mRNA was collected from ~50,000 TFH cells with the Qiagen miRNeasy Mini Kit (Qiagen) following kit instructions. mRNA was prepared following QC via an Illumina TruSeq Stranded RNA Sample Prep with RiboZero treatment (human, mouse, rat, etc.) and analyzed with Illumina HiSeq Sequencing.
Disease model-TNBS colitis
100μL of a 50/50 (v/v) mixture of EtOH and TNBS was administered intra-rectally on days 0 and 5. Weights were collected daily for the first five days and tissue was collected on day 7. Animals were scored as described in detail in the supplement and as reported in (Round and Mazmanian, 2010)
Germ Free model
Germ free mice were maintained in sterile isolators and assayed monthly for germ-free status by plating and PCR. Balb/c germ free mice were compared to SPF mice to measure TFH cells. Germ free Rag1−/− mice from the C57/B6 background were given bone marrow from WT or T-MyD88−/− mice and allowed to reconstitute for 8 weeks. Mice were either given Pam3CSK in their drinking water or were gavaged with B. fragilis expressing OVA.
Quantification of IgA+ fecal bacteria
Fecal microbiota were stained with SYBR green dye and a PE-conjugated IgA antibody to enumerate IgA-coated bacteria via Flow Cytometry. Please see detailed supplementary methods.
Sequencing of microbiota communities
DNA was extracted with bead beating methods from fecal, mucosal and IgA-bound enriched samples from each animal. The V3 and V4 regions of the bacterial 16S rRNA gene was amplified and sequenced on an Illumina MiSeq with 300 cycles from each end. Sequences were processed and analyzed with mothur 1.32 (Schloss et al., 2009) and Qiime 1.8.0 (Caporaso et al., 2010). Illumina MiSeq 16S rRNA gene sequences have been deposited in the NCBI sequence read archive (SRA) under the accession SRP050978.
Statistical Analysis
Figure creation and statistical analysis was performed with the Prism6.0 statistical software, R 3.1.0, STAMP 2.0.5 (Parks and Beiko, 2010), and utilities within the Qiime software. Unpaired two-tailed Student’s t-tests were used for statistical comparisons unless otherwise noted (see Supplementary Materials). Non-parametric t-tests were performed with 9,999 Monte Carlo simulations. All statistical tests, and estimates of dispersion, are referenced in Figure legends.
Animal housing/Cohousing experiments
SPF mice in our facility are housed under identical conditions and fed the same diet which minimizes the effect of environmental variability on host phenotypes and microbial communities. In an effort to standardize any potential effect of isolation (across cages and between genotypes) on microbiota communities multiple independent homozygous WT and T-MyD88−/− breeder pairs were set up at the same time from initial heterozygous crosses. Clustering of genotypes in 16S microbiota analysis confirms that host genotype and not isolation is the primary driver of differences among individuals in their communities. For co-housed experiments pregnant homozygous WT or T-MyD88−/− animals gave birth to litters in the same cage (births occurred within one week of each other). Animals remained in the same cage until weaning (4 weeks of age) and were subsequently housed separately until analysis (8 weeks of age).
Lamina Propria Lymphocyte Isolation
Lamina propria lymphocytes (LPLs) were isolated with a combination of two previously described methods (Atarashi et al., 2011; Round et al., 2011). Prior to LPL isolations of small intestines (SI), all visible peyer’s patches were removed. Colons and SI tissue were opened longitudinally and mucus was removed by gently scraping and then rinsing in sterile 1X HBSS. The tissue was cut into small pieces and incubated in sterile 1X HBSS (without Ca2+ and Mg2+) containing 5mM EDTA (Cellgro) and 1 mM of DL-Dithiothreitol (DTT)(Sigma) for 45 minutes at 37°C on a shaker. Supernatant was removed by filtering through a 100μM filter and remaining tissue was incubated in a solution containing sterile 1X HBSS containing 5% (v/v) fetal bovine serum (Gibco BRL), 50 units/ml Dispase(Roche), 0.5 mg/ml of Collagenase D (Roche), and 0.5 mg/mL of DNaseI (Sigma) for 45 minutes at 37°C on a shaker. The supernatant was filtered over a 40mm cell strainer into ice cold sterile 1X HBSS. Cells were passed through a Percoll (GE Healthcare) gradient (40%/80% (v/v) gradient) and spun at 620 × g for 20 minutes with no brake. Cells at the 40/80 interface were collected and washed twice with supplemented HBSS (10mM Hepes (Cellgro), 2mM EDTA (Cellgro), and 0.5% (v/v) fetal bovine serum (Gibco BRL)) and prepared for flow cytometry analysis. While this isolation strategy is widely used as an approach for isolating lamina propria tissue-resident lymphocytes, it does not prohibit contamination by lymphocytes residing within the microenvironment of isolated lymphoid follicles (ILFs), which are organized lymphoid structures found throughout the lamina propria, therefore we cannot completely exclude the possibility that some of the cells we are assaying reside with ILFs.
