

Abstract
The present study examined the heme oxygenase (HO) system in an in vivo murine model of pathological shear stress induced by partial carotid artery ligation. In this model, along with upregulation of vasculopathic genes, HO-1 is induced in the endothelium and adventitia, whereas HO-2 is mainly upregulated in the endothelium. Within minutes of ligation, NF-κB, a transcription factor that upregulates vasculopathic genes and HO-1, is activated. Failure to express either HO-1 or HO-2 exaggerates the reduction in carotid blood flow and exacerbates vascular injury. After artery ligation, comparable induction of HO-2 occurred in HO-1+/+ and HO-1−/− mice, whereas HO-1 induction was exaggerated in HO-2−/− mice compared with HO-2+/+ mice. Upregulation of HO-1 by an adeno-associated viral vector increased vascular HO-1 expression and HO activity and augmented blood flow in both ligated and contralateral carotid arteries. Acute inhibition of HO activity decreased flow in the ligated carotid artery, whereas a product of HO, carbon monoxide (CO), delivered by CO-releasing molecule-3, increased carotid blood flow. In conclusion, in the partial carotid artery ligation model of pathological shear stress, this study provides the first demonstration of 1) upregulation and vasoprotective effects of HO-1 and HO-2 and the vasorelaxant effects of CO as well as 2) vascular upregulation of HO-1 in vivo by an adeno-associated viral vector that is attended by a salutary vascular response. Induction of HO-1 may reside in NF-κB activation, and, along with induced HO-2, such upregulation of HO-1 provides a countervailing vasoprotective response in pathological shear stress in vivo.
Keywords: vascular injury, heme oxygenase, shear stress, inflammation, cytokines
insults to the vasculature instigate an interplay of vasculopathic and vasoprotective processes. Exploring the basis for such vasoprotective responses is clinically relevant because of the possibility of uncovering novel therapeutic strategies for cardiovascular diseases (35). In this regard, substantial interest centers on heme oxygenase (HO)-1, the inducible isoform of HO, as the induction of HO-1 often confers salutary effects in models of tissue injury (1, 41). HO degrades heme, generates the gaseous product carbon monoxide (CO), produces the bile pigment biliverdin, and releases iron from the tetrapyrrole heme ring. While the induction of HO-1 is often viewed as a cytoprotective response, all products resulting from HO-based degradation of heme are potentially toxic, and, indeed, injurious effects have been ascribed to induced HO-1 (23, 27, 50). The other HO isoform, constitutively expressed HO-2, is also of interest in tissue injury since, depending on the experimental setting, protective or injurious effects may emanate from HO-2 (30, 42, 53).
The present study examined whether the HO system is induced in the vasculature subjected to pathologic forms of shear stress. Low laminar and oscillatory shear stress are the main types of pathologic shear stress, and both are increasingly implicated in the initiation and progression of vascular disease because of their proinflammatory, proliferative, and other effects on the vessel wall (6, 7, 14). This study examined whether such alterations in shear stress upregulate HO-1 and HO-2, and the functional significance of any such induction.
To address this question, we used a model of vascular injury induced by selective ligation of the major branches of the common carotid artery, the partial carotid artery ligation (PCAL) model (32, 40). In this model, blood flow in the main trunk of the carotid artery diminishes and is accompanied by vascular remodeling and an attendant increase in intimal-media thickness (32, 40). Increased intimal-media thickness of the carotid artery in humans presages the risk for cardiovascular disease, both in general and in specific vascular beds such as the coronary artery, and has been used as an index for such risk and disease in clinical trials (46, 51).
The specific choice of the PCAL model in this study resided, however, not just in its clinical relevance but also because this model induces low laminar and oscillatory shear stress in the main trunk of the carotid artery, as definitively documented in a previous study (40). While pathological shear stress is presumed to exist in models of vascular injury, in very few studies has such shear stress been demonstrated or confirmed. Because of prior documentation of the presence of low laminar and oscillatory shear stress in the PCAL model, this clinically relevant in vivo model was thus used to examine the behavior of the HO system in response to pathological shear stress.
MATERIALS AND METHODS
Experiments using the PCAL model in mice.
All experiments were approved by the Institutional Animal Care and Use Committee of Mayo Clinic and were performed in accordance with National Institutes of Health guidelines. These experiments primarily used 10- to 12-wk-old male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME). These experiments also used age- and sex-matched HO-1−/− mice, HO-2−/− mice, and their respective control mice generated from relevant colonies maintained as we have previously described (20, 30, 44). HO-1+/+ and HO-1−/− mice were in the age range of 20 to 35 wk, and HO-2+/+ and HO-2−/− mice were in the age range of 10 to 50 wk.
The PCAL model was performed as previously described (32, 40). Briefly, after mice were anesthetized [pentobarbital (60 mg/kg ip)], three of the four branches (external carotid artery, internal carotid artery, and occipital artery) of the left carotid artery (LCA) were ligated using 6-0 silk suture, leaving flow through the superior thyroid artery. Sham operation for control mice included anesthesia, ventral neck incision, and exposure of the LCA. Buprenorphine (0.1 mg/kg sc) was administered for these procedures as an analgesic. At time points ranging from 15 min to 2 wk after PCAL or sham surgery, the LCA and right carotid artery (RCA) were harvested for assessment of transcription factor activation, analysis of gene and protein expression, or histological/immunohistochemical experiments. Two weeks after PCAL or sham surgery, blood flow through the carotid arteries was measured using a perivascular flow probe (catalog no. 0.5 PSB, Transonic Systems, Ithaca, NY), as described in our previous study (31).
For some experiments in C57BL/6J mice, HO-1 expression was upregulated using adeno-associated viral (AAV) vectors, whereas in other experiments, the HO product CO was delivered via a CO emitting compound [CO-releasing molecule (CORM)-3, tricarbonylchloro[glycinato]ruthenium [II], Sigma-Aldrich, St. Louis, MO]. AAV9(HO-1) vector (1 × 1012 vg/mouse) or PBS was administered intravenously, and, 3 wk later, PCAL or sham surgery was performed. After 2 additional weeks, blood flow was measured. CORM-3 was administered to mice via intraperitoneal injections (40 mg/kg) (12, 60), with doses given 1 day before, the day of, and 1 day after PCAL surgery. Control mice received equivalent doses of inactivated (i)CORM-3, which was prepared by allowing CO to evolve from a solution of CORM-3 at room temperature for 24 h; nitrogen gas was bubbled through the solution for 10 min to remove residual CO. One day after PCAL surgery, blood flow through the carotid arteries was assessed 45–60 min after the final dose of CORM-3 or iCORM-3 was administered.
In additional experiments in C57BL/6J mice, the effect of inhibition of HO activity using tin protoporphyrin (SnPP; catalog no. Sn749-9, Frontier Scientific, Logan, UT) was assessed (29). At 13 days after PCAL surgery, mice were administered SnPP (50 μmol/kg ip) or saline vehicle. The next day, mice were given a second dose of SnPP or saline, and blood flow through the carotid arteries was measured 45–60 min later using a perivascular flow probe as described above.
AAV9(HO-1) vector construction and production.
The self-complementary HO-1 transgene used in these experiments was made by subcloning HO-1 from pcDNA3.1/HO-1(55) into pTRS-KS/CBh-GFP (NGVB) (21) in the place of GFP. AAV vectors were produced in 293T cells as previously described (24). Briefly, a triple transfection of AAV and helper plasmids was performed using 25-kDa linear polyethylenimine. AAV vectors were purified by iodixanol density gradient ultracentrifugation followed by AAV vector concentration, dialysis, filtration, and storage at −80°C. DNaseI-resistant vector genomes were titered with a custom Taqman primer/probe set (Applied Biosystems, Foster City, CA) specific to the bovine growth hormone polyadenylation signal found in the AAV9(HO-1) vector transgene.
Assessment of mRNA expression by quantitative real-time RT-PCR.
Gene expression in carotid arteries was assessed by quantitative real-time RT-PCR as previously described (49). Total RNA was extracted from carotid arteries using the TRIzol method (Invitrogen, Carlsbad, CA) and further purified with an RNeasy Mini kit (Qiagen, Valencia, CA) according to each manufacturer's protocol. Purified RNA was subsequently used in reverse transcription reactions (Transcriptor First Strand cDNA Synthesis kit, Roche Applied Science, Indianapolis, IN) using random hexamers. The resulting cDNA was used for quantitative real-time PCR analysis performed on an ABI Prism 7900HT (Applied Biosystems). FastStart Universal Probe Master mastermix reagent (catalog no. 04914058001, Roche Applied Science) was used for these reactions using probes and primers obtained as assay sets (TaqMan Gene Expression Assays, Applied Biosystems). Parameters for quantitative PCR were as follows: 10 min at 95°C followed by 40 cycles of amplification for 15 s at 95°C and 1 min at 60°C. Expression of 18S rRNA was used for normalization of the expression of each target gene.
Western blot analysis.
Western blot analysis for HO-1 and HO-2 protein expression in carotid arteries was performed as previously described (43, 49). Two carotid arteries per sample were combined and homogenized in RIPA buffer consisting of 10 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1% Nonindet P-40 substitute, 0.5% sodium deoxycholate, 0.1% SDS, and protease/phosphatase inhibitors (catalog no. 1861281, Thermo Scientific, Waltham, MA). After quantitation by the Lowry method, 10 μg protein aliquots were separated on 12.5% Tris·HCl gels and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were block in 5% nonfat milk for 1 h and incubated overnight with rabbit polyclonal primary antibody for HO-1 or HO-2 (catalog nos. ADI-SPA-895 and ADI-SPA-897, respectively, Enzo Life Sciences, Farmingdale, NY). The next day, horseradish peroxidase-conjugated goat anti-rabbit polyclonal secondary antibody (Bio-Rad Laboratories) was used in a 1-h room temperature incubation followed by detection using chemiluminescence methods (GE Healthcare, Buckinghamshire, UK). Equivalency of protein loading was assessed by immunoblot analysis for β-actin using rabbit polyclonal antibody (catalog no. 612656, BD Biosciences, San Jose, CA). Expression of p65, phospho-p65, and IκBα was analyzed in nuclear and cytosolic protein fractions prepared as described below. Rabbit monoclonal antibody against p65 (catalog no. 8242, Cell Signaling Technology, Danvers, MA) and rabbit polyclonal antibodies against phospho-p65 and IκBα (catalog nos. 3031 and 9242, respectively, Cell Signaling Technology) were used as primary antibodies with goat polyclonal secondary antibody.
Preparation of nuclear/cytoplasmic fractions from mouse carotid arteries.
Nuclear and cytoplasmic protein fractions were prepared from mouse carotid arteries for assessment of activation of NF-κB by Western blot analysis of total and phosphorylated p65 subunits as well as IκBα. Fractionation was performed using a nuclear extraction kit (catalog no. 40010, Active Motif, Carlsbad, CA). For each sample to be fractioned, three to four sham-operated or three to four PCAL carotid arteries were combined and processed according to the manufacturer's protocol with the substitution of RIPA buffer [10 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1% Nonindet P-40 substitute, 0.5% sodium deoxycholate, 1% SDS, and protease/phosphatase inhibitors] for the kit's complete nuclear lysis buffer. Cytosolic fractions derived with this kit method were also diluted in RIPA buffer. Nuclear and cytosolic protein aliquots were then analyzed by Western blot analysis as described above.
Immunohistochemical localization of HO-1 and HO-2 expression.
Immunohistochemical analysis of HO-1 and HO-2 expression was performed as previously described (31) on 5-μm sections prepared from formalin-fixed paraffin-embedded carotid arteries from mice subjected to either sham or PCAL surgery. Briefly, immunohistochemical staining was carried out using a Bond III automated immunohistochemical staining system (Leica Microsystems, Buffalo Grove, IL) by the Mayo Pathology Research Core using the following parameters. After peroxidase quenching (catalog no. PX968M, Biocare Medical, Concord, CA) for 5 min, antigen retrieval in 10 mM EDTA (pH 9, 10 min, 95°C) was performed, and sections were blocked for 15 min with Rodent Block M (catalog no. RBM961, Biocare Medical). Sections were then incubated with primary antibodies for HO-1 and HO-2 (as described above for Western blot analysis, 1:200 for HO-1 and 1:100 for HO-2) for 60 min at room temperature followed by a 15-min incubation in anti-rabbit secondary antibody conjugated with a horseradish peroxidase-labeled polymer (EnVision+ System-HRP, catalog no. 4010, Dako, Carpinteria, CA). Staining was visualized with diaminobenzidine substrate and counterstained with hematoxylin.
HO activity in mouse aortas.
HO activity in mouse aortas was determined as described in our previous study (45) in which the rate of generation of bilirubin from hemin was measured. Briefly, aortas were homogenized in 0.2 ml of 0.1 M potassium phosphate (pH 7.4) containing 2 mM MgCl2, and homogenates were centrifuged at 12,000 g for 15 min. Aliquots (200 μg) of supernatant protein were incubated in 0.2-ml reaction mixtures containing 0.1 M potassium phosphate (pH 7.4), mouse liver cytosol (2 mg cytosolic protein), hemin (20 μM), glucose-6-phosphate (2 mM), glucose-6-phosphate-dehydrogenase (0.2 units), and NADPH (0.8 mM) for 90 min at 37°C in the dark. A control reaction omitting NADPH, glucose-6-phosphate, and glucose-6-phoshate-dehydrogense was also performed for each lysate. Bilirubin was extracted from each reaction with 0.2 ml chloroform and quantitated by the measurement of optical density at 464–530 nm. Using the extinction coefficient for bilirubin (40 mM−1·cm−1), HO activity was calculated and expressed as picomoles of bilirubin formed per 60 minutes per milligram of protein.
Serum bilirubin.
Measurement of serum bilirubin concentration was performed using a commercially available kit (catalog no. MAK126, Sigma-Aldrich), which was based on the Jendrassik-Grof method.
Statistical analysis.
Results are expressed as means ± SE and were considered statistically significant for P < 0.05. Student's t-test was used for parametric data, and the Mann-Whitney U-test was used for nonparametric data. Vascular patency in HO-1+/+ and HO-1−/− mice as well as in HO-2+/+ and HO-2−/− mice after PCAL was assessed using Fisher's exact test.
RESULTS
Characterization of the PCAL model.
Two weeks after the creation of the PCAL model, histological and functional parameters were assessed. As previously described, this model leads to mild thickening of the vessel wall along with the reduplication and splitting of the internal elastic lamina (data not shown). Two weeks after the creation of this model, blood flow in the LCA was markedly decreased compared with the sham-operated group (Fig. 1); in contrast, blood flow in the contralateral RCA of animals subjected to partial ligation of the LCA was increased compared with the sham-operated group. Additional evidence that PCAL is a valid model for vascular injury was provided by the expression of vasculopathic candidate genes. Genes such as monocyte chemoattractant protein (MCP)-1, IL-6, chemokine (C-C motif) ligand (CCL)5, and TNF-α are well recognized as vasculopathic but have not been evaluated, to date, in this model. These genes were significantly induced after PCAL (Fig. 2), along with other genes relevant to vascular injury (Table 1).
Fig. 1.

Blood flow assessment in carotid arteries in mice 2 wk after partial carotid artery ligation (PCAL) or sham operation (sham). Blood flow was measured in the left carotid artery (LCA; ipsilateral) and right carotid artery (RCA; contralateral) using a perivascular flow probe; n = 11 in each group. *P < 0.05 vs. sham LCA; †P < 0.05 vs. sham RCA.
Fig. 2.

Gene expression in carotid arteries of sham mice and mice subjected to PCAL. mRNA expression of monocyte chemoattractant protein (MCP)-1 (A), IL-6 (B), chemokine (C-C motif) ligand (CCL)5 (C), and TNF-α (D) was assessed by quantitative real-time RT-PCR in LCAs 1 wk after PCAL or sham surgery; n = 9 and 10 in sham and PCAL groups, respectively. *P < 0.05 vs. sham.
Table 1.
mRNA expression in carotid arteries of sham and PCAL mice at 1 week after PCAL
| Sham Mice | PCAL Mice | Fold Change | P Value | |
|---|---|---|---|---|
| Transforming growth factor-β1 | 2.6 ± 0.2 | 7.7 ± 3.1 | 3.0 | 0.0021 |
| Collagen type I | 7.2 ± 0.8 | 18.7 ± 5.2 | 2.6 | 0.028 |
| Collagen type III | 2.1 ± 0.3 | 5.8 ± 1.6 | 2.8 | 0.0057 |
| Collagen type IV | 6.7 ± 0.7 | 9.1 ± 1.4 | 1.4 | NS |
| Cyclooxygenase 1 | 9.6 ± 0.5 | 7.6 ± 1.1 | ↓0.8 | 0.017 |
| Cyclooxygenase 2 | 4.0 ± 0.9 | 9.3 ± 3.3 | 2.3 | NS |
| PDGF-A | 4.6 ± 0.4 | 6.4 ± 0.5 | 1.4 | 0.016 |
| PDGF-B | 6.7 ± 0.5 | 11.1 ± 2.4 | 1.7 | 0.0101 |
| Endothelial nitric oxide synthase | 7.1 ± 0.4 | 5.0 ± 0.7 | ↓0.7 | 0.0152 |
| Inducible nitric oxide synthase | 2.7 ± 0.6 | 5.2 ± 1.4 | 1.9 | NS |
Data are expressed as means ± SE; n = 9 and 10 for sham-operated (sham) and partial carotid artery ligation (PCAL) mice, respectively. mRNA expression was assessed by quantitative real-time RT-PCR. The above values are the result of relative quantification performed against a standard curve constructed for each mRNA target, normalized for expression of 18S rRNA, and expressed in arbitrary units.
NS, not significant.
Induction of HO-1 after PCAL.
Because induction of vasculopathic genes is often accompanied by countervailing responses, we examined one such possible response, namely, expression of HO-1. One week after PCAL, HO-1 mRNA and protein were both markedly induced, and, indeed, such induction of HO-1 protein was detectable as early as 12 h after PCAL (Fig. 3, A and B).
Fig. 3.

Heme oxygenase (HO)-1 expression in carotid arteries of sham mice and mice subjected to partial ligation of the LCA (PCAL). A: HO-1 mRNA expression 1 wk after PCAL or sham surgery as assessed by quantitative real-time RT-PCR; n = 9 and 10 for sham and PCAL groups, respectively. *P < 0.05 vs. sham. B: Western blot analysis of HO-1 protein expression 1 wk and 12 h after PCAL and sham surgery. Each lane represents a sample obtained from two carotid arteries with equivalency of loading assessed by immunoblot analysis for β-actin.
Immunohistochemical experiments were also performed to localize such HO-1 expression; very weak expression of HO-1 was observed in the endothelium in sham-operated arteries, whereas in the PCAL model, HO-1 was prominently expressed in the endothelium, adventitial cells, and, to a lesser extent, smooth muscle cells (Fig. 4, A–D).
Fig. 4.
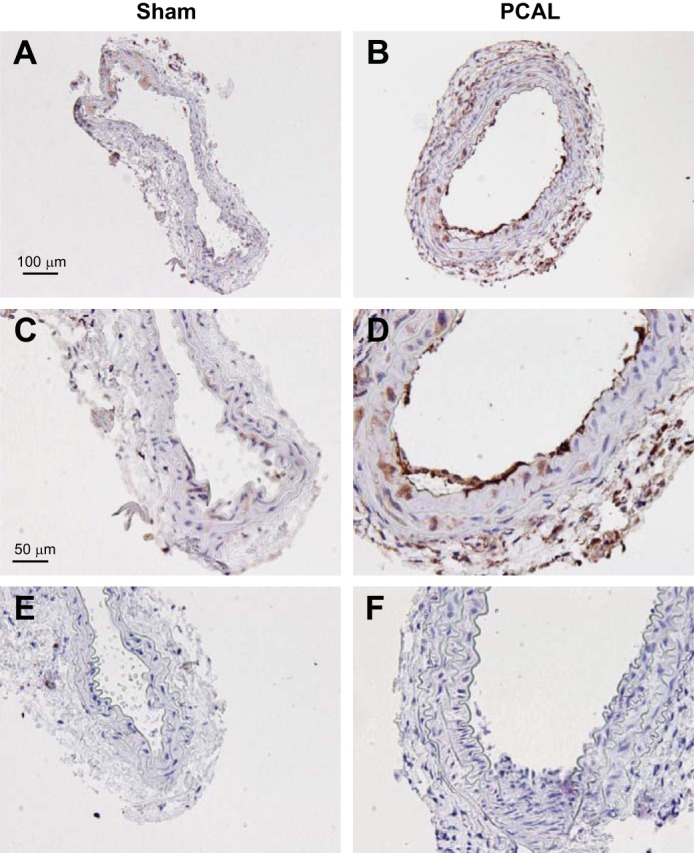
Immunohistochemical analysis of HO-1 protein expression in LCAs of mice 2 wk after sham operation or partial ligation of the LCA (PCAL). A and C: low-power (A) and high-power (C) views of HO-1 expression in a carotid artery of a mouse after sham surgery. B and D: low-power (B) and high-power (D) views of HO-1 expression in a carotid artery of a mouse after PCAL surgery. E and F: isotype control staining for sham (E) and PCAL (F) groups. Scale bar = 100 μm for A and B and 50 μm for C–F.
Because HO-1 gene expression is regulated by NF-κB, we examined whether NF-κB activation occurred at an early time point in this model. In this regard, using Western blot analysis of cellular extracts of murine carotid arteries, we examined the expression of p65 and IkBα protein in cytosolic fractions and the expression of phospho-p65 protein in nuclear fractions. Because of the low yield, carotid arteries from three mice subjected to PCAL were pooled in each lane shown in Fig. 5A and four arteries were pooled in each lane shown in Fig. 5C. As shown in Fig. 5, expression of p65 and IkBα was diminished in the cytosol, and this was accompanied by increased expression of phospho-p65, the active form of p65, in the nuclear fraction 15 min after the creation of the PCAL model.
Fig. 5.

Assessment of NF-κB activation in the PCAL model. A: Western blot analysis of cytosolic expression of IκBα and total p65 in LCAs of mice 15 min after sham or PCAL surgery. Each lane represents cytosolic protein obtained from three pooled carotid arteries. B: densitometric assessment for expression of IκBα (left) and p65 (right) by Western blot analysis shown in A; n = 5 in each group. *P < 0.05 vs. sham. C: Western blot analysis of nuclear expression of phosphorylated (p-)p65 in LCAs of mice 15 min after sham or PCAL surgery. Shown are results of two separate experiments with each lane representing nuclear protein obtained from four pooled carotid arteries. D: densitometric assessment of p-p65 Western blot analysis shown in C; n = 4 in each group. *P < 0.05 vs. sham.
PCAL in HO-1+/+ and HO-1−/− mice.
To determine the functional significance of the induction of HO-1 in this model, we performed PCAL in HO-1+/+ and HO-1−/− mice. HO-1−/− mice compared with HO-1+/+ mice, subjected to this model, exhibited even more marked reduction in carotid artery blood flow and an increased number of arteries with complete loss of patency (Fig. 6, A and B). Histological examination showed thickening of the arterial wall in HO-1−/− mice compared with HO-1+/+ mice after PCAL as well as the presence of intravascular thrombus formation in some vessels (Fig. 6C).
Fig. 6.

Assessment of the partial ligation of the LCA (PCAL) model in HO-1-deficient (HO-1−/−) mice. A: blood flow in the LCA (ipsilateral) and RCA (contralateral) in HO-1−/− and wild-type (HO-1+/+) mice 2 wk after PCAL surgery. Blood flow was measured using a perivascular flow probe. B: patency rates of carotid arteries 2 wk after PCAL surgery in HO-1−/− and HO-1+/+ mice. n = 9 and 8 in HO-1+/+ and HO-1−/− groups, respectively, for both analyses. *P < 0.05 vs. HO-1+/+ LCA. C: histological examination of PCAL and contralateral (control) carotid arteries in HO-1+/+ and HO-1−/− mice 2 wk after PCAL surgery. Representative views of contralateral arteries from HO-1+/+ mice (i) and HO-1−/− mice (ii) and of PCAL in HO-1+/+ mice (iii) and HO-1−/− mice (iv) are shown. All sections were stained with hematoxylin and eosin and displayed at an original magnification of ×100. Scale bar = 100 μm.
Effect of increased HO-1 expression mediated by AAV9 on carotid artery blood flow after PCAL.
To determine whether increased expression of HO-1 could exert functional effects in this model, we delivered HO-1 cDNA using an AAV9 vector by intravenous injection. Immunohistochemistry demonstrated upregulation of HO-1 in the endothelium and smooth muscle cells in the carotid artery when studied 5 wk after the administration of AAV9(HO-1) (Fig. 7, A–D). Such administration markedly upregulated expression of HO-1 protein in the carotid artery (Fig. 7, E and F) and increased vascular (aorta) HO activity, the aorta providing adequate amounts of the vasculature so as to undertake such measurements of HO activity (Fig. 7G). Administration of AAV9(HO-1) led to the appearance of increased amounts of full-length (32 kDa) HO-1 protein in plasma (Fig. 7H). We also measured total bilirubin levels in serum after the administration of AAV9(HO-1), as elevated serum bilirubin levels often occur in concert with, and reflect, induction of HO-1 in tissues (13, 26). After the administration of AAV9(HO-1), serum levels of bilirubin were significantly increased (3.7 ± 0.4 vs 2.3 ± 0.2 μM, n = 7 and 6 respectively, P < 0.05).
Fig. 7.

Analysis of HO-1 expression and aortic HO activity after the administration of adeno-associated viral (AAV)9(HO-1). A and B: HO-1 protein expression in carotid arteries assessed by immunohistochemistry in PBS-treated (A) and AAV9(HO-1)-treated (B) mice 5 wk after injection. C and D: isotype control staining for PBS-treated (C) and AAV9(HO-1)-treated (D) mice. All sections are displayed at an original magnification of ×400. Scale bar = 100 μm. E: Western blot analysis of HO-1 protein expression in carotid arteries of PBS-treated and AAV9(HO-1)-treated mice 3 wk after injection. F: densitometric assessment of Western blot analysis shown in E; n = 5 in each group. *P < 0.05 vs. PBS. G: HO activity measured in thoracic aortas of PBS-treated and AAV9(HO-1)-treated mice 3 wk after injection; n = 5 in each group. *P < 0.05 vs. PBS. H: Western blot analysis of HO-1 protein expression in plasma of AAV9(HO-1)-treated and PBS-treated mice 3 wk after injection.
This upregulation of HO-1 by AAV9(HO-1) exerted a protective effect in the PCAL model, as demonstrated by greater blood flow in the ipsilateral, partially ligated LCA (Fig. 8). Interestingly, prior upregulation of HO-1 by AAV9 led to significantly increased carotid artery blood flow not only on the ligated side but also in the intact contralateral RCA (Fig. 8).
Fig. 8.
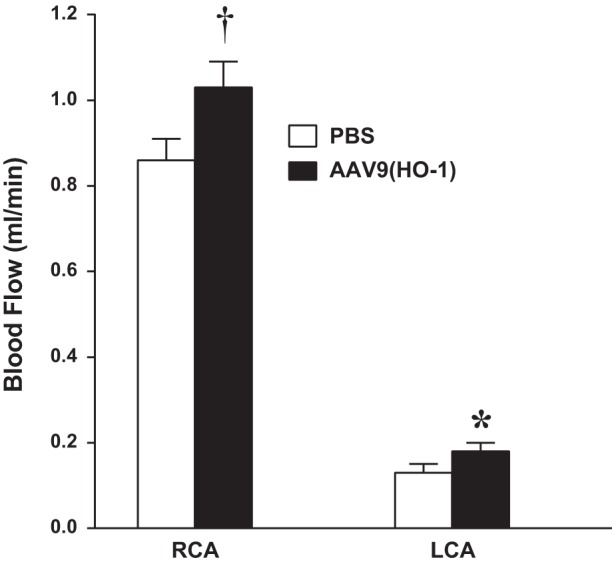
Effect of the administration of AAV9(HO-1) on blood flow in the PCAL model. Blood flow was measured in the LCA (ipsilateral) and RCA (contralateral) in PBS-treated and AAV9(HO-1)-treated mice 2 wk after PCAL surgery. Blood flow was measured using a perivascular flow probe; n = 12 and 10 in PBS-treated and AAV9(HO-1)-treated groups, respectively. *P < 0.05 vs. PBS-treated LCA; †P < 0.05 vs. PBS-treated RCA.
Effect of CO on carotid artery blood flow after PCAL.
Because carotid artery blood flow after PCAL was decreased in the absence of HO-1 and increased after the upregulation of HO-1 by AAV9, experiments were undertaken to determine if a product of HO activity, namely, CO, could improve blood flow. As shown in Fig. 9, the administration of CORM-3, an effective delivery system for CO, increased carotid artery blood flow when measured 24 h after PCAL.
Fig. 9.
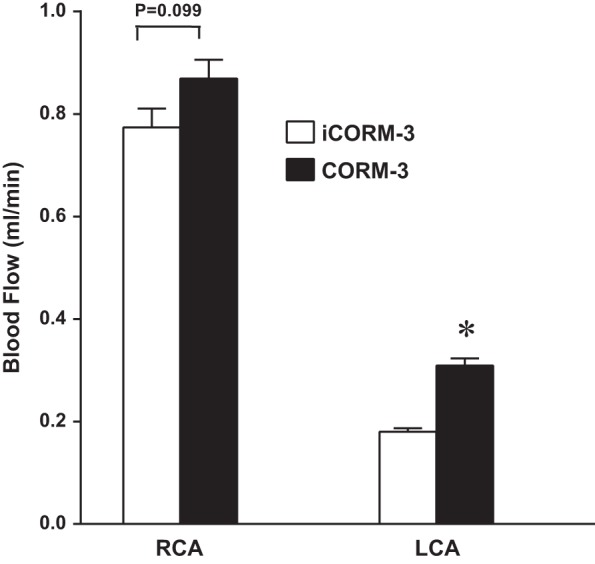
Effect of carbon monoxide-releasing molecule (CORM)-3 treatment on blood flow in the PCAL model. Blood flow in the LCA (ipsilateral) and RCA (contralateral) was measured in inactivated (i)CORM-3-treated and CORM-3-treated mice 2 wk after partial ligation of the LCA. Flow measurements were made using a perivascular flow probe; n = 7 for each group. *P < 0.05 vs. iCORM-3-treated LCA.
PCAL in HO-2+/+ and HO-2−/− mice.
Constitutively expressed HO-2 contributes to basal production of CO in the vasculature and other tissues. We thus assessed the expression and functional significance of this HO isoform in the PCAL model. After PCAL, HO-2 mRNA and protein expression were both increased in the carotid artery (Fig. 10, A–C), and immunohistochemical experiments demonstrated that such upregulation of HO-2 in the PCAL model essentially occurred in the endothelium (Fig. 10, D–G). In experiments undertaken in HO-2+/+ and HO-2−/− mice, carotid artery blood flow was significantly decreased in HO-2−/− mice compared with HO-2+/+ mice on the ipsilateral side after PCAL (Fig. 11A); blood flow in the contralateral carotid artery was not significantly different in HO-2+/+ and HO-2−/− mice (Fig. 11A). There was no significant difference in patency rates of the carotid artery after PCAL in HO-2+/+ and HO-2−/− mice (100 vs. 78%, P = not significant). Histological analyses demonstrate focal neointimal hyperplasia in three of nine carotid arteries in HO-2−/− mice subjected to PCAL (Fig. 11B), whereas such lesions were not seen in any of the carotid arteries from HO-2+/+ mice subjected to PCAL. Thus, both HO isoforms, HO-1 and HO-2, exert vasoprotective effects in the PCAL model.
Fig. 10.

HO-2 expression in mice in the PCAL model. A: Western blot analysis of HO-2 protein expression 1 wk after PCAL and sham surgery. Each lane represents a sample obtained from two carotid arteries with equivalency of loading assessed by immunoblot analysis for β-actin. B: densitometric assessment of HO-2 Western blot analysis shown in A; n = 5 in each group. *P < 0.05 vs. sham. C: HO-2 mRNA expression in LCAs 1 wk after PCAL or sham surgery as assessed by quantitative real-time RT-PCR; n = 9 and 10 for sham and PCAL groups, respectively. *P < 0.05 vs. sham. D–G: immunohistochemical analysis of HO-2 protein expression in the RCA (contralateral; D) and LCA (ligated, PCAL; E) of wild-type mice 2 wk after partial ligation of the LCA. F and G: isotype control staining for contralateral (F) and PCAL (G) carotid arteries. Scale bar = 50 μm.
Fig. 11.

Assessment of the partial ligation of the LCA (PCAL) model in HO-2-deficient (HO-2−/−) mice. A: blood flow in the LCA (ipsilateral) and RCA (contralateral) in HO-2−/− and wild-type (HO-2+/+) mice 2 wk after PCAL surgery. Blood flow was measured using a perivascular flow probe; n = 9 for each group. *P < 0.05 vs. HO-2+/+ LCA. B: histological examination of PCAL and contralateral (control) carotid arteries in HO-2+/+ and HO-2−/− mice 2 wk after PCAL surgery, demonstrating focal neointimal hyperplasia in HO-2−/− mice. Shown are sections of contralateral arteries from HO-2+/+ mice (i) and HO-2−/− mice (ii) and of PCAL in HO-2+/+ mice (iii) and HO-2−/− mice (iv). All sections were stained with hematoxylin and eosin and displayed at an original magnification of ×100. Scale bar = 100 μm.
Expression of HO-1 in HO-2+/+ and HO-2−/− mice and expression of HO-2 in HO-1+/+ and HO-1−/− mice after PCAL.
The mutant murine models used in this study involved global knockout of the specific HO gene. Such specific deficiency may influence the nature of the response in the expression of the other HO gene after PCAL. We thus determined whether the deficiency of one isoform influenced the degree of expression of the other isoform after PCAL. As shown in Fig. 12, the induction of HO-1 mRNA in the carotid artery was exaggerated in HO-2−/− mice compared with HO-2+/+ mice after PCAL. Thus, the exacerbation of vascular injury when HO-2 is deficient occurred despite greater induction of HO-1 in HO-2−/− mice subjected to PCAL. The results shown in Fig. 12 also demonstrate that HO-2 mRNA was comparably induced in HO-1+/+ and HO-1−/− mice subjected to PCAL. Thus, the exacerbation of vascular injury after PCAL in HO-1−/− mice cannot be ascribed to less induction of HO-2.
Fig. 12.

Expression of HO-1 in HO-2+/+ and HO-2−/− mice and expression of HO-2 in HO-1+/+ and HO-1−/− mice after PCAL. A: HO-1 mRNA expression in the LCA (ligated, PCAL) and RCA (contralateral) carotid artery in HO-2+/+ and HO-2−/− mice 1 wk after PCAL surgery; n = 9 in each group. *P < 0.05 vs. HO-2+/+ PCAL; †P < 0.05 vs. HO-2+/+ RCA. B: HO-2 mRNA expression in the LCA (ligated, PCAL) and RCA (contralateral) in HO-1+/+ and HO-1−/− mice 1 wk after PCAL surgery; n = 10 in each group.
Effect of acute inhibition of HO activity on carotid blood flow after PCAL.
The beneficial effects of each isoform of HO may arise from vasorelaxant effects of HO activity emanating from that isoform, effects on vascular remodeling, or a combination of both. To determine the extent to which HO activity exerts vasorelaxant effects in the PCAL model, we examined carotid blood flow after the acute inhibition of HO activity by the competitive inhibitor SnPP; such an inhibitor effectively blocks activity arising from both HO-1 and HO-2. Twenty-four hours after such inhibition (compared with vehicle-treated mice), carotid blood flow was significantly reduced on the ipsilateral side in which PCAL was performed (0.11 ± 0.01 vs. 0.07 ± 0.01 ml/min, n = 4 and 5, respectively, P < 0.05) but was not significantly altered on the contralateral, intact carotid artery (0.91 ± 0.05 vs. 0.92 ± 0.06 ml/min, n = 4 and 5, respectively, P = not significant). Thus, HO activity, arising in aggregrate from HO-1 and HO-2 isoforms, exerts vasorelaxant effects since the acute inhibition of such activity leads to a reduction in carotid blood flow in the partially ligated carotid artery.
DISCUSSION
PCAL led to vascular injury, as evidenced by a prompt reduction in ipsilateral carotid artery blood flow, vascular remodeling, and the induction of vasculopathic genes. In this setting, marked induction of HO-1 mRNA and protein occurred, the significance of which was assessed in HO-1−/− mice: the imposition of the PCAL model in HO-1−/− mice caused a further reduction in ipsilateral carotid blood flow, the complete loss of patency in some arteries, and exacerbation of histological vascular injury. We pursued additional approaches in examining the countervailing, protective effects of the HO system using carotid artery blood flow as a readout of injury because this index is readily quantified and functionally significant. Such experiments demonstrated that carotid artery blood flow in the PCAL model increased when HO-1 was upregulated by AAV9 and when the HO product CO was delivered by CORM-3; conversely, carotid artery blood flow decreased further in the absence of the constitutive HO isoform, HO-2.
Previous studies have clearly demonstrated that vascular injury induces HO-1 and that such an induction may exert vasoprotective effects (17, 36, 48, 52). However, the expression and significance of HO-1 have not been studied, to date, in the PCAL model. We used this specific model because, as shown in a previous study (40), low laminar and oscillatory shear stress have been documented in the main trunk of the carotid artery. We suggest that the present findings provide needed information in the field of HO-1 and shear stress because prior analyses of HO-1 expression in response to shear stress have been undertaken, almost exclusively, in vitro. An in vitro approach raises at least three issues, the first of which centers on the lack of consistency in conclusions so obtained. Shear stress includes high laminar shear stress, which is vasoprotective, and low laminar and oscillatory shear stress, both of which are vasculopathic (6, 7, 14). HO-1 is induced in endothelial cells exposed in vitro to conditions intended to recapitulate high (physiological) laminar shear stress (5, 8, 11, 16). However, there are very few in vitro studies that have examined HO-1 induction in response to what is regarded as a model for low (pathological) laminar shear stress (61), perhaps because of the uncertainty of the threshold for flow rates in vitro that demarcates high from low laminar shear stress. Studies of oscillatory stress in vitro have yielded quite divergent findings: compared with laminar shear stress, oscillatory shear stress-elicited induction of HO-1 in endothelial cells has been described as exaggerated (15), equivalent (56), or nonexistent (25); induction of HO-1 in statin-exposed endothelial cells is attenuated by oscillatory shear stress but amplified by laminar shear stress (3).
In addition to this issue of consistency of findings, a second consideration is that an in vitro approach often leaves unresolved the question of the functional significance of HO-1 when shear stress is altered. The third consideration centers on the fidelity with which studies in vitro truly reflect what exists in vivo. Endothelial cells in vitro differ phenotypically from endothelial cells in vivo, and, in certain microvascular beds, >40% of the proteins expressed in endothelial cells in vivo may not be detectable in vitro (19). Additionally, gene expression profiles in in vitro systems of shear stress may not recapitulate the responses observed in vivo (6, 7, 14). For example, of the mechanosensitive genes altered in in vivo models of perturbed stress, some 50%, when examined in in vitro systems, are not expressed, remain unaltered, or are altered in the opposite direction (47). Such uncertainties and vagaries observed in in vitro models of pathological shear stress systems may be circumvented by a valid, reproducible in vivo model, the latter provided by the PCAL model. Based on the present findings, we suggest that this model affords an appealing and robust approach in analyzing the basis for the induction of HO-1 in response to pathological shear stress.
In regard to a mechanism relevant to the induction of HO-1 in PCAL, we examined activation of NF-κB, one of the main transcriptional activators of HO-1 expression (2, 34). We observed that within minutes of PCAL, activation of NF-κB occurred in the main trunk of the carotid artery. NF-κB regulates the induction of multiple vasculopathic genes, especially the genes highlighted in this study (IL-6, MCP-1, TNF-α, and CCL5), and NF-κB is increasingly regarded as an instigator of pathological shear stress, as indicated by studies conducted exclusively in vitro. We speculate that in the PCAL model, NF-κB orchestrates vasculopathic processes on the one hand (arising from IL-6, MCP-1, TNF-α, CCL5, and others shown in Table 1) and a vasoprotective response emanating from HO-1 on the other hand. The expression of vasculopathic genes in the PCAL model when HO-1 and HO-2 are deficient would be of interest.
The present findings are germane to the current interest in the HO system and its products as a therapeutic strategy. Compounds that deliver products of HO activity, in particular, CO, are under active development for clinical use because of their potential to confer vasorelaxant and other vasoprotective effects (18, 39, 54). Additionally, other products of HO activity, such as biliverdin and bilirubin, possess vasoprotective properties, including the capacity to scavenge the vasoconstricting oxidant superoxide anion; epidemiologic observations (37) in humans have demonstrated that plasma levels of bilirubin in the high-normal or mildly elevated range are associated with a reduced risk for cardiovascular disease and lower mortality. Finally, a number of drugs currently used in the treatment of cardiovascular disease (statins, nitrates, aspirin, niacin, and sildenafil) and a number of salutary dietary constituents (curcumin and resveratrol) can all induce HO-1, and, for some of these, evidence is available that such induction of HO-1 contributes to their protective effects in experimental models (58). Indeed, there is substantial effort in drug discovery programs targeted to the identification of clinically approved drugs that induce HO-1 and to the screening of chemical libraries for compounds and chemical motifs that elicit HO-1 expression.
To the best of our knowledge, the present findings are the first to demonstrate vascular and systemic upregulation of functionally active HO-1 after the utilization of an AAV delivery system. While there is clear prior evidence that upregulation of HO-1 by adenoviral gene therapy vectors can be vasoprotective (28, 36, 59), there are concerns using this immunostimulatory vector in humans. In contrast, AAV vectors are less immunogenic and are currently the lead vector platform for human gene therapy. Indeed, some 100 trials in humans using AAV vectors have already been conducted (4, 22). Our experiments confirmed the effective upregulation of HO-1 in the carotid artery along with a functional effect of this upregulation, namely, improvement in carotid artery blood flow. Such vascular upregulation of HO-1 by AAV9 also increased carotid artery blood flow in the contralateral, intact carotid artery, an effect likely reflecting HO-1 upregulation in that vascular segment. The administration of AAV9(HO-1) led to substantial amounts of HO-1 protein in the systemic circulation and increased vascular HO activity. The effect of AAV9(HO-1) on vasculopathic gene expression and histological injury in the PCAL model is of interest.
The reduction in carotid artery blood flow after PCAL was alleviated by the administration of CO, with the latter administered as CORM-3. Thus, a product of HO (in this case, CO) is capable of increasing blood flow in the ligated carotid artery. It should be emphasized, however, that these experiments were performed with the objective of demonstrating that a given product of HO may increase carotid blood flow; it should not be inferred that these experiments identified CO as the basis for the vasorelaxant effects of HO. At least three additional issues may be germane to the findings from these experiments. First, CORMs may exert cardiac and systemic effects, and not just local vasorelaxant effects. Second, the findings from these experiments do not lead us to specify whether this is a vasorelaxant effect, a remodeling effect, or a combination of both; however, as the carotid blood flow experiments were performed 48 h after the administration of CO, the former may more likely underlie the increase in blood flow. Third, several mechanisms besides lesser amounts of CO may be relevant to the observed effects of HO-1 deficiency. These include 1) the loss of the antioxidant effects of the enzyme; 2) the lack of generation of bilirubin/biliverdin, products of HO that are antioxidant, anti-inflammatory, and vasoprotective; and 3) upregulation of proliferative, proinflammatory, and procoagulant pathways in the setting of HO-1 deficiency.
Our findings also demonstrate a beneficial effect of HO-2 in the PCAL model, as assessed both by functional indexes and histologic analyses. In one-third of the HO-2−/− mice subjected to PCAL, focal neointimal hyperplasia was observed; as our analyses did not involve serial sectioning of the entire length of the trunk of the carotid artery, the true frequency of this focal histological lesion may be higher than 33%. These findings in HO-2−/− mice raise the following three considerations regarding HO-2 and vascular injury. First, the presence of neointimal hyperplasia after PCAL when HO-2 is deficient is notable, as this lesion is not a usual finding in the PCAL model when the latter is induced in otherwise unmanipulated, wild-type mice; vascular expression of HO-2 may thus represent an endogenous defense mechanism that guards against the development of neointimal hyperplasia in the injured vasculature. Second, the present findings, in conjunction with our recent study (30) demonstrating that HO-2 is required in maintaining patency and blood flow in an arteriovenous fistula, underscore the vasoprotective properties of HO-2. Finally, the HO-2 gene is readily induced by corticosteroids, with the latter commonly used in the treatment of inflammatory vasculopathies. Based on our present findings, we speculate that the beneficial effects of corticosteroids in vasculitides may involve the induction of HO-2.
The present experiments using the PCAL model underscore the usefulness of this model in the study of cardiovascular disease. In addition to demonstrating carotid intimal-media thickening and the presence of pathological shear stress (32, 40), this model exhibits endothelial dysfunction and oxidative stress (40), accelerates the progression of atherosclerosis (40), and provides important insights regarding the roles of such diverse proteins as Axl (a receptor tyrosine kinase) (33), fibronectin (10), fibroblast growth factor 2 (57), platelet/endothelial cell adhesion molecule-1 (9), and IL-17 (38) in vascular remodeling.
In summary, the present study demonstrates the induction and functional significance of the HO system in the vasculature subjected to pathological shear stress, demonstrating, in the process, protective effects conferred by the observed induction of HO-1 and the administration of CORMs, efficacy in achieving vascular upregulation of HO-1 by AAV9 along with a vasorelexant effect, and the vasoprotective effects of HO-2. Finally, we provide evidence for the activation of NF-κB in this model. As this transcription factor is an inducer of HO-1, such NF-κB activation may not only underlie genetic responses accounting for pathological shear stress but also those that mitigate the severity of these pathological responses.
GRANTS
This work was supported by National Institutes of Health Grants DK-70124, DK-47060, HL-91867, and T32-DK-007013. This work was also supported by the Mayo Clinic Center for Regenerative Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.K., M.L.H., A.J.C., M.A.B., Z.S.K., and K.A.N. conception and design of research; L.K., M.L.H., and A.J.C. performed experiments; L.K., M.L.H., J.P.G., A.J.C., M.A.B., G.F., Z.S.K., and K.A.N. analyzed data; L.K., A.J.C., and K.A.N. prepared figures; L.K., M.L.H., A.J.C., and K.A.N. drafted manuscript; L.K., M.L.H., J.P.G., A.J.C., M.A.B., G.F., Z.S.K., and K.A.N. edited and revised manuscript; L.K., M.L.H., J.P.G., A.J.C., M.A.B., G.F., Z.S.K., and K.A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the technical expertise of Allan Ackerman and the secretarial expertise of Kara Zelinske in the preparation of this work.
REFERENCES
- 1.Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med 39: 1–25, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36: 166–174, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Ali F, Zakkar M, Karu K, Lidington EA, Hamdulay SS, Boyle JJ, Zloh M, Bauer A, Haskard DO, Evans PC, Mason JC. Induction of the cytoprotective enzyme heme oxygenase-1 by statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by disturbed flow. J Biol Chem 284: 18882–18892, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20: 699–708, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braam B, de Roos R, Bluyssen H, Kemmeren P, Holstege F, Joles JA, Koomans H. Nitric oxide-dependent and nitric oxide-independent transcriptional responses to high shear stress in endothelial cells. Hypertension 45: 672–680, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Bryan MT, Duckles H, Feng S, Hsiao ST, Kim HR, Serbanovic-Canic J, Evans PC. Mechanoresponsive networks controlling vascular inflammation. Arterioscler Thromb Vasc Biol 34: 2199–2205, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Cecchi E, Giglioli C, Valente S, Lazzeri C, Gensini GF, Abbate R, Mannini L. Role of hemodynamic shear stress in cardiovascular disease. Atherosclerosis 214: 249–256, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J Biol Chem 278: 703–711, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Chen Z, Tzima E. PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol 29: 1067–1073, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol 29: 1074–1079, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu TJ, Peters DG. Serial analysis of the vascular endothelial transcriptome under static and shear stress conditions. Physiol Genomics 34: 185–192, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 93: e2–e8, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Czibik G, Sagave J, Martinov V, Ishaