

Abstract
The thyroid hormone plays a significant role in diverse processes related to growth, development, differentiation, and metabolism. Thyroid hormone signaling modulates energy expenditure through both central and peripheral pathways. At the cellular level, the thyroid hormone exerts its effects after concerted mechanisms facilitate binding to the thyroid hormone receptor. In the hypothalamus, signals from a range of metabolic pathways, including appetite, temperature, afferent stimuli via the autonomic nervous system, availability of energy substrates, hormones, and other biologically active molecules, converge to maintain plasma thyroid hormone at the appropriate level to preserve energy homeostasis. At the tissue level, thyroid hormone actions on metabolism are controlled by transmembrane transporters, deiodinases, and thyroid hormone receptors. In the modern environment, humans are susceptible to an energy surplus, which has resulted in an obesity epidemic and thus understanding the contribution of the thyroid hormone to cellular and organism metabolism is increasingly relevant.
Keywords: energy homeostasis, deiodinases, thyroid hormone, basal metabolic rate, cellular metabolism
Introduction
Metabolism is regulated by the hypothalamus through a complex neural circuitry that controls the amount of energy that is ingested and utilized at any given time. Important components of this system include sensation of hunger, satiety, autonomic nervous system activity, and the endocrine system. Through clinical observation of experimental and pathological conditions it became clear that the thyroid hormone (TH) is a main regulator of metabolic rate.1 For example, in severe hypothyroidism the total body energy expenditure (EE) can slow down by as much as 50%.2 Under normal physiological conditions the intact hypothalamic-pituitary-thyroid (HPT) axis maintains stable serum TH levels, resulting in a steady contribution of TH to EE and energy homeostasis.3 In contrast, during fasting or caloric restriction, there is an adaptive decrease in EE, much of which is determined by a fall in circulating TH levels.4,5
Availability of energy substrates, either from the environment or internal stores, is of high priority to the organism to ensure adequate supply needed to perform the multitude of cellular processes inherent to life. In a resting individual, the collective sum of the energy utilized in these processes feeds the basal metabolic rate (BMR), and the resulting heat produced is the obligatory thermogenesis. Of course, BMR can be accelerated by physical activity or stimulation of metabolic processes, which will result in production of more heat (i.e., adaptive thermogenesis).6 At rest, adaptive thermogenesis is largely controlled by the sympathetic nervous system (SNS) as seen during cold exposure or excessive caloric intake. In both conditions the hypothalamus activates the SNS to accelerate EE and increase heat production. The main site of adaptive thermogenesis in small mammals and infants is in brown adipose tissue (BAT), a specialized adipose tissue that produces heat by physiologic uncoupling of mitochondria.7 The mitochondrial uncoupling in BAT allows for acceleration in substrate oxidation without the parallel increase in adenosine triphosphate (ATP), such that energy is lost as heat. This process is mediated by the mitochondrial uncoupling protein 1 (UCP1) and is stimulated by the SNS and TH. Interest in BAT has surged in recent years due to incidental imaging findings of metabolically active BAT in adult humans.8 This has propelled clinical interest in the field; thus, our knowledge of the importance of TH’s contribution to adaptive thermogenesis and energy homeostasis has become more well-defined.9
Understanding the role of the SNS and TH in energy homeostasis is particularly relevant given the worldwide epidemic of obesity. Obesity is the result of chronic caloric intake in excess of EE, a common occurrence in the modern environment that facilitates consumption of hypercaloric diets and a sedentary lifestyle. Although obesity itself is associated with increased total and free serum triiodothyronine (T3) levels,10 which may reflect increased leptin levels, TH in conjunction with the SNS regulates metabolism and EE through multiple mechanisms and thus represents a potential target for obesity therapies. Current therapeutic standards in obesity either aim to decrease energy input or increase EE and thus tip the energy balance toward a net loss, leading to weight reduction. Both such strategies have limitations. In particular, increasing EE represents a challenging lifestyle with respect to patient compliance, as most such strategies involve increased physical activity.
The actions of TH in the target cell are achieved after a series of of highly regulated intracellular processes that allow TH to bind to thyroid hormone receptors (TRs) and influence gene transcription. These processes include transport of TH into the cell, activation or inactivation by the deiodinases, and expression of TR isoforms, nuclear corepressors, and coactivators.11 Recent scientific advances involving these regulatory processes have improved our understanding of the multifaceted contribution of TH to energy homeostasis and energy metabolism.
Central pathways involved in TH signaling and energy metabolism: role of the deiodinases
Neurons in the hypothalamus are affected by circulating TH as well as by TH that is locally activated by enzymes known as deiodinases.12 Glial cells including the tanycytes (specialized ependymal cells that line the walls of the third ventricle) express high levels of the type 2 deiodinase (D2) that converts the pro-hormone thyroxine (T4) to its active metabolite, T3.13,14 D2 is an endoplasmic reticulum–resident protein, and thus D2-generated T3 has regional access to the TR-containing nuclei.12 In addition, hypothalamic neurons also express the type 3 deiodinase (D3)—an enzyme located in the axonal neurosecretory vesicles and nuclear membrane—which inactivates both T4 and T3.15-17 There is an exchange of TH between these different cell types; D2 localized in glial cells produces T3 that acts in a paracrine fashion to induce T3-responsive genes in the neurons and, in turn, D3 activity in the neurons modulates these effects.18 This paracrine pathway is furthermore regulated by other signals, such as hypoxia, hedgehog molecules, and inflammation. Given that cell membranes are not permeable to TH, there is the obvious role played by transporters in TH signaling. The previous paradigm that proposed that TH, a hydrophobic molecule, passively diffused into the intracellular compartment was abandoned upon the discovery of monocarboxylate transporter 8 (MCT8), a TH transporter, and its association with the X-linked Allan-Herndon-Dudley syndrome (AHDS). It is now widely accepted that TH enters the cells through a group of tissue-specific transporter proteins, including the MCT family and organic anion transporters (OATPs).19,20 MCT8 is highly expressed in the liver, kidney, heart, and brain, where it also plays a role in hypothalamic TH feedback and gene regulation.21 For example, during development, there is coordination in the expression of D2, D3, and MCT8 in the embryonic hypothalamus, suggesting that regulation of TH availability is crucial to hypothalamic development.22
Thyrotropin-releasing hormone (TRH)-producing neurons in the paraventricular nucleus (PVN) define the set point of thyroid gland function by regulating pituitary thyroid-stimulating hormone (TSH) secretion and thus the circulating levels of TH (Fig. 1). During caloric restriction or disease states, central hypothyroidism results, at least in part, from the elevation in local D2 expression.23-25 A similar phenotype is also observed in mice with deletion of the fatty acid amide hydrolase; these mice develop peroxisome proliferator-activated receptor gamma (PPARg)-mediated hypothalamic D2 activation, resulting in suppressed TRH.26 At the same time, mice with targeted inactivation of the D3 gene (Dio3) exhibit neonatal elevation of hypothalamic T3 that later in life results in central hypothyroidism.27 Fasting not only reduces HPT function but also accelerates TH clearance from the circulation through conjugation to glucuronic acid and sulfation.4 These liver pathways are controlled by the hypothalamus through mechanisms modulated by leptin and mediated by agouti-related protein (AgRP)/neuropeptide Y (NPY) and pro-opiomelanocortin (POMC). This implies that the hypothalamus controls not only activation but also inactivation and clearance of TH, thus explaining the reduced EE observed in fasting states. Liver pathways are also implicated in TH-mediated glucose production in rats where, independent of circulating glucoregulatory hormones, stimulation of T3-sensitive neurons in the PVN increases sympathetic outflow to the liver, resulting in endogenous glucose production.28
Figure 1.
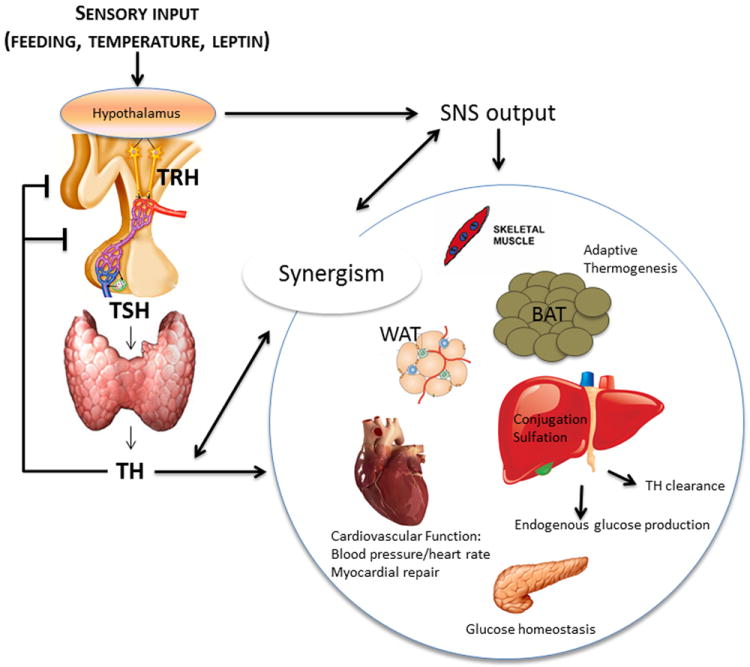
The role of thyroid hormone in energy homeostasis. In response to environmental stimuli (such as feeding or temperature) orto hormonal stimuli (such as leptin), hypothalamic pathways modulate sympathetic outflow and TH secretion through the hypothalamic-pituitary-thyroid axis. TH and sympathetic output act in multiple organ systems to affect energy metabolism and thus regulate whole-body energy homeostasis. In particular, TH signaling and SNS stimulation promote adaptive thermogenesis in BAT, regulate cardiovascular functions including blood pressure and heart rate, modulate glucose homeostasis through actions in the pancreatic β cell, regulate systemic TH clearance and endogenous glucose production in the liver, and effect other tissues including WAT and skeletal muscle.TRH: thyrotropin-releasing hormone; TH: thyroid hormone; SNS: sympathetic nervous system; BAT: brown adipose tissue; WAT: white adipose tissue.
The relative contributions of hypothalamic and pituitary D2 pathways in the HPT feedback loop were studied with two mouse strains: pituitary- and astrocyte-specific D2 knockdowns (pit-D2 KO and astro-D2 KO mice, respectively).29 The pit-D2 KO mice were systemically euthyroid with a normal serum T3 level but had increased serum TSH (approximately threefold) and serum T4 levels; the elevated serum T4 resulted in greater D2-mediated T3 production in the medial basal hypothalamus (MBH), thus decreasing TRH mRNA. A similar phenotype is seen under the pharmacologic administration of amiodarone, a nonselective D2 inhibitor.30 The astro-D2 KO mice had near-complete loss of brain D2, save for tanycyte D2, which was preserved at levels sufficient to maintain both the T4-dependent negative feedback loop and the systemic thyroid economy. Thus, tanycytes, not astrocytes, are the cells within the MBH that mediate T4-to-T3 conversion. A corollary of these studies is that coordination in TH signaling between the hypothalamus and the pituitary gland ensures steady serum T3 levels.29,31
Central pathways involved in TH signaling and energy metabolism: appetite regulation
In addition to mediating feedback mechanisms regulating thyroid economy, TH signaling in the hypothalamus also regulates energy homeostasis by influencing appetite and sympathetic activity, possibly via adenosine monophosphate (AMP)-activated protein kinase (AMPK). It is assumed that D2-generated T3 in the MBH is important for the orexigenic response during fasting, by increasing appetite and decreasing BAT sympathetic activity.24 In contrast, stereotaxic administration of T3 directly into the ventromedial nucleus of rats increases BAT sympathetic activity and at the same time maintains appetite as compared to controls.32 Similarly, a population of parvalbuminergic neurons in the anterior hypothalamus exhibits intrinsic temperature sensitivity and can modulate sympathetic activity in response to TH. These neurons require TH for adequate development as demonstrated in mice heterozygous for a point mutation in TRα1, which exhibits a phenotype consistent with receptor-mediated hypothyroidism with underdevelopment of these neurons. Mice with stereotaxic ablation of these neurons display hypertension and temperature-dependent tachycardia compared with controls. Although the TR-mediated mechanisms influencing the development of these neurons remain to be elucidated, the presence of multiple central mechanisms regulating energy homeostasis in response to temperature underscores its physiologic importance.33
Deiodinase-mediated TH activation/inactivation controls metabolism
Individual cells outside of the central nervous system (CNS) are also actively involved in the regulation/modulation of TH signaling through deiodinase expression and activity. This can create a local tissue-specific state of hypothyroidism or thyrotoxicosis even in the setting of systemic euthyroidism. In many cases there is coordinated expression of D2 and D3, such that TH signaling can be modified accordingly. For example, D2 transcription is positively regulated by cyclic AMP (cAMP) and thus its expression is stimulated by the SNS,34 bile acids (via TGR5),35 flavonols (Kaempferol),36 and chemical chaperones.37 In mice treated with an α-glucosidase inhibitor, bile acid concentrations in the feces and portal circulation increase, resulting in increased D2 expression in BAT. When these mice are on a high-fat diet (HFD), they demonstrate decreased body weight, glucose intolerance, insulin resistance, and adipokine regulation compared to controls.38 D2 is also regulated post-transcriptionally by ubiquitination/deubiquitination, whereby conjugation to ubiquitin inactivates D239 that can be reactivated via deubiquitination.40 At the same time, hypoxia-inducible factor 1α (HIF-1α) and TGFβ-SMADs stimulate D3 expression,41,42 whereas hedgehog molecules not only stimulate D3 but also inactivate D2 via ubiquitination.43,44
TH stimulates Dio3 expression via TRα; in turn, D3 negatively regulates the effects of TH in energy metabolism.45 Neuronal cells and cardiomyocytes expressing D3 exhibit an acceleration in EE a few hours after exposure to iopanoic acid, a non-competitive inhibitor of D3.41 Such a mechanism has important pathophysiological implications given that the heart ectopically expresses D3 in the acute phase that follows myocardial infarction (MI).46 In fact, after MI, there is a limited hypertrophic response of the myocardium with partial expression of the fetal gene program. Microarray of mouse myocardium after MI compared with sham mice demonstrated enrichment of the Dlk1-Dio3 region and there was a six-fold increase in D3 in this region by quantitative polymerase chain reaction (qPCR).47 A similar mechanism has been observed in the brain 24 h after ischemic ligation of the internal carotid artery in rats. In this case, hypoxia-induced D3 expression was linked to reduced metabolic effects of TH in neuroblastoma cells18 and isolated hippocampal neurons.16
It is well accepted that deiodinases modulate metabolism and EE by creating cell-specific states of hypothyroidism or thyrotoxicosis in tissues that are metabolically relevant, such as the hypothalamus, BAT, and skeletal muscle.9 However, it is becoming increasingly understood that deiodinase-mediated TH signaling can also affect the proper differentiation, development, and function of many tissues. For example, synchronized changes in deiodinase activity regulate TH signaling to promote BAT development. BAT develops in the mouse embryo between E16.5 and E18.5 during which time local TH signaling is increased as a consequence of increased Dio2 expression and decreased Dio3 expression.48 The importance of D2 as a key regulator of BAT development was confirmed in mice with a targeted disruption of Dio2, D2 KOs, which display defective mature brown adipocytes with impaired expression of key molecules involved in the adipogenic program (aP2, Cidea, and ACSL5), as well as an impaired expression of markers of thermogenic identity (UCP1, PGC-1α, and D2).48 Along these lines, D3 activity was found to be increased several fold within 1–2 days of partial hepatectomy, and this increase correlated with cell proliferation as assessed by 5-bromo-2’-deoxyuridine incorporation.49
Transmembrane TH transporters and receptors
The X-linked AHDS is characterized by truncal hypotonia, spasticity, mental retardation, and is explained by mutations in the MCT8 gene, which also results in abnormal serum TH levels (normal to high TSH, elevated serum T3, decreased serum T4).20,50 These patients exhibit accelerated metabolism, as evidenced by normal linear growth with poor weight gain in the affected child,51,52 that could reflect elevated serum T3 or decreased TH signaling in discrete brain areas involved in metabolic control. Increased EE despite adequate caloric intake is also demonstrated in MCT8KO mice, although these mice do not exhibit the severe neuromuscular phenotype associated with AHDS in humans.53 When these mice have the additional defect of Dio1KO (i.e., MCT8+D1KO), EE is stabilized and matches that of D1KO littermate controls. This implicates the high T3 level as the etiology of the increased EE observed in individuals with AHDS.53 In a small study that administered the MCT8-independent TH analog diiodothyropropionic acid (DITPA) to four human patients with AHDS, TH signaling was restored, thyroid function tests were normalized, and metabolic rate stabilized.54 Although these advancements in AHDS treatment are promising, the results are preliminary and future studies will address whether the neurologic deficits can be improved, or even prevented, with earlier administration of DITPA.
Once inside cells, TH diffuses to the nucleus and signaling occurs after binding with TRs, which exist in two different isoforms: TRα and TRβ.11 The genes encoding the TRs, TRα and TRβ, and the TR proteins exhibit varying expression both developmentally and spatially within TH target tissues, suggesting a specific tissue-dependent role for each TR isoform. Studies of TR-specific mutations and administration of TR-specific agonists have been used to clarify the roles of the isoforms in central and peripheral regulation of metabolism by TH and thus provide an alternate method of intracellular control of TH signaling.
Several mutations have been described in the TRα gene, which result in a recently described human phenotype associated with constipation, developmental delay, and short stature, with normal TSH, free T4, rT3, and T3,55-57 where response to treatment with T4 is tissue-variable. Mice heterozygous for a point mutation in TRα1 display increased EE as a consequence of increased SNS stimulation of BAT, although they maintain normal core body temperatures. These mice exhibit altered heat conservation and dissipation due to impaired SNS-induced vaso-constriction/dilatation, leading to compensatory BAT stimulation. Administration of Midodrine, a selective α1-adrenergic agonist, restores appropriate vascular function, leading to normalization of BAT activity and EE. This illustrates another aspect to the diverse thermoregulatory roles played by TH.58
A different phenotype is displayed in humans with mutations in TRβ; these individuals display elevated serum TH levels with a nonsuppresed TSH and goiter, yet lack stigmata of thyrotoxicosis.59 For this reason, this condition was initially termed “general resistance to thyroid hormone,” although this was revised upon more recent discovery of the TRα mutants. Homozygotes for TRβ mutations display a more severe phenotype; they exhibit higher T3 and TSH, hearing loss, goiter, cognitive delay, and growth retardation60 in a syndrome more reminiscent of TRα deficiency, suggesting that mutant TRβ can antagonize the action of TRα.11 Accordingly, mice with dominant-negative TRβ PV mutation exhibit impaired adaptive thermogenesis and reduced UCP1 expression in brown adipocytes.61
Adverse effects of supraphysiologic doses of TH make it an inappropriate therapy to induce EE, but given the tissue specificity of the TR isoforms, isoform-specific TRβ-specific TH analogs (thyromimetics) have been proposed as a way of targeting the TH actions to a specific tissue (e.g., lowering cholesterol due to actions in the liver, while limiting systemic side effects in the cardiovascular and skeleton systems). Some first-generation thyromimetics showed promise for potential benefit in the treatment of obesity, glucose metabolism, and non-alcoholic fatty liver disease (NAFLD), but they have failed to obtain U.S. Food and Drug Administration (FDA) approval.62
TRs interact with a group of nuclear proteins known as co-regulators, which can promote or inhibit transcription (i.e., coactivators or corepressors, respectively). For example, mutant TRα and TRβ both exhibit dominant-negative activity, consistent with findings in mouse models that show that these mutant receptors do not adequately recruit the nuclear corepressor NCoR1, contributing to the hypothyroidism in these individuals.63,64 At the same time, PGC-1 is a TR coactivator that has been shown to cooperate with SIRT1 to enhance the response of TRβ1 to T3.65 The steroid receptor coactivators (SRCs) bind to liganded TRs and also play a major role in TH signaling.66 Notably, molecular interactions between TR and comodulators play an important role in TH signaling, leading to important phenotypes, such as the opposing roles in adipocyte differentiation exhibited by alternative mRNA splicing of the two TR corepressors SMRT and NCoR.67
Thyroid hormone signaling in glucose homeostasis and insulin resistance
The role of TH in glucose homeostasis has been the subject of many studies68,69 and is clinically relevant given that the prevalence of diabetes mellitus is increased in both hypo- and hyperthyroidism.70 In hyperthyroid rats, reduced glucose tolerance and reduced insulin-secretory capacity of β cells are demonstrated in addition to increased hepatic insulin resistance,71-73 but similarly, hypothyroidism promotes glucose intolerance such that hypothyroid non-diabetic mice exhibit fasting hyperglycemia and reduced insulin levels in response to glucose stimulation.74 This evidence was not supported in a recent study of overtly hypothyroid adult humans in which, aside from elevated triglycerides, there was no evidence of glucose intolerance in the hypothyroid state and no improvement in other glucose parameters upon treatment of the hypothyroidism.75
At least some of the controversial findings in this area could be explained by the fact that studies did not take into account the fact that D3 is expressed in embryonic and adult β cells in humans and mice.76 Neonatal D3 KO mice have reduced β cell mass and deceased insulin content, suggesting a role of D3 in islet development. The D3 KO phenotype includes impaired glucose-stimulated insulin secretion, resulting in glucose intolerance. Therefore, D3 is an important pathway in islet development, insulin secretion, and glucose homeostasis.76 Further evidence of the importance of TH in β cell development is demonstrated in the postnatal rat where TR isoforms and deiodinases are expressed in an age-dependent fashion. Metabolic development is delayed in rats after neonatal conditions of TH excess or deficiency. TH induces Mafa and thus physiologically regulates β cell maturation.77
Aside from its role in β cell development and insulin secretion, TH also affects insulin action. For example, in streptozotocin-induced diabetic mice treated with TH after MI, there was improved serum glucose levels, improved cardiac functional markers including left ventricular ejection fraction, and increased Akt/mTOR and AMPK, compared with untreated mice.78 Improvements in glucose utilization due to TH can also be insulin independent: in transgenic mice expressing D2 in the heart, there is a six-fold increase in basal cardiac glucose uptake that is insulin independent. This change in metabolism seems to be beneficial given that mortality caused by doxorubicin cardiotoxicity was reduced by 50% in mice expressing D2 in the myocardium.79 These findings echo previous observations that mice with D2 expression in the myocardium exhibit improved cardiac functionality and, in the setting of pressure overload, impairment in contractility and elevation of markers of pathological hypertrophy are prevented.80
Adaptive thermogenesis is thyroid hormone–dependent
For decades BAT has been known to be the primary site of adaptive thermogenesis for rodents, hibernators, and neonates, but its role in the adult human was dismissed due to the lack of distinct anatomic deposits at autopsy.81 This concept has evolved in recent years due to the incidental findings of active BAT in adult humans, using advanced functional images techniques.82,83 These findings have stimulated research in the field as BAT may represent a site of targetable thermogenesis for disposal of excess energy in the obese adult human. BAT physiology is uniquely tied to TH signaling as TH is needed not only for BAT differentiation and development,48 but is required for normal BAT thermogenic function as it induces UCP1 expression.84 For example, hypothyroid rats develop profound hypothermia and may succumb rapidly to cold exposure,85 largely because BAT responds insufficiently to norepinephrine (NE) stimulation in the absence of adequate TH,86,87 even when accounting for the fact that cold-exposed hypothyroid rats have a much greater increase in BAT sympathetic activity.88,89 Thyroid hormone also plays a role in adaptive BAT thermogenesis in response to a hypercaloric diet. For example, D2 KO mice are able to compensate and maintain normal body weight at room temperature by increasing BAT sympathetic activity,90,91 similarly to the UCP1 KO mice.92 However, when acclimated at thermoneutrality, D2 KO mice become obese and develop glucose intolerance with worsening liver steatosis.90
TH also regulates thermogenesis in other tissues, namely the skeletal muscle, using mechanisms that are less dependent on sympathetic activity. For example, hypothyroidism downregulates genes in the soleus muscle but not BAT thermogenic programs, without affecting daily EE.93 BAT thermogenic programs are kept intact because of the compensatory increase in SNS activity.88,89 Only at thermoneutrality (30 °C), a temperature at which a compensatory increase in SNS is no longer present, do hypothyroid mice exhibit a slower rate of EE.93 A byproduct of this compensatory increase in BAT SNS activity at room temperature (22 °C) is that hypothyroid mice are protected against diet-induced obesity (i.e., only at thermoneutrality did hypothyroid mice become obese when placed on an HFD.93
Susceptibility to diet-induced obesity is a function of the animal’s ability to accelerate EE, largely in BAT. This mechanism is diminished in mice treated with dexamethasone, given that glucocorticoids reduce UCP1 expression94 and EE while predisposing to diet-induced obesity.95 In fact, dexamethasome-treated mice have a decreased metabolic rate, lower VO2 response to β3 stimulation, decreased UCP1 mRNA in BAT, and higher body fat compared to mice on an HFD not treated with simultaneous dexamethasone.95 Rodents induce BAT in response to hypercaloric diets, but unfortunately a response of the same magnitude is not observed in humans where short-term overeating does not appropriately stimulate adaptive thermogenesis, a finding that is independent of caloric density or macronutrient composition of the food consumed.96
Interestingly, aside from the distinct populations of white adipocytes and brown adipocytes clearly identified in humans by their microscopic characteristics, there is a population of adipocytes that are able to take on brown fat characteristics under certain stimuli. These cells, termed beige or brite cells, are the site at which “browning” of the white fat can occur, and exemplifies the considerable plasticity within the adipose tissue.97 In obese humans with large stores of white adipocytes, the concept of browning is intriguing and is attracting attention in obesity research.98 The role of TH in browning is an evolving topic, but evidence for its importance is suggested by the expression of D2 in human preadipocytes99 and by findings that T3 induces UCP1 expression, mitochondrial biogenesis, and increases oxygen consumption in human multipotent adipose-derived stem cells.100
Aside from mitochondrial uncoupling in BAT, TH also accelerates EE in other tissues via UCP1-independent mechanisms.9 One such mechanism long thought to underlie the thermogenic effects of TH is an acceleration of fatty acid β-oxidation.101 Given the powerful effects of TH on the rate of β-oxidation, an interesting question is, Which cellular source provides fatty acid to the mitochondria for β-oxidation? Former studies have revealed that TH also strongly accelerates fatty acid synthesis coupled to the stimulation of β-acid oxidation, constituting a substrate cycle that per se contributes to energy expenditure.101 However, new data indicate that TH stimulation of fatty acid β-oxidation is also coupled with induction of hepatic autophagy, which channels cellular fatty acids into β-oxidation. Evidence of this mechanism has been obtained in cultured cells as well as in livers of thyrotoxic mice.102
Thyronamines, a product of the thyroid gland, may provide balance in energy homeostasis
Thyronamines (TAM) are decarboxylated TH molecules. The extent of their physiologic role has yet to be defined but when given to animals they show metabolic and behavioral effects,103 some of which oppose those of T3, indicating that they provide balance in energy homeostasis. For example, dosing of TAM to mice results in decreased body temperature and cardiac output104-106 and reduced metabolic rate.107 Using isotope labeling to identify T4 metabolites, it has been found that 3-iodothyronamine (T1AM) is synthesized in the thyroid gland.108 In addition, studies in thyroidectomized patients treated with levothyroxine indicate that TAM can also be produced outside of the thyroid parenchyma.109 In fact, in vitro studies have shown that TAM can serve as a substrate and be deiodinated by the iodothyronine deiodinases.110
Conclusion
TH has broad contributions to energy homeostasis through its effects in a range of metabolically relevant tissues, and its actions depend upon concerted regulation through both central and peripheral pathways. The multitude of signals that are centrally integrated to define the set point at which TH acts systemically underscores its physiologic importance in maintenance of energy homeostasis. As research continues to understand the role of TH signaling in tissues such as the hypothalamus and BAT, new contributions to the basic cellular physiology of TH, such as data on TH transporters, deiodinases, and receptors, propel the field further toward understanding the role of TH in energy homeostasis. Recent discoveries have contributed to the treatment of conditions of improper TH signaling, including AHDS and TRα resistance. It remains promising that developments in this field may also contribute to a therapeutic target to restore energy homeostasis in obesity.
Acknowledgments
Studies included in this article were funded by the NIDDK.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Hollenberg AN. The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid. 2008;18:131–139. doi: 10.1089/thy.2007.0251. [DOI] [PubMed] [Google Scholar]
- 2.Oppenheimer JH, Schwartz HL, Mariash CN, Kinlaw WB, Wong NCW, Freake HC. Advances in our understanding of thyroid hormone action at the cellular level. Endocrine Reviews. 1987;8:288–308. doi: 10.1210/edrv-8-3-288. [DOI] [PubMed] [Google Scholar]
- 3.Andersen S, Bruun NH, Pedersen KM, Laurberg P. Biologic variation is important for interpretation of thyroid function tests. Thyroid. 2003;13:1069–1078. doi: 10.1089/105072503770867237. [DOI] [PubMed] [Google Scholar]
- 4.Vella KR, Ramadoss P, Lam FS, Harris JC, Ye FD, Same PD, O’Neill NF, Maratos-Flier E, Hollenberg AN. NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. 2011;14:780–790. doi: 10.1016/j.cmet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E, Leibel RL. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115:3579–3586. doi: 10.1172/JCI25977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov. 2010;9:465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 8.Nedergaard J, Cannon B. The changed metabolic world with human brown adipose tissue: therapeutic visions. Cell Metab. 2010;11:268–272. doi: 10.1016/j.cmet.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Bianco AC, Mcaninch EA. The role of thyroid hormone and brown adipose tissue in energy homeostasis. The Lancet Diabetes & Endocrinology. 2013;1:250–258. doi: 10.1016/S2213-8587(13)70069-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Pergola G, Ciampolillo A, Paolotti S, Trerotoli P, Giorgino R. Free triiodothyronine and thyroid stimulating hormone are directly associated with waist circumference, independently of insulin resistance, metabolic parameters and blood pressure in overweight and obese women. Clin Endocrinol (Oxf) 2007;67:265–269. doi: 10.1111/j.1365-2265.2007.02874.x. [DOI] [PubMed] [Google Scholar]
- 11.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122:3035–3043. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, Lechan RM. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138:3359–3368. doi: 10.1210/endo.138.8.5318. [DOI] [PubMed] [Google Scholar]
- 14.Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A. 1997;94:10391–10396. doi: 10.1073/pnas.94.19.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kallo I, Mohacsik P, Vida B, Zeold A, Bardoczi Z, Zavacki AM, Farkas E, Kadar A, Hrabovszky E, Arrojo EDR, et al. A novel pathway regulates thyroid hormone availability in rat and human hypothalamic neurosecretory neurons. PLoS One. 2012;7:e37860. doi: 10.1371/journal.pone.0037860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jo S, Kallo I, Bardoczi Z, Arrojo EDR, Zeold A, Liposits Z, Oliva A, Lemmon VP, Bixby JL, Gereben B, et al. Neuronal hypoxia induces hsp40-mediated nuclear import of type 3 deiodinase as an adaptive mechanism to reduce cellular metabolism. J Neurosci. 2012;32:8491–8500. doi: 10.1523/JNEUROSCI.6514-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, Wiersinga WM, Swaab DF, Visser TJ, Fliers E. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab. 2005;90:4322–4334. doi: 10.1210/jc.2004-2567. [DOI] [PubMed] [Google Scholar]
- 18.Freitas BC, Gereben B, Castillo M, Kallo I, Zeold A, Egri P, Liposits Z, Zavacki AM, Maciel RM, Jo S, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120:2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visser WE, Friesema EC, Visser TJ. Thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. 2011;25:1–14. doi: 10.1210/me.2010-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun D, Lelios I, Krause G, Schweizer U. Histidines in potential substrate recognition sites affect thyroid hormone transport by monocarboxylate transporter 8 (MCT8) Endocrinology. 2013;154:2553–2561. doi: 10.1210/en.2012-2197. [DOI] [PubMed] [Google Scholar]
- 22.Friesema EC, Visser TJ, Borgers AJ, Kalsbeek A, Swaab DF, Fliers E, Alkemade A. Thyroid hormone transporters and deiodinases in the developing human hypothalamus. Eur J Endocrinol. 2012;167:379–386. doi: 10.1530/EJE-12-0177. [DOI] [PubMed] [Google Scholar]
- 23.Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–235. doi: 10.1016/S0079-6123(06)53012-2. [DOI] [PubMed] [Google Scholar]
- 24.Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, et al. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boelen A, Kwakkel J, Fliers E. Beyond low plasma T3: local thyroid hormone metabolism during inflammation and infection. Endocr Rev. 2011;32:670–693. doi: 10.1210/er.2011-0007. [DOI] [PubMed] [Google Scholar]
- 26.Brown WH, Gillum MP, Lee HY, Camporez JP, Zhang XM, Jeong JK, Alves TC, Erion DM, Guigni BA, Kahn M, et al. Fatty acid amide hydrolase ablation promotes ectopic lipid storage and insulin resistance due to centrally mediated hypothyroidism. Proc Natl Acad Sci U S A. 2012;109:14966–14971. doi: 10.1073/pnas.1212887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest. 2006;116:476–484. doi: 10.1172/JCI26240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klieverik LP, Janssen SF, van Riel A, Foppen E, Bisschop PH, Serlie MJ, Boelen A, Ackermans MT, Sauerwein HP, Fliers E, et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci U S A. 2009;106:5966–5971. doi: 10.1073/pnas.0805355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fonseca TL, Correa-Medina M, Campos MP, Wittmann G, Werneck-de-Castro JP, Drigo RA, Mora-Garzon M, Ueta CB, Caicedo A, Fekete C, et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J Clin Invest. 2013;123:1492–1500. doi: 10.1172/JCI61231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosene ML, Wittmann G, Arrojo e Drigo R, Singru PS, Lechan RM, Bianco AC. Inhibition of the type 2 iodothyronine deiodinase underlies the elevated plasma TSH associated with amiodarone treatment. Endocrinology. 2010;151:5961–5970. doi: 10.1210/en.2010-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christoffolete MA, Arrojo e Drigo R, Gazoni F, Tente SM, Goncalves V, Amorim BS, Larsen PR, Bianco AC, Zavacki AM. Mice with impaired extrathyroidal thyroxine to 3,5,3’-triiodothyronine conversion maintain normal serum 3,5,3’-triiodothyronine concentrations. Endocrinology. 2007;148:954–960. doi: 10.1210/en.2006-1042. [DOI] [PubMed] [Google Scholar]
- 32.Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, Morgan DA, Schoenmakers E, Agassandian K, Lage R, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16:1001–1008. doi: 10.1038/nm.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mittag J, Lyons DJ, Sallstrom J, Vujovic M, Dudazy-Gralla S, Warner A, Wallis K, Alkemade A, Nordstrom K, Monyer H, et al. Thyroid hormone is required for hypothalamic neurons regulating cardiovascular functions. J Clin Invest. 2013;123:509–516. doi: 10.1172/JCI65252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva JE, Larsen PR. Adrenergic activation of triiodothyronine production in brown adipose tissue. Nature. 1983;305:712–713. doi: 10.1038/305712a0. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 36.da-Silva WS, Harney JW, Kim BW, Li J, Bianco SD, Crescenzi A, Christoffolete MA, Huang SA, Bianco AC. The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes. 2007;56:767–776. doi: 10.2337/db06-1488. [DOI] [PubMed] [Google Scholar]
- 37.da-Silva WS, Ribich S, Arrojo e Drigo R, Castillo M, Patti ME, Bianco AC. The chemical chaperones tauroursodeoxycholic and 4-phenylbutyric acid accelerate thyroid hormone activation and energy expenditure. FEBS Lett. 2011;585:539–544. doi: 10.1016/j.febslet.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamada Y, Nagasaki H, Fuchigami M, Furuta S, Seino Y, Nakamura J, Oiso Y. The alpha-glucosidase inhibitor miglitol affects bile acid metabolism and ameliorates obesity and insulin resistance in diabetic mice. Metabolism. 2013;62:734–742. doi: 10.1016/j.metabol.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 39.Sagar GD, Gereben B, Callebaut I, Mornon JP, Zeold A, da Silva WS, Luongo C, Dentice M, Tente SM, Freitas BC, et al. Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol. 2007;27:4774–4783. doi: 10.1128/MCB.00283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curcio-Morelli C, Zavacki AM, Christofollete M, Gereben B, de Freitas BC, Harney JW, Li Z, Wu G, Bianco AC. Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest. 2003;112:189–196. doi: 10.1172/JCI18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simonides WS, Mulcahey MA, Redout EM, Muller A, Zuidwijk MJ, Visser TJ, Wassen FW, Crescenzi A, da-Silva WS, Harney J, et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest. 2008;118:975–983. doi: 10.1172/JCI32824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang SA, Mulcahey MA, Crescenzi A, Chung M, Kim BW, Barnes C, Kuijt W, Turano H, Harney J, Larsen PR. Transforming growth factor-beta promotes inactivation of extracellular thyroid hormones via transcriptional stimulation of type 3 iodothyronine deiodinase. Mol Endocrinol. 2005;19:3126–3136. doi: 10.1210/me.2005-0173. [DOI] [PubMed] [Google Scholar]
- 43.Dentice M, Luongo C, Huang S, Ambrosio R, Elefante A, Mirebeau-Prunier D, Zavacki AM, Fenzi G, Grachtchouk M, Hutchin M, et al. Sonic hedgehoginduced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc Natl Acad Sci U S A. 2007;104:14466–14471. doi: 10.1073/pnas.0706754104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeold A, et al. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7:698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peeters RP, Hernandez A, Ng L, Ma M, Sharlin DS, Pandey M, Simonds WF, St Germain DL, Forrest D. Cerebellar abnormalities in mice lacking type 3 deiodinase and partial reversal of phenotype by deletion of thyroid hormone receptor alpha1. Endocrinology. 2013;154:550–561. doi: 10.1210/en.2012-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olivares EL, Marassi MP, Fortunato RS, da Silva AC, Costa-e-Sousa RH, Araujo IG, Mattos EC, Masuda MO, Mulcahey MA, Huang SA, et al. Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology. 2007;148:4786–4792. doi: 10.1210/en.2007-0043. [DOI] [PubMed] [Google Scholar]
- 47.Janssen R, Zuidwijk M, Muller A, Mulders J, Oudejans CB, Simonides WS. Cardiac expression of deiodinase type 3 (Dio3) following myocardial infarction is associated with the induction of a pluripotency microRNA signature from the Dlk1-Dio3 genomic region. Endocrinology. 2013;154:1973–1978. doi: 10.1210/en.2012-2017. [DOI] [PubMed] [Google Scholar]
- 48.Hall JA, Ribich S, Christoffolete MA, Simovic G, Correa-Medina M, Patti ME, Bianco AC. Absence of thyroid hormone activation during development underlies a permanent defect in adaptive thermogenesis. Endocrinology. 2010;151:4573–4582. doi: 10.1210/en.2010-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kester MH, Toussaint MJ, Punt CA, Matondo R, Aarnio AM, Darras VM, Everts ME, de Bruin A, Visser TJ. Large induction of type III deiodinase expression after partial hepatectomy in the regenerating mouse and rat liver. Endocrinology. 2009;150:540–545. doi: 10.1210/en.2008-0344. [DOI] [PubMed] [Google Scholar]
- 50.Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- 51.Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, Ramirez P, Iorcansky S, Rivarola MA, Belgorosky A. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2007;67:1–6. doi: 10.1159/000095805. [DOI] [PubMed] [Google Scholar]
- 52.Wemeau JL, Pigeyre M, Proust-Lemoine E, d’Herbomez M, Gottrand F, Jansen J, Visser TJ, Ladsous M. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084–2088. doi: 10.1210/jc.2007-2719. [DOI] [PubMed] [Google Scholar]
- 53.Di Cosmo C, Liao XH, Ye H, Ferrara AM, Weiss RE, Refetoff S, Dumitrescu AM. Mct8-deficient mice have increased energy expenditure and reduced fat mass that is abrogated by normalization of serum t3 levels. Endocrinology. 2013;154:4885–4895. doi: 10.1210/en.2013-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verge CF, Konrad D, Cohen M, Di Cosmo C, Dumitrescu AM, Marcinkowski T, Hameed S, Hamilton J, Weiss RE, Refetoff S. Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J Clin Endocrinol Metab. 2012;97:4515–4523. doi: 10.1210/jc.2012-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, Henning E, Reinemund J, Gevers E, Sarri M, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med. 2012;366:243–249. doi: 10.1056/NEJMoa1110296. [DOI] [PubMed] [Google Scholar]
- 56.van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, Tsatsoulis A, Peeters R, Visser TJ. Clinical phenotype and mutant TRalpha1. N Engl J Med. 2012;366:1451–1453. doi: 10.1056/NEJMc1113940. [DOI] [PubMed] [Google Scholar]
- 57.Moran C, Schoenmakers N, Agostini M, Schoenmakers E, Offiah A, Kydd A, Kahaly G, Mohr-Kahaly S, Rajanayagam O, Lyons G, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. J Clin Endocrinol Metab. 2013;98:4254–4261. doi: 10.1210/jc.2013-2215. [DOI] [PubMed] [Google Scholar]
- 58.Warner A, Rahman A, Solsjo P, Gottschling K, Davis B, Vennstrom B, Arner A, Mittag J. Inappropriate heat dissipation ignites brown fat thermogenesis in mice with a mutant thyroid hormone receptor alpha1. Proc Natl Acad Sci U S A. 2013;110:16241–16246. doi: 10.1073/pnas.1310300110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakurai A, Takeda K, Ain K, Ceccarelli P, Nakai A, Seino S, Bell GI, Refetoff S, DeGroot LJ. Generalized resistance to thyroid hormone associated with a mutation in the ligand-binding domain of the human thyroid hormone receptor beta. Proc Natl Acad Sci U S A. 1989;86:8977–8981. doi: 10.1073/pnas.86.22.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. Homozygous thyroid hormone receptor beta-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab. 2012;97:1328–1336. doi: 10.1210/jc.2011-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ribeiro MO, Bianco SD, Kaneshige M, Schultz JJ, Cheng SY, Bianco AC, Brent GA. Expression of uncoupling protein 1 in mouse brown adipose tissue is thyroid hormone receptor-beta isoform specific and required for adaptive thermogenesis. Endocrinology. 2010;151:432–440. doi: 10.1210/en.2009-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meruvu S, Ayers SD, Winnier G, Webb P. Thyroid Hormone Analogues: Where Do We Stand in 2013? Thyroid. 2013;23:1333–1344. doi: 10.1089/thy.2012.0458. [DOI] [PubMed] [Google Scholar]
- 63.Fozzatti L, Kim DW, Park JW, Willingham MC, Hollenberg AN, Cheng SY. Nuclear receptor corepressor (NCOR1) regulates in vivo actions of a mutated thyroid hormone receptor alpha. Proc Natl Acad Sci U S A. 2013;110:7850–7855. doi: 10.1073/pnas.1222334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fozzatti L, Lu C, Kim DW, Park JW, Astapova I, Gavrilova O, Willingham MC, Hollenberg AN, Cheng SY. Resistance to thyroid hormone is modulated in vivo by the nuclear receptor corepressor (NCOR1) Proc Natl Acad Sci U S A. 2011;108:17462–17467. doi: 10.1073/pnas.1107474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suh JH, Sieglaff DH, Zhang A, Xia X, Cvoro A, Winnier GE, Webb P. SIRT1 is a direct coactivator of thyroid hormone receptor beta1 with gene-specific actions. PLoS One. 2013;8:e70097. doi: 10.1371/journal.pone.0070097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goodson ML, Mengeling BJ, Jonas BA, Privalsky ML. Alternative mRNA splicing of corepressors generates variants that play opposing roles in adipocyte differentiation. J Biol Chem. 2011;286:44988–44999. doi: 10.1074/jbc.M111.291625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chidakel A, Mentuccia D, Celi FS. Peripheral metabolism of thyroid hormone and glucose homeostasis. Thyroid. 2005;15:899–903. doi: 10.1089/thy.2005.15.899. [DOI] [PubMed] [Google Scholar]
- 69.Lenzen S, Bailey CJ. Thyroid hormones, gonadal and adrenocortical steroids and the function of the islets of Langerhans. Endocr Rev. 1984;5:411–434. doi: 10.1210/edrv-5-3-411. [DOI] [PubMed] [Google Scholar]
- 70.Mouradian M, Abourizk N. Diabetes mellitus and thyroid disease. Diabetes Care. 1983;6:512–520. doi: 10.2337/diacare.6.5.512. [DOI] [PubMed] [Google Scholar]
- 71.Lenzen S, Panten U, Hasselblatt A. Thyroxine treatment and insulin secretion in the rat. Diabetologia. 1975;11:49–55. doi: 10.1007/BF00422818. [DOI] [PubMed] [Google Scholar]
- 72.Ximenes HM, Lortz S, Jorns A, Lenzen S. Triiodothyronine (T3)-mediated toxicity and induction of apoptosis in insulin-producing INS-1 cells. Life Sci. 2007;80:2045–2050. doi: 10.1016/j.lfs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Klieverik LP, Sauerwein HP, Ackermans MT, Boelen A, Kalsbeek A, Fliers E. Effects of thyrotoxicosis and selective hepatic autonomic denervation on hepatic glucose metabolism in rats. Am J Physiol Endocrinol Metab. 2008;294:E513–520. doi: 10.1152/ajpendo.00659.2007. [DOI] [PubMed] [Google Scholar]
- 74.Taguchi Y, Tasaki Y, Terakado K, Kobayashi K, Machida T, Kobayashi T. Impaired insulin secretion from the pancreatic islets of hypothyroidal growth-retarded mice. J Endocrinol. 2010;206:195–204. doi: 10.1677/JOE-09-0465. [DOI] [PubMed] [Google Scholar]
- 75.Nada AM. Effect of treatment of overt hypothyroidism on insulin resistance. World J Diabetes. 2013;4:157–161. doi: 10.4239/wjd.v4.i4.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Medina MC, Molina J, Gadea Y, Fachado A, Murillo M, Simovic G, Pileggi A, Hernandez A, Edlund H, Bianco AC. The thyroid hormone-inactivating type III deiodinase is expressed in mouse and human beta-cells and its targeted inactivation impairs insulin secretion. Endocrinology. 2011;152:3717–3727. doi: 10.1210/en.2011-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, Bonner-Weir S. Thyroid Hormone Promotes Postnatal Rat Pancreatic beta-Cell Development and Glucose-Responsive Insulin Secretion Through MAFA. Diabetes. 2013;62:1569–1580. doi: 10.2337/db12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mourouzis I, Giagourta I, Galanopoulos G, Mantzouratou P, Kostakou E, Kokkinos AD, Tentolouris N, Pantos C. Thyroid hormone improves the mechanical performance of the post-infarcted diabetic myocardium: A response associated with up-regulation of Akt/mTOR and AMPK activation. Metabolism. 2013;62:1387–1393. doi: 10.1016/j.metabol.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 79.Hong EG, Kim BW, Young Jung D, Hun Kim J, Yu T, Seixas Da Silva W, Friedline RH, Bianco SD, Seslar SP, Wakimoto H, et al. Cardiac Expression of Human Type 2 Iodothyronine Deiodinase Increases Glucose Metabolism and Protects Against Doxorubicin-Induced Cardiac Dysfunction in Male Mice. Endocrinology. 2013;154:3937–3946. doi: 10.1210/en.2012-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trivieri MG, Oudit GY, Sah R, Kerfant BG, Sun H, Gramolini AO, Pan Y, Wickenden AD, Croteau W, Morreale de Escobar G, et al. Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc Natl Acad Sci U S A. 2006;103:6043–6048. doi: 10.1073/pnas.0601072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heaton JM. The distribution of brown adipose tissue in the human. J Anat. 1972;112:35–39. [PMC free article] [PubMed] [Google Scholar]
- 82.Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, Huang TL, Roberts-Toler C, Weiner LS, Sze C, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19:635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sacks H, Symonds ME. Anatomical locations of human brown adipose tissue: functional relevance and implications in obesity and type 2 diabetes. Diabetes. 2013;62:1783–1790. doi: 10.2337/db12-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bianco AC, Kieffer JD, Silva JE. Adenosine 3’,5’-monophosphate and thyroid hormone control of uncoupling protein messenger ribonucleic acid in freshly dispersed brown adipocytes. Endocrinology. 1992;130:2625–2633. doi: 10.1210/endo.130.5.1374009. [DOI] [PubMed] [Google Scholar]
- 85.Sellers EA, You SS. Role of the thyroid in metabolic responses to a cold environment. The American Journal of Physiology. 1950;163:81–91. doi: 10.1152/ajplegacy.1950.163.1.81. [DOI] [PubMed] [Google Scholar]
- 86.Bianco AC, Silva JE. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. J Clin Invest. 1987;79:295–300. doi: 10.1172/JCI112798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ribeiro MO, Lebrun FL, Christoffolete MA, Branco M, Crescenzi A, Carvalho SD, Negrao N, Bianco AC. Evidence of UCP1-independent regulation of norepinephrine-induced thermogenesis in brown fat. Am J Physiol Endocrinol Metab. 2000;279:E314–322. doi: 10.1152/ajpendo.2000.279.2.E314. [DOI] [PubMed] [Google Scholar]
- 88.Young JB, Saville E, Rothwell NJ, Stock MJ, Landsberg L. Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. Journal of Clinical Investigation. 1982;69:1061–1071. doi: 10.1172/JCI110541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Young JB, Saville E, Landsberg L. Effect of thyroid state on norepinephrine (NE) turnover in rat brown adipose tissue (BAT): potential importance of the pituitary. Clin Res. 1982;32:407. Abstract. [Google Scholar]
- 90.Castillo M, Hall JA, Correa-Medina M, Ueta C, Won Kang H, Cohen DE, Bianco AC. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes. 2011;60:1082–1089. doi: 10.2337/db10-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Christoffolete MA, Linardi CC, de Jesus L, Ebina KN, Carvalho SD, Ribeiro MO, Rabelo R, Curcio C, Martins L, Kimura ET, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53:577–584. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]
- 92.Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 93.Ueta CB, Olivares EL, Bianco AC. Responsiveness to thyroid hormone and to ambient temperature underlies differences between brown adipose tissue and skeletal muscle thermogenesis in a mouse model of diet-induced obesity. Endocrinology. 2011;152:3571–3581. doi: 10.1210/en.2011-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moriscot A, Rabelo R, Bianco AC. Corticosterone inhibits uncoupling protein gene expression in brown adipose tissue. Am J Physiol. 1993;265:E81–87. doi: 10.1152/ajpendo.1993.265.1.E81. [DOI] [PubMed] [Google Scholar]
- 95.Poggioli R, Ueta CB, ED RA, Castillo M, Fonseca TL, Bianco AC. Dexamethasone reduces energy expenditure and increases susceptibility to diet-induced obesity in mice. Obesity (Silver Spring) 2013 doi: 10.1002/oby.20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Apolzan JW, Bray GA, Hamilton MT, Zderic TW, Han H, Champagne CM, Shepard D, Martin CK. Short-term overeating results in incomplete energy intake compensation regardless of energy density or macronutrient composition. Obesity (Silver Spring) 2013 doi: 10.1002/oby.20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peschechera A, Eckel J. “Browning” of adipose tissue - regulation and therapeutic perspectives. Arch Physiol Biochem. 2013;119:151–160. doi: 10.3109/13813455.2013.796995. [DOI] [PubMed] [Google Scholar]
- 98.Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nomura E, Toyoda N, Harada A, Nishimura K, Ukita C, Morimoto S, Kosaki A, Iwasaka T, Nishikawa M. Type 2 iodothyronine deiodinase is expressed in human preadipocytes. Thyroid. 2011;21:305–310. doi: 10.1089/thy.2010.0068. [DOI] [PubMed] [Google Scholar]
- 100.Lee JY, Takahashi N, Yasubuchi M, Kim YI, Hashizaki H, Kim MJ, Sakamoto T, Goto T, Kawada T. Triiodothyronine induces UCP-1 expression and mitochondrial biogenesis in human adipocytes. Am J Physiol Cell Physiol. 2012;302:C463–472. doi: 10.1152/ajpcell.00010.2011. [DOI] [PubMed] [Google Scholar]
- 101.Oppenheimer JH, Schwartz HL, Lane JT, Thompson MP. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J Clin Invest. 1991;87:125–132. doi: 10.1172/JCI114961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Piehl S, Hoefig CS, Scanlan TS, Kohrle J. Thyronamines--past, present, and future. Endocr Rev. 2011;32:64–80. doi: 10.1210/er.2009-0040. [DOI] [PubMed] [Google Scholar]
- 104.Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- 105.Chiellini G, Frascarelli S, Ghelardoni S, Carnicelli V, Tobias SC, DeBarber A, Brogioni S, Ronca-Testoni S, Cerbai E, Grandy DK, et al. Cardiac effects of 3-iodothyronamine: a new aminergic system modulating cardiac function. FASEB J. 2007;21:1597–1608. doi: 10.1096/fj.06-7474com. [DOI] [PubMed] [Google Scholar]
- 106.Klieverik LP, Coomans CP, Endert E, Sauerwein HP, Havekes LM, Voshol PJ, Rensen PC, Romijn JA, Kalsbeek A, Fliers E. Thyroid hormone effects on whole-body energy homeostasis and tissue-specific fatty acid uptake in vivo. Endocrinology. 2009;150:5639–5648. doi: 10.1210/en.2009-0297. [DOI] [PubMed] [Google Scholar]
- 107.Braulke LJ, Klingenspor M, Debarber A, Tobias SC, Grandy DK, Scanlan TS, Heldmaier G. 3-Iodothyronamine: a novel hormone controlling the balance between glucose and lipid utilisation. J Comp Physiol [B] 2008;178:167–177. doi: 10.1007/s00360-007-0208-x. [DOI] [PubMed] [Google Scholar]
- 108.Hackenmueller SA, Marchini M, Saba A, Zucchi R, Scanlan TS. Biosynthesis of 3-iodothyronamine (T1AM) is dependent on the sodium-iodide symporter and thyroperoxidase but does not involve extrathyroidal metabolism of T4. Endocrinology. 2012;153:5659–5667. doi: 10.1210/en.2012-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hoefig CS, Kohrle J, Brabant G, Dixit K, Yap B, Strasburger CJ, Wu Z. Evidence for extrathyroidal formation of 3-iodothyronamine in humans as provided by a novel monoclonal antibody-based chemiluminescent serum immunoassay. J Clin Endocrinol Metab. 2011;96:1864–1872. doi: 10.1210/jc.2010-2680. [DOI] [PubMed] [Google Scholar]
- 110.Piehl S, Heberer T, Balizs G, Scanlan TS, Smits R, Koksch B, Kohrle J. Thyronamines Are Isozyme Specific Substrates of Deiodinases. Endocrinology. 2008;149:3037–3045. doi: 10.1210/en.2007-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]