Flow Cytometry Staining of Isolated Lymphocytes
Lymphocytes were isolated from the colonic and small intestinal lamina propria as mentioned above. Spleen, mesenteric lymph node, or Peyer’s patch tissues were gently pushed through a 40μM filter to obtain white blood cells. Spleen cells were additionally treated with 1X RBC Lysis Buffer (Biolegend) to lyse and remove red blood cells. Surface staining for lymphocytes was done in sterile 1X HBSS (Corning) supplemented with 10mM Hepes (Cellgro), 2mM EDTA (Cellgro), and 0.5% (v/v) fetal bovine serum (Gibco BRL) for 20 minutes at 4°C. Cells were then washed twice in supplemented 1X HBSS and then enumerated via flow cytometry. The following antibodies were used: anti-CD4 (Biolegend: clone GK1.5 APC/Pacific Blue, clone RM4-5 FITC; eBioscience clone RM4-5 PerCP-Cy5.5), anti-CD3 (Biolegend: 145-2C11 Pacific Blue), anti-B220 (Biolegend: clone RA3-6B2 PerCP-Cy5.5), anti-CD138 (Biolegend: clone 281-2 PE), anti-CXCR5 (eBioscience: clone SPRCL5 PE), anti-PD-1 (Biolegend: RMP1-30 Pe/Cy7), anti-ICOS (eBioscience: clone 7E.17G9 FITC), anti-GL-7 (eBioscience: clone GL-7 Alexa Fluor 488), anti-Fas (BD Biosciences: clone Jo2 PE-Cy7), anti-IgD (Biolegend: clone 11-26c.2a Alexa Fluor 647), or anti-IgA (Southern Biotech goat anti-mouse IgA FITC). For intracellular staining, cells were first stimulated with ionomycin (500ng/mL), PMA (5ng/mL), and Brefeldin A (5μg/mL) (Biolegend) for 4hrs at 37°C. Cells were surface stained, washed, and then permeabilized and fixed in 100μL of Perm/Fix buffer (eBiosciences) at 4°C overnight. Cells were washed twice in Perm/Wash buffer (eBioscience) and then stained for intracellular cytokines with the following antibodies: anti-Foxp3 (eBioscience: clone FJK-16s APC/PerCP-Cy5.5), anti-IL-17A (eBioscience: clone eBio17B7 eFluor 660), or anti-IFNγ (Biolegend: clone XMG1.2 PE). Cells were again washed twice in Perm/Wash buffer and then placed with supplemented HBSS (10mM Hepes (Cellgro), 2mM EDTA (Cellgro), and 0.5% (v/v) fetal bovine serum (Gibco BRL)) and enumerated via flow cytometry. Gating and analysis of positive cell populations was done utilizing respective isotype controls for antibodies against ICOS, CXCR5, GL-7 and PD-1. Other cell populations were identified with single stain controls. These data were collected with a BD LSR Fortessa and analyzed with FLOWJO software.
Supplementary Material
Acknowledgments
We would like to thank members of the Round and O’Connell labs for their critical review of the manuscript. The B. fragilis-OVA strain was created by Dr. Yue Shen and Dr. Sarkis Mazmanian (California Institute of Technology, Pasadena, CA). We thank James Marvin for assistance with flow sorting. The Flow cytometry core is supported by the National Center for Research Resources of the National Institutes of Health under Award Number 1S10RR026802-01. Some of the germfree mice used in this publication were provided from UNC’s Gnotobiotic Facility which is supported by grants 5-P39-DK034987 AND 5-P40-OD010995. J.L.K. and C.P. are supported by a T32 fellowship in microbial pathogenesis (AI-055434). R.S. was supported by a T32 genetics training grant (GM007464). R.M.O. is supported by the NIH New Innovator Award DP2GM111099-01, the NHLBI R00HL102228-05, an American Cancer Society Research Grant, and a Kimmel Scholar Award. Support for this project comes from the Edward Mallinckrodt Jr. Foundation, Pew Scholars Program, NSF CAREER award (IOS-1253278), Packard Fellowship in Science and Engineering and NIAID K22 (AI95375) and NIAID (AI107090, AI109122) and an NIH innovator award DP2AT008746-01 to J.L.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes supplemental experimental procedures, five supplemental figures and two supplemental tables and can be found with this article online at
AUTHOR CONTRIBUTION
J.L.R surpervised the study. J.L.R., J.L.K and C.P designed the study. C.P and J.L.K performed the experiments. C.P. performed most of the immunological experiments including microbiota transfers, disease induction and germfree mouse studies. J.L.K. performed some of the initial immunology experiments, developed the IgA bacterial purification protocol and helped analyze the microbiota data. W.Z.S. performed the 16S sequencing and W.Z.S and J.L.K analyzed the data. R.S. assisted during mouse harvests and provided comments during the study. J.L.R, J.L.K, C.P, and W.Z.S wrote the manuscript.
References
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemark M, Boysen P, Lycke NY. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann N Y Acad Sci. 2012;1247:97–116. doi: 10.1111/j.1749-6632.2011.06378.x. [DOI] [PubMed] [Google Scholar]
- Berry D, Schwab C, Milinovich G, Reichert J, Ben Mahfoudh K, Decker T, Engel M, Hai B, Hainzl E, Heider S, et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. Isme J. 2012;6:2091–2106. doi: 10.1038/ismej.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtzaeg P. Secretory IgA: Designed for Anti-Microbial Defense. Front Immunol. 2013;4:222. doi: 10.3389/fimmu.2013.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A, Cols M, Gentile M, Cassis L, Barra CM, He B, Puga I, Chen K. Regulation of mucosal IgA responses: lessons from primary immunodeficiencies. Ann N Y Acad Sci. 2011;1238:132–144. doi: 10.1111/j.1749-6632.2011.06266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang CH. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci U S A. 2013;110:2270–2275. doi: 10.1073/pnas.1206048110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- Hansen SG, Skov MN, Justesen US. Two cases of Ruminococcus gnavus bacteremia associated with diverticulitis. J Clin Microbiol. 2013;51:1334–1336. doi: 10.1128/JCM.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen HJ, Pouwels SD, Funke A, Bos NA, Dijkstra G. Crohn’s disease patients have more IgG-binding fecal bacteria than controls. Clin Vaccine Immunol. 2012;19:515–521. doi: 10.1128/CVI.05517-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, Fagarasan S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- Kubinak JL, Round JL. Toll-like receptors promote mutually beneficial commensal-host interactions. PLoS Pathog. 2012;8:e1002785. doi: 10.1371/journal.ppat.1002785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner C, Wahl B, Fohse L, Suerbaum S, Macpherson AJ, Prinz I, Pabst O. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J Exp Med. 2012;209:365–377. doi: 10.1084/jem.20111980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Liston A, Vinuesa CG. T-follicular helper cell differentiation and the co-option of this pathway by non-helper cells. Immunol Rev. 2012;247:143–159. doi: 10.1111/j.1600-065X.2012.01121.x. [DOI] [PubMed] [Google Scholar]
- Lycke NY, Bemark M. The role of Peyer’s patches in synchronizing gut IgA responses. Front Immunol. 2012;3:329. doi: 10.3389/fimmu.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantis NJ, Rol N, Corthesy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011;4:603–611. doi: 10.1038/mi.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- McFall-Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet-Loso T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, et al. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A. 2013;110:3229–3236. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin a coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715–721. doi: 10.1093/bioinformatics/btq041. [DOI] [PubMed] [Google Scholar]
- Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, Mirpuri J, Vaishnava S, Hou B, Defranco AL, et al. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-gamma-dependent elimination of Paneth cells. Nat Immunol. 2013;14:136–142. doi: 10.1038/ni.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten D, Nish SA, Yu S, Yan X, Lee HK, Brodsky I, Pasman L, Yordy B, Wunderlich FT, Bruning JC, et al. Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity. 2014;40:78–90. doi: 10.1016/j.immuni.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD. T follicular helper cell dynamics in germinal centers. Science. 2013;341:673–677. doi: 10.1126/science.1241680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack E, Balmer ML, Fritz JH, Hapfelmeier S. Functional flexibility of intestinal IgA – broadening the fine line. Front Immunol. 2012;3:100. doi: 10.3389/fimmu.2012.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder TF, Hardt WD, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens WZ, Round JL. IgA targets the troublemakers. Cell Host Microbe. 2014;16:265–267. doi: 10.1016/j.chom.2014.08.012. [DOI] [PubMed] [Google Scholar]
- van der Waaij LA, Kroese FG, Visser A, Nelis GF, Westerveld BD, Jansen PL, Hunter JO. Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2004;16:669–674. doi: 10.1097/01.meg.0000108346.41221.19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.