

Abstract
Elevated plasma free fatty acids (FFA) induce insulin resistance in skeletal muscle. Previously, we have shown that experimental insulin resistance induced by lipid infusion prevents the dispersion of insulin through the muscle, and we hypothesized that this would lead to an impairment of insulin moving from the plasma to the muscle interstitium. Thus, we infused lipid into our anesthetized canine model and measured the appearance of insulin in the lymph as a means to sample muscle interstitium under hyperinsulinemic euglycemic clamp conditions. Although lipid infusion lowered the glucose infusion rate and induced both peripheral and hepatic insulin resistance, we were unable to detect an impairment of insulin access to the lymph. Interestingly, despite a significant, 10-fold increase in plasma FFA, we detected little to no increase in free fatty acids or triglycerides in the lymph after lipid infusion. Thus, we conclude that experimental insulin resistance induced by lipid infusion does not reduce insulin access to skeletal muscle under clamp conditions. This would suggest that the peripheral insulin resistance is likely due to reduced cellular sensitivity to insulin in this model, and yet we did not detect a change in the tissue microenvironment that could contribute to cellular insulin resistance.
Keywords: insulin, interstitium, glucose, lipid, skeletal muscle
the association between obesity, insulin resistance, and diabetes has long been reported, but the mechanisms linking them are still under investigation, and some have suggested that insulin resistance might be a common factor between all aspects of the metabolic syndrome (47). Although cellular insulin resistance can be induced by free fatty acids (FFA) (4, 39), another major contributor to insulin resistance is thought to be reduced insulin access, which may account for 30% of the metabolic deficit observed in diabetes (31).
Several studies have shown impaired access of insulin to skeletal muscle in obese humans (38, 41), yet a recent study using lipid infusion to induce insulin resistance observed no reduction in the transport of insulin across the vessel wall (45). However, this human study did not report actual levels of interstitial insulin but instead reported change from basal. Access to muscle is a major component of insulin sensitivity (1, 11) likely due to the microvascular effects of insulin, which increase distribution of blood through muscle, thus increasing delivery to the insulin-sensitive tissues.
These microvascular effects of insulin are endothelium dependent. Insulin relies on a functioning endothelium for the vasodilation and vasoconstriction required to increase muscle perfusion and thus increase delivery (35), and insulin's metabolic effects are greater in subjects that have good microvascular responses to insulin (15). Impairments in insulin signaling in knockout models have demonstrated impaired insulin access and lowered insulin-mediated glucose uptake, an effect that is reversed by restoring vasodilation (30). Insulin's microvascular effects are blocked by hyperlipidemia, and we have shown that hyperlipidemia impairs insulin dispersion in canine muscle (10), similar to that in diet-induced insulin resistance (28). Thus we anticipated that plasma hyperlipidemia would impair endothelial function and insulin access, leading to tissue insulin resistance, although this is contrary to a recently published study (45). Therefore, we investigated the effect of increasing systemic plasma FFA on the lymph concentration of insulin in skeletal muscle and the relationship to insulin sensitivity of muscle.
Initially, we began the hyperlipidemia at the same time as hyperinsulinemia to allow us to observe the temporal effect of hyperlipidemia on insulin's access to lymph and metabolic effects. We used the results from this study in selecting a 3-h preinfusion of lipid before hyperinsulinemia in an effort to establish endothelial dysfunction prior to exposure to insulin. Furthermore, due to the minor changes in lymph FFA, we incorporated a more complete assessment of lipid within the hind leg. We expected that a longer time of systemic hyperlipidemia would reduce the access of insulin to muscle.
MATERIALS AND METHODS
Animals.
Experiments were performed on anesthetized male mongrel dogs. Animals were housed in the University of Southern California (USC) Medical School Vivarium under controlled kennel conditions (12:12-h light-dark) and fed standard chow once/day (49% carbohydrate, 25% protein, and 9% fat; Alfred Mills, Chicago, IL). Dogs were used for experiments only if they were judged to be in good health as determined by visual observation, body weight, hematocrit, and body temperature. Eight animals are presented in each group. Protocols were conducted in conformity with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the USC Institutional Animal Care and Use Committee, where the experiments were performed.
Surgery.
Animals were fasted 15 h before the morning of the experiment at 0600. Dogs were preanesthetized with acepromazine maleate (0.22 mg/kg, Prom-Ace; Aueco, Fort Dodge, IA) and atropine sulfate (0.11 ml/kg; Western Medical, Arcadia, CA). Anesthesia was induced with pentobarbital sodium (0.5 ml/kg; Western Medical) or propofol (6 mg/kg; Western Medical) and maintained with isofluorane (Western Medical). Dogs were placed on heating pads to maintain body temperature. An indwelling catheter was implanted in the left carotid artery for sampling and blood pressure monitoring (model no. 90603A; Space Labs, Issaquah, WA). Intracatheters were inserted into the left cephalic vein for variable glucose infusion and the right cephalic vein for insulin, somatostatin, lipid, and tracer infusion. Saline was infused into the jugular or cephalic vein during surgery and administered at ∼1 liter during the surgery and at a slow drip until the end of surgery. Indwelling catheters were also placed into both the left femoral artery and vein for sampling. The hindlimb lymphatic vessel was cannulated by placing a polyethylene catheter (PE10) into the afferent lymphatic vessel of the deep inguinal lymph node. Lymph was collected by gently massaging the leg directly above the popliteal area, which has been shown to instantaneously increase lymph drainage without affecting lymph or plasma oncotic pressures or the lymph/plasma protein concentration ratio (25). Blood pressure, heart rate, O2 saturation, and CO2 were monitored continuously. At the conclusion of these experiments, animals were euthanized with an overdose of pentobarbital sodium (65 mg/kg, Eutha-6; Western Medical).
Study 1.
Immediately after surgical procedures, a basal insulin euglycemic clamp was started (t = −180 min) with a primed tracer infusion of [3-3H]glucose (priming dose 25 μCi, infusion 0.25 μCi/min; DuPont-NEN, Boston, MA) into the right cephalic. Somatostatin was infused (1 μg·min−1·kg−1; Bachem) and basal insulin replaced systemically (0.2 mU·min−1·kg−1; Novo Nordisk, Bagsvaerd, Denmark) and continuously for the remainder of the study. Exogenous 20% glucose labeled with [3-3H]glucose (2.7 μCi/g glucose) was infused into the left cephalic vein at variable rates to clamp arterial glucose to the basal level (established during the surgery prior to −180 min) throughout the entire experimental period. Intralipid (20%, Fresenius Kabi, with 16 U/ml heparin) or saline (control) was infused at a rate of 1.5 ml/min beginning at time 0. Samples were taken from the left carotid artery, and lymph vessels were sampled by gently massaging the hindlimb distal to the site of catheterization from 2 min prior to 2 min after each blood sample point. After 180 min of basal insulin infusion (time 0), the insulin concentration was increased to 1.2 mU·min−1·kg−1. This was continued for the remainder of the study.
Study 2.
Based on results from study 1, we sampled from the femoral artery and femoral vein and began lipid infusion 180 min prior to hyperinsulinemia. The experimental procedure was otherwise similar to study 1. Briefly, immediately after surgery was completed, a primed tracer infusion was initiated, somatostatin and intralipid were infused, and basal insulin replacement was initiated (−180 to 0 min). At time 0, insulin infusion was increased to 1 mU·min−1·kg−1. Exogenous glucose was infused to clamp arterial glucose to the basal level, and samples were taken from femoral artery, femoral vein, and lymph vessels.
Assays.
Plasma and lymph samples were assayed for insulin, glucose, [3-3H]glucose, and FFA. Arterial, venous, and lymph samples were collected in microtubes that were precoated with lithium-heparin (Becton Dickinson, Franklin Lakes, NJ) or in microtubes without lithium-heparin for lipid measurement. Arterial and venous tubes also contained 50 μl of EDTA (Sigma Chemical). Blood samples were centrifuged immediately, and the supernatant was transferred and stored at −20°C until further assay. Femoral artery plasma samples were assayed immediately for glucose with a YSI 2700 autoanalyzer (Yellow Springs Instruments, Yellow Springs, OH) before freezing at −20°C. Lymph samples were immediately stored at −20°C after sampling. Insulin was measured in plasma and lymph with an ELISA adapted for dog plasma. Samples for measuring [3-3H]glucose were deproteinized with barium hydroxide and zinc sulfate, evaporated to remove radiolabeled water, and counted in Ready Safe scintillation fluid (Beckman Instruments, Fullerton, CA). Plasma FFAs were measured using the NEFA C kit (Wako Pure Chemical Industries, Richmond, VA), and these results were validated with an independent assay, the Cayman Free Fatty Acid Assay kit (Cayman Chemical, Ann Arbor, MI). Although no paraoxon was added to the tubes to suppress lipolysis, our studies show that with immediate centrifugation and placement on ice, there is no significant FFA difference between samples treated with paraoxon and those without (Fig. 1).
Fig. 1.
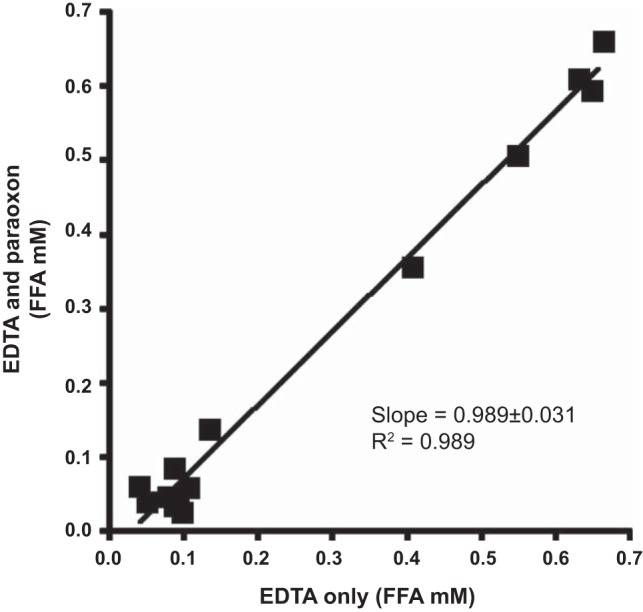
Comparison between free fatty acid (FFA) concentrations in samples treated with EDTA and EDTA plus paraoxon (both nonheparinized tubes). Not shown is the comparison between EDTA and paraoxon plus EDTA in heparinized tubes provided a slope of 0.0989 ± 0.031 and an r2 value of 0.989 when processed as described in materials and methods.
Tissue insulin sensitivity.
Tracer-determined whole body Rd (glucose disposal) and EGP (endogenous glucose production) were calculated using Steele's equation modified for use with labeled glucose infusion according to Finegood et al. (16) after smoothing plasma glucose and tracer data by optimal segments. Steady state was defined as the final 40 min of the hyperinsulinemic clamp.
Cellular insulin sensitivity.
Cellular insulin sensitivity (Scell) can be calculated from the peripheral insulin action (ΔRd), the change in lymph insulin (ΔInsI), and the glucose concentration (GlucSS) by the equation Scell = ΔRd/(ΔInsI × GlucSS).
Statistical analyses.
All experimental data are shown in the figures as means ± SE. Statistical analyses were performed with paired or unpaired Student's t-tests, one-way ANOVAs, or two-way repeated-measures ANOVAs with Tukey's pairwise comparisons as appropriate.
RESULTS
Study 1: lipid coinfusion.
Our first study used lipid infusion beginning at the same time as insulin, at time 0, to enable us to see the time course of onset of lipid-induced insulin resistance. As can be seen in Fig. 2, insulin increased immediately in the plasma and more slowly in the lymph, and there was no difference between the plasma insulin in the control (saline-infused) or lipid groups. The FFA levels in the plasma increased rapidly with lipid infusion; however, a much slower and smaller increase was observed in lymph FFA, reaching significance only after 90 min.
Fig. 2.

Insulin and FFA concentrations with coinfusion of lipid or saline. A: arterial plasma insulin concentrations. B: insulin concentrations in lymph. C: plasma FFA concentrations in lipid-infused animals. D: lymph FFA concentrations in lipid-infused animals. Dotted vertical line indicates start of insulin and lipid infusion. *Significantly different from time 0 (P < 0.05, 1-way ANOVA).
Rd in the control animals increased gradually with insulin infusion, reaching a maximal rate at ∼90 min. Endogenous glucose production was quickly decreased immediately after insulin infusion and was inhibited for the remainder of the clamp. In the lipid-infused animals, insulin's normal effects to prevent EGP were blocked (Fig. 3). Lipid infusion caused a much slower effect to suppress insulin-mediated increases in peripheral glucose usage (Rd), as glucose uptake originally increased similarly to control animals and was inhibited only after ∼90 min.
Fig. 3.

Glucose infusion (GINF; A), arterial glucose (B), glucose disposal (Rd; C), and endogenous glucose production (EGP; D) with coinfusion of insulin and lipid over time. Dotted vertical line indicates start of insulin and lipid infusion. *Significantly different from control at the same time point (P < 0.05, 2-way ANOVA).
Study 2: lipid preinfusion.
Study 2 investigated insulin access in established insulin resistance. Since the lipid-induced insulin resistance was observed only in the periphery after 90 min in study 1, we infused lipid for 3 h prior to establishing insulin resistance and endothelial dysfunction prior to hyperinsulinemia and sampled vein plasma to better measure the removal of insulin and FFA across the leg. Artery and vein insulin levels increased rapidly with insulin infusion (Fig. 4 A and B). There was slightly less insulin in the vein compared with the artery in each group (control: artery 80.55 ± 10.37 mU/l, vein 77.55 ± 9.51 mU/l, P = 0.10; lipid: artery 95.06 ± 5.74 mU/l, vein 87.58 ± 3.98 mU/l, P = 0.065). Whereas the plasma insulin concentration reached its maximum ∼30 min after initiation of the insulin infusion, the increase in lymph insulin was much slower, requiring ∼120 min to reach its maximum value (Fig. 4C). The lymph insulin concentration was much lower than that in plasma in both groups, and there was no significant difference between the lymph insulin concentrations in control or lipid-infused animals or in the time required to reach the maximum concentration.
Fig. 4.
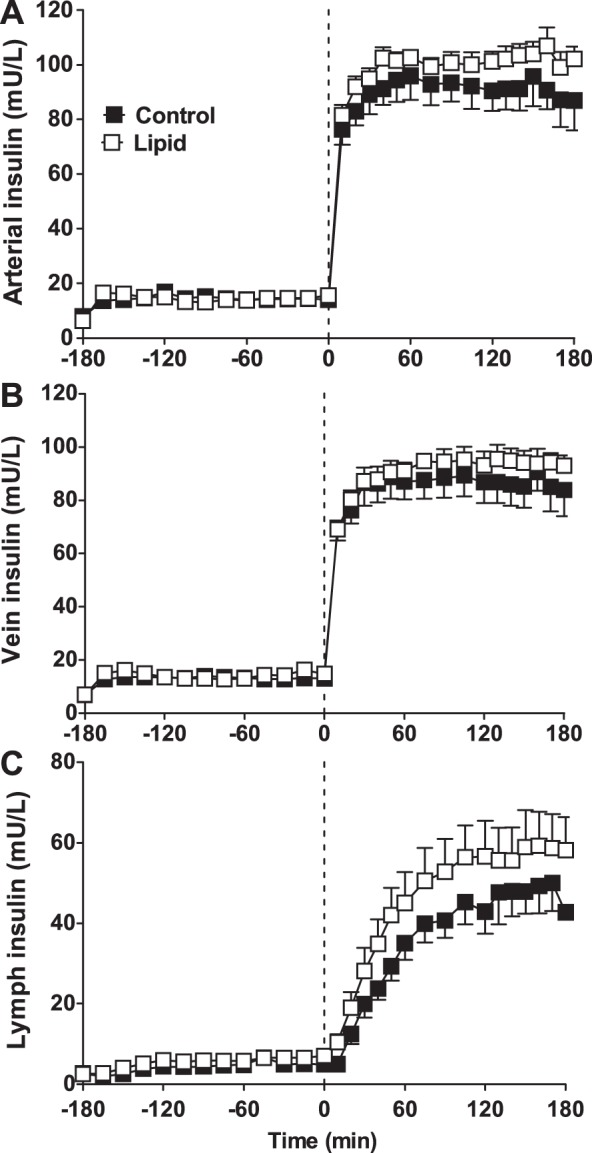
Insulin concentrations in artery (A), vein (B), and lymph (C) for control (saline; ■) and lipid-infused (□) animals. Lymph insulin is significantly lower than arterial insulin throughout. Vein insulin is slightly lower than arterial insulin. Vertical dotted line represents initiation of hyperinsulinemia at time 0. There were no significant differences between control and lipid at any time point.
Control lipid levels were low throughout the experiment (Fig. 5, A and B). Lipid infusion increased plasma lipid levels in both artery and vein (Fig. 5A), and similar effects were observed for glycerol (data not shown). Interestingly, whereas plasma FFA increased with lipid infusion, reaching a maximum within 90 min, lymph FFAs, which were similar to plasma prior to lipid infusion (control 0.62 ± 0.17 vs. lipid 0.65 ± 0.21 mmol/l), remained at basal levels (0.54 ± 0.22 mmol/l at 180 min), and there was no significant effect of lipid infusion on lymph FFA concentration compared with control animals (P = 0.18, 2-way repeated-measures ANOVA). Rather than elevating lymph FFA concentrations, plasma hyperlipidemia appeared to prevent the reduction in lymph FFA that occurred in control animals. Lymph glycerol concentrations did increase in response to lipid infusion (Fig. 5D), although they were still significantly lower than the plasma glycerol levels observed (Fig. 5C).
Fig. 5.

FFA (A and B) and glycerol (C and D) in control and lipid-infused animals showing artery (■), vein (□), and lymph. Vertical dotted line represents initiation of hyperinsulinemia. *Significantly different from control at the same time point (P < 0.05, 2-way ANOVA).
The glucose infusion rate required to maintain euglycemia increased upon insulin infusion but was significantly lower in the lipid-infused animals (control 10.38 ± 1.51, lipid 3.75 ± 0.55 mg·min−1·kg−1, P = 0.0008; Fig. 6A). Arterial glucose levels were not significantly different between groups and were maintained throughout the experiment by infusing exogenous glucose (Fig. 6B). Hyperlipidemia induced insulin resistance, as measured by rate of glucose disposal (Fig. 6C), demonstrating peripheral insulin resistance. Endogenous glucose production by the liver was suppressed by insulin in control animals. Basal glucose production tended to increase with lipid infusion (control 1.74 ± 0.44, lipid 2.83 ± 0.30 mg·min−1·kg−1, P = 0.051), and the effect of insulin to suppress glucose production was lost (Fig. 6D). The impaired Rd and no change in lymph insulin combine to show that cellular insulin sensitivity is significantly impaired with lipid infusion (control 3.62 ± 0.69, lipid: 1.38 ± 0.25 mg·min−1·kg−1, P < 0.05).
Fig. 6.
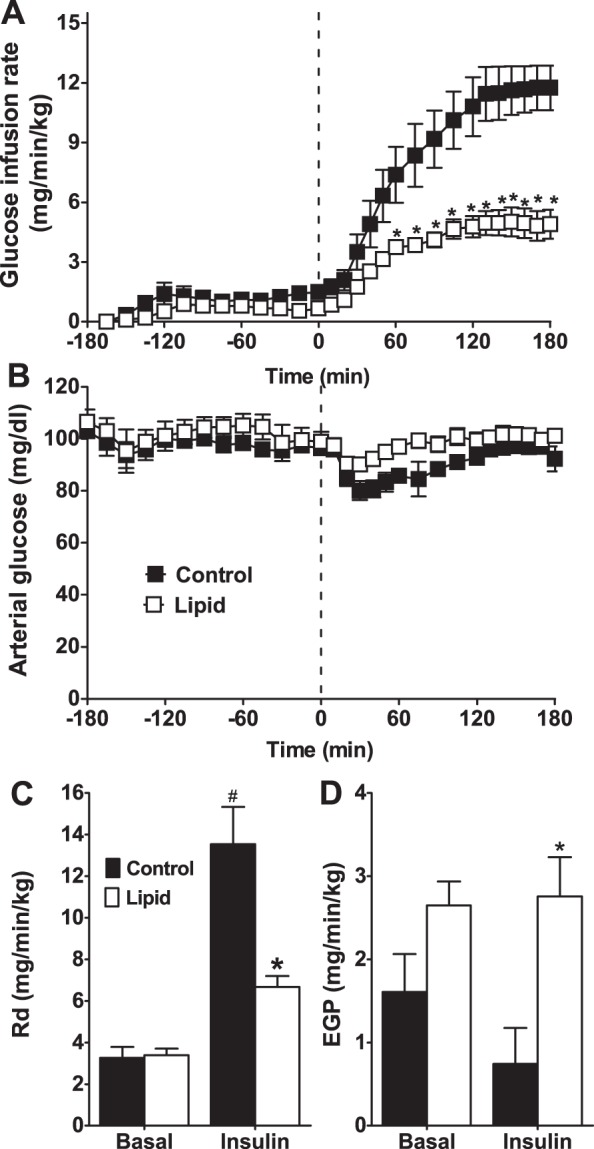
Glucose infusion rate (A), arterial glucose (B), Rd (C), and EGP (D) for control (black bars and ■) and lipid-infused (open bars and □) animals. Basal value indicates the 45-min steady state immediately prior to hyperinsulinemia (time = −45 to 0). Insulin indicates steady state at the end of hyperinsulinemia (time = 140–180 min). #Significantly different from corresponding basal value (P < 0.05); *significantly different from control (P < 0.05).
DISCUSSION
We show that lipid infusion, which elevates plasma lipid levels, caused peripheral insulin resistance but did not reduce insulin transport into the muscle lymph. Initial studies demonstrated that lipid infusion led to an impairment of peripheral insulin action after ∼90 min, which coincided with the peak in lymph FFA. This would suggest that the decrease in Rd may be due to the small increase in lymph FFA. We repeated this experiment with lipid infusion for 3 h to increase lymph FFA levels and induce cellular resistance and endothelial dysfunction prior to the hyperinsulinemic clamp. However, we see no impairment in insulin access to the lymph by lipid infusion. Therefore, we conclude that acute lipid infusion-induced insulin resistance does not impair insulin access to skeletal muscle but instead reduces cellular insulin sensitivity.
In study 1, we found that peripheral insulin resistance began after ∼90 min of lipid infusion, coinciding with the modest increase in lymph FFA levels. However, the hepatic insulin resistance occurred much quicker, as the normal effect of insulin to suppress endogenous glucose production was immediately blocked. Thus, although coinfusion of lipid is useful for studying temporal responses to a change in plasma or a tissue, preinfusion of lipid is required to ensure that the muscle is insulin resistant prior to analysis. This result guided our choice in using a preinfusion of lipid for study 2 in an effort to induce both insulin resistance and endothelial dysfunction prior to hyperinsulinemia.
Endothelial function is a major component of insulin action, increasing the delivery of insulin to muscle and other tissues (3), and endothelial action can be impaired in various aspects of the metabolic syndrome. Changes in blood flow distribution can improve insulin sensitivity (36) and insulin-mediated glucose disposal (48) by increasing the delivery of insulin to the cell surface (11). Systemically elevated plasma FFAs impair insulin-mediated changes in blood flow distribution (12), and similar results have been shown in obese humans (13). Some studies have proposed that impaired insulin access due to endothelial dysfunction may account for ≤30% of the metabolic deficit observed in diabetes (31). Hyperlipidemia causes both insulin resistance and endothelial dysfunction, and therefore, we had expected our preinfusion of lipid to impair the movement of insulin from the plasma into lymph. This could be observed as either a longer time for lymph insulin to reach steady state in lipid-induced insulin resistance or a lower steady state compared with control animals, yet neither of these outcomes was observed in this study.
Our results showing that insulin access is not impaired by lipid infusion-induced insulin resistance are consistent with previous studies (45) that demonstrated that lipid infusion in humans induced insulin resistance but did not alter the rate of movement of insulin into the interstitium. Szendroedi et al. (45) did not measure actual concentrations of insulin within muscle but used microdialysis to measure the change in insulin levels from baseline. We confirmed that there was no change in the time to steady state but also that the insulin concentration in the lymph (as a measure of the interstitial fluid) was not different from healthy animals. In contrast, long-term physiological models of human insulin resistance do show impaired insulin access (41), and obese patients with postprandial hyperglycemia require a higher level of insulin in the plasma to attain similar interstitial insulin levels as those in healthy patients (38). Thus, it is possible that insulin clearance rates in models of insulin resistance are altered to keep the supply of insulin to muscle and other insulin-sensitive tissues at an optimum level.
Peripheral insulin resistance was observed with lipid infusion in both studies. In study 2, we were able to confirm that this is the result of cellular insulin resistance. Because insulin was able to access the lymph, but insulin-mediated glucose uptake was blocked, the ability of cells to respond to insulin is impaired. We show that peripheral insulin action (Rd) is impaired after ∼90 min of FFA infusion, whereas effects on the liver are almost instant (study 1). This temporal difference can likely be attributed to the endothelial structure in each of these tissues (2, 27). The muscle has a continuous endothelium, which provides an effective barrier slowing the movement of nutrients and hormones from the plasma into the tissue. In contrast, liver has a discontinuous endothelium with large fenestrations, effectively allowing more rapid exchange between the plasma and tissue. The liver could detect and then respond to increases in plasma lipids or insulin much more quickly than muscle. Because the elevation of FFA in the lymph, although small, coincided with peripheral insulin resistance when insulin and lipid were coinfused, cellular insulin resistance was likely responsible, as no defect in insulin access could be detected (study 1). FFAs cause cellular insulin resistance by directly inhibiting insulin-stimulated glucose transport in human skeletal muscle (14, 33, 37), which is due possibly to PKC activation (46) altering the insulin signaling cascade (17). With preinfusion of lipid (study 2), insulin sensitivity was similarly reduced, yet there was no impairment of insulin access to muscle, consistent with previous studies (45).
We found that the small increase in lymph FFA observed with lipid coinfusion was lost when lipid was preinfused, possibly suggesting that insulin was involved in the transport of FFA from the plasma to the lymph. Basal lipid levels were different in studies 1 and 2, and potential explanations for the different lymph FFA levels may include a group effect (several years between study 1 and study 2) or the length of the surgical procedure; anesthesia alone can decrease lipid levels, and the lipid infusion prevents the decrease observed in controls. Interestingly, even with coinfusion of lipid, the increase in lymph lipid was extremely small (0.11–0.17) compared with that in plasma (0.3–3.0 mM), and there was a trend for the reduction in lymph FFA in control animals to be prevented by lipid infusion. Similarly, we observed no significant difference between arterial and vein levels of FFAs, suggesting a low level of lipid removal from plasma. In contrast, glycerol levels were increased in lymph, although not enough to match plasma levels. Glycerol alone does not affect glucose oxidation or induce insulin resistance (14) but may reflect that there was FFA entering the muscle at a similar rate. Because there was no significant increase in lymph FFAs, the interstitium may have a very effective buffering capacity, whereby any FFA moving into the interstitial space are immediately incorporated into the cell membrane or internalized into the cell. The myocytes or adipocytes that line the lymph vessels may effectively scavenge any lipid before the interstitial fluid enters the lymph and may contribute to the cellular insulin resistance observed in our study. This uptake would lead to the accumulation of intramyocellular fat, as measured previously (9, 29), although this often occurs only after several hours (6).
Therefore, our finding of no increase in lymph FFA is consistent with previous studies (6, 18), and further measures of intramyocellular lipid levels are required. Studies have shown a slightly higher level of albumin, a carrier of FFA, in the lymph than in the interstitium (24). This should indicate that availability of albumin allows FFA movement from the interstitium to the lymph. However, plasma albumin levels are approximately double those in the interstitium, which may impede the movement of FFA from the plasma to the interstitium. Further studies are required to determine the fate of the infused lipid and may require tracer studies of FFA, histological investigation to demonstrate the presence of adipocytes around the lymph vessels, and measurements of FFA uptake into these adipocytes. Other methods of assessing interstitial lipid concentrations have been questioned, as microdialysis can be impaired by lipid (26), and other interstitial measurements are crude, often limited to accessible tissues such as skin, and typically reflect an end point analysis rather than a dynamic measurement (51).
The findings of this study and our previous lymph studies are dependent on lymph being a good representation of the interstitial environment. The comparison of lymph and other interstitial measures for insulin are quite good between studies, as both microdialysis (19, 42) and lymph (10, 28) have shown a similar gradient between insulin in plasma and the interstitium, and the large pores in the lymph vessel walls permit large particles to enter the lymph vessel (40). However, the location of lymph vessels is closely associated with blood vessels; therefore, lymph sampling may be more indicative of transendothelial transport than the deep interstitium (8). Lymphatic vessels are typically found adjacent to arterioles and venules, and as such they are located close to blood vessels if not directly in contact with the capillaries, where nutrient and hormone exchange typically occur (43). Furthermore, molecules the size of lipoproteins appear to be excluded from ≤50% of the interstitial space (44), and total protein and albumin concentrations of lymph are greater than those in the interstitial space (24). Yet lymph sampling remains one of the better methods for interstitial fluid isolation (51). However, our results showing that injected insulin does not appear in the lymph of insulin-resistant muscle (10, 28) could demonstrate that the lymph vessel itself suffers from dysfunction in insulin-resistant muscle, and the similarities between blood and lymph vessels could support this argument. Thus, the ability of lymph to drain skeletal muscle interstitial fluid needs to be more fully investigated in both health and disease (52). However, since our insulin results are consistent with those of Szendroedi et al. (45), and our FFA results are supported by both the absence of arterio-venous FFA difference and the time requirement for FFA incorporation into tissue (6, 18), we believe that lymph is a good representation of the interstitial space in acute hyperlipidemia.
As discussed, lipid infusion was expected to induce endothelial dysfunction, reducing insulin's microvascular effects (12) and theoretically decreasing access to muscle as well as reducing cellular insulin sensitivity. We found that insulin delivery to the cell surface in the presence of elevated plasma FFAs was not impaired. This could be seen as a contrast to our previous studies, where insulin injected into muscle of a lipid-infused animal was not detected in the lymph (10). Although the assumption is that lymph accurately represents the interstitial environment, there could be substantial regional differences based in part on the location of the lymph vessels as discussed, and whether they represent deep interstitium or an area surrounding the blood vessels. Our theory was that the local endothelial dysfunction prevents dissemination of the injected insulin through the muscle (10), and we calculated that a large proportion of the injected insulin is not washed out but remains in muscle. Mechanisms of dispersing insulin through the interstitial space appear to include blood vessels, lymph vessels, or passive diffusion, and thus it remains possible that lipid infusion prevents the capillary recruitment necessary to distribute injected insulin through the interstitial space, and lymph function may also be impaired by FFA. In contrast, systemic insulin infusion during the clamp exposes a greater amount of blood vessels to a higher level of plasma insulin (approximately double that of injection) for a prolonged period, and thus there is greater blood vessel surface area for insulin transport to the whole muscle tissue and greater potential for capillary recruitment, even if impaired by FFA. In addition, intramuscular injection may not deliver the insulin to an area adjacent to a lymph vessel and would not be detected, yet infusing insulin through the blood vessels, as we have done in the present study, represents a more physiological route of insulin access to muscle, induces substantially higher plasma insulin levels than injection, and causes a systemic elevation of insulin affecting all areas of muscle and presumably lymph. Therefore, FFA should be causing the same endothelial or transport defect in each system; however, the method and amount of insulin administration differentially affect insulin dispersion and transport. It is also possible that there is directionality of active insulin transport such that insulin is preferentially transported from the plasma to the tissue and does not move as readily from tissue to plasma (49). However, because venous insulin levels after insulin injection are higher in lipid-infused animals compared with controls (10, 11), this may suggest that there is no directionality of transport or that there is increased permeability with FFA, allowing insulin to move more readily into the plasma from tissues. Thus, we believe our two experimental models, insulin injection and insulin infusion, may in fact be testing different aspects of insulin access, interstitial dynamics and transendothelial transport, respectively. By engaging different areas and proportions of the muscle and blood vessels, and with substantially different insulin levels and exposure times in each case, our current and previous (10, 11, 28) studies may test different aspects of insulin access and dispersion.
Potential methods for the contribution of fat to endothelial dysfunction and insulin resistance include inflammation, as some studies demonstrate that infused lipid upregulates Toll-like receptors (22, 23), although other studies do not support the involvement of inflammation (20) but cite a metabolic feedback mechanism, as proposed by Randle (34). Lipids have previously been shown to induce insulin resistance by inhibiting glucose transport (37) and reducing nonoxidative glucose disposal (33). Although mitochondrial function impairments are often suspected (32), and decreased mitochondrial production of ATP is detected with lipid infusion (7), we cannot conclude from our study whether mitochondrial function was affected or whether any other FFA metabolites such as diacylglycerol may be directly causing cellular insulin resistance either by directly affecting insulin-signaling cascades (46) or through mitochondrial function (7). Recent studies have suggested that mitochondrial dysfunction may be a consequence rather than a cause of insulin resistance (21) and unlikely to produce insulin resistance (5); however, the role of PKC and diacylglycerol in the reduced peripheral glucose uptake shown here should be investigated (33, 46).
Elevated lipid has effects on microvascular function that may contribute to insulin resistance. Endothelial function is thought to be a major factor in delivery of insulin to the myocyte, and FFA block insulin signaling in endothelial cells (50), which is required for vasodilation and insulin transport (49). Therefore, we used hyperlipidemia to induce both endothelial dysfunction and insulin resistance in skeletal muscle. We found that insulin access to muscle was not impaired by acute hyperlipidemia, and FFA levels are not substantially elevated in the skeletal muscle lymph in canines exposed to an infusion of lipid, even though skeletal muscle insulin resistance is detected. This is consistent with results showing a delay in accumulation of intramyocellular lipid (6, 18). Further studies are required to investigate whether other lipid metabolites may contribute to insulin resistance at the cellular level and whether lymph is an appropriate measure of the interstitial space in both health and disease. We conclude that acute lipid-induced insulin resistance does not impair the access of insulin to the skeletal muscle.
GRANTS
This research was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (DK-029867). C. M. Kolka and A. V. B. Castro were supported by the American Diabetes Association Mentor-based Fellowship.
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, to declare.
AUTHOR CONTRIBUTIONS
C.M.K. and J.M.R. conception and design of research; C.M.K., J.M.R., A.V.B.C., and J.L.B. performed experiments; C.M.K. and J.M.R. analyzed data; C.M.K., J.M.R., J.L.B., V.I., and R.N.B. interpreted results of experiments; C.M.K. prepared figures; C.M.K. drafted manuscript; C.M.K., J.M.R., A.V.B.C., J.L.B., V.I., and R.N.B. edited and revised manuscript; C.M.K., J.M.R. J.L.B, V.I., and R.N.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Rita Thomas and Young Jeng for assays and Edgardo Paredes and Edward Zuniga for experimental assistance (all from University of Southern California).
Present address of C. M. Kolka, V. Ionut, J. L. Broussard, and R. N. Bergman: Diabetes and Obesity Research Institute, Department of Biomedical Science, Cedars-Sinai Medical Center, 8700 Beverly Blvd., Los Angeles, CA 90048.
Present address of A. V. B. Castro: Centro Universitario Barao de Maua and Internal Medicine Department, Endocrinology e Metabolism, Ribeirao Preto Medical School of University of Sao Paulo, Ribeirao Preto, Sao Paulo, Brazil.
REFERENCES
- 1.Ader M, Bergman RN. Importance of transcapillary insulin transport to dynamics of insulin action after intravenous glucose. Am J Physiol Endocrinol Metab 266: E17–E25, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 100: 174–190, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Barrett EJ, Wang H, Upchurch CT, Liu Z. Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab 301: E252–E263, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, Defronzo RA, Cusi K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 54: 1640–1648, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Boden G. Fatty acids and insulin resistance. In: Diabetes Mellitus: A Fundamental and Clinical Text, edited by LeRoith D, Olefsky JM, and Taylor SI. Philadelphia, PA: Lippincott, Williams, & Wilkins, 2004, p. 979–986. [Google Scholar]
- 6.Brechtel K, Dahl DB, Machann J, Bachmann OP, Wenzel I, Maier T, Claussen CD, Haring HU, Jacob S, Schick F. Fast elevation of the intramyocellular lipid content in the presence of circulating free fatty acids and hyperinsulinemia: a dynamic 1H-MRS study. Magn Reson Med 45: 179–183, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhausl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes 55: 136–140, 2006. [PubMed] [Google Scholar]
- 8.Castillo C, Bogardus C, Bergman R, Thuillez P, Lillioja S. Interstitial insulin concentrations determine glucose uptake rates but not insulin resistance in lean and obese men. J Clin Invest 93: 10–16, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavez AO, Kamath S, Jani R, Sharma LK, Monroy A, Abdul-Ghani MA, Centonze VE, Sathyanarayana P, Coletta DK, Jenkinson CP, Bai Y, Folli F, Defronzo RA, Tripathy D. Effect of short-term free Fatty acids elevation on mitochondrial function in skeletal muscle of healthy individuals. J Clin Endocrinol Metab 95: 422–429, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu JD, Kolka CM, Richey JM, Harrison LN, Zuniga E, Kirkman EL, Bergman RN. Experimental hyperlipidemia dramatically reduces access of insulin to canine skeletal muscle. Obesity (Silver Spring) 17: 1486–1492, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu JD, Richey JM, Harrison LN, Zuniga E, Kolka CM, Kirkman EL, Ellmerer M, Bergman RN. Direct administration of insulin into skeletal muscle reveals that the transport of insulin across the capillary endothelium limits the time course of insulin to activate glucose disposal. Diabetes 57: 828–835, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Clerk LH, Rattigan S, Clark MG. Lipid infusion impairs physiologic insulin-mediated capillary recruitment and muscle glucose uptake in vivo. Diabetes 51: 1138–1145, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Clerk LH, Vincent MA, Jahn LA, Liu Z, Lindner JR, Barrett EJ. Obesity blunts insulin-mediated microvascular recruitment in human forearm muscle. Diabetes 55: 1436–1442, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103: 253–259, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eggleston EM, Jahn LA, Barrett EJ. Early microvascular recruitment modulates subsequent insulin-mediated skeletal muscle glucose metabolism during lipid infusion. Diabetes Care 36: 104–110, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 36: 914–924, 1987. [DOI] [PubMed] [Google Scholar]
- 17.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 48: 1270–1274, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi H, Nutting DF, Fujimoto K, Cardelli JA, Black D, Tso P. Transport of lipid and apolipoproteins A-I and A-IV in intestinal lymph of the rat. J Lipid Res 31: 1613–1625, 1990. [PubMed] [Google Scholar]
- 19.Herkner H, Klein N, Joukhadar C, Lackner E, Langenberger H, Frossard M, Bieglmayer C, Wagner O, Roden M, Muller M. Transcapillary insulin transfer in human skeletal muscle. Eur J Clin Invest 33: 141–146, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Hoeg LD, Sjoberg KA, Jeppesen J, Jensen TE, Frosig C, Birk JB, Bisiani B, Hiscock N, Pilegaard H, Wojtaszewski JF, Richter EA, Kiens B. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes 60: 64–73, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoeks J, van Herpen NA, Mensink M, Moonen-Kornips E, van Beurden D, Hesselink MK, Schrauwen P. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes 59: 2117–2125, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–1870, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hussey SE, Lum H, Alvarez A, Cipriani Y, Garduno-Garcia J, Anaya L, Dube J, Musi N. A sustained increase in plasma NEFA upregulates the Toll-like receptor network in human muscle. Diabetologia 57: 582–591, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huxley VH, Scallan J. Lymphatic fluid: exchange mechanisms and regulation. J Physiol 589: 2935–2943, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikomi F, Hunt J, Hanna G, Schmid-Schonbein GW. Interstitial fluid, plasma protein, colloid, and leukocyte uptake into initial lymphatics. J Appl Physiol (1985) 81: 2060–2067, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Jensen SM, Hansen HS, Johansen T, Malmlof K. In vivo and in vitro microdialysis sampling of free fatty acids. J Pharm Biomed Anal 43: 1751–1756, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Kolka CM, Bergman RN. The barrier within: endothelial transport of hormones. Physiology (Bethesda) 27: 237–247, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolka CM, Harrison LN, Lottati M, Chiu JD, Kirkman EL, Bergman RN. Diet-induced obesity prevents interstitial dispersion of insulin in skeletal muscle. Diabetes 59: 619–626, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krssak M, Falk PK, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42: 113–116, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, Kumagai K, Kawai T, Hashimoto S, Kobayashi T, Sato M, Tokuyama K, Nishimura S, Tsunoda M, Ide T, Murakami K, Yamazaki T, Ezaki O, Kawamura K, Masuda H, Moroi M, Sugi K, Oike Y, Shimokawa H, Yanagihara N, Tsutsui M, Terauchi Y, Tobe K, Nagai R, Kamata K, Inoue K, Kodama T, Ueki K, Kadowaki T. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 13: 294–307, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest 85: 1844–1852, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 307: 384–387, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Nowotny B, Zahiragic L, Krog D, Nowotny PJ, Herder C, Carstensen M, Yoshimura T, Szendroedi J, Phielix E, Schadewaldt P, Schloot NC, Shulman GI, Roden M. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes 62: 2240–2248, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14: 263–283, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Rattigan S, Richards SM, Keske MA. Microvascular contributions to insulin resistance. Diabetes 62: 343–345, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rattigan S, Zhang L, Mahajan H, Kolka CM, Richards SM, Clark MG. Factors influencing the hemodynamic and metabolic effects of insulin in muscle. Curr Diabetes Rev 2: 61–70, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97: 2859–2865, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandqvist M, Strindberg L, Schmelz M, Lonnroth P, Jansson PA. Impaired delivery of insulin to adipose tissue and skeletal muscle in obese women with postprandial hyperglycemia. J Clin Endocrinol Metab 96: E1320–E1324, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Schmitz-Peiffer C. Signalling aspects of insulin resistance in skeletal muscle: mechanisms induced by lipid oversupply. Cell Signal 12: 583–594, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Sherman AI, Ter-Pogossian M. Lymph-node concentration of radioactive colloidal gold following interstitial injection. Cancer 6: 1238–1240, 1953. [DOI] [PubMed] [Google Scholar]
- 41.Sjöstrand M, Gudbjörnsdottir S, Holmäng A, Lönn L, Strindberg L, Lönnroth P. Delayed transcapillary transport of insulin to muscle interstitial fluid in obese subjects. Diabetes 51: 2742–2748, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Sjöstrand M, Holmäng A, Lönnroth P. Measurement of interstitial insulin in human muscle. Am J Physiol Endocrinol Metab 276: E151–E154, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Skalak TC, Schmid-Schonbein GW, Zweifach BW. New morphological evidence for a mechanism of lymph formation in skeletal muscle. Microvasc Res 28: 95–112, 1984. [DOI] [PubMed] [Google Scholar]
- 44.Sloop CH, Dory L, Roheim PS. Interstitial fluid lipoproteins. J Lipid Res 28: 225–237, 1987. [PubMed] [Google Scholar]
- 45.Szendroedi J, Frossard M, Klein N, Bieglmayer C, Wagner O, Pacini G, Decker J, Nowotny P, Müller M, Roden M. Lipid-induced insulin resistance is not mediated by impaired transcapillary transport of insulin and glucose in humans. Diabetes 61: 3176–3180, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, Jelenik T, Müller J, Herder C, Nowotny P, Shulman GI, Roden M. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci USA 111: 9597–9602, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utsunomiya K. Treatment strategy for type 2 diabetes from the perspective of systemic vascular protection and insulin resistance. Vasc Health Risk Manag 8: 429–436, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S, Barrett EJ. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53: 1418–1423, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Liu Z, Li G, Barrett EJ. The vascular endothelial cell mediates insulin transport into skeletal muscle. Am J Physiol Endocrinol Metab 291: E323–E332, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, Coselli JS, Shen YH. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 55: 2301–2310, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev 92: 1005–1060, 2012. [DOI] [PubMed] [Google Scholar]
- 52.Zawieja SD, Wang W, Wu X, Nepiyushchikh ZV, Zawieja DC, Muthuchamy M. Impairments in the intrinsic contractility of mesenteric collecting lymphatics in a rat model of metabolic syndrome. Am J Physiol Heart Circ Physiol 302: H643–H653, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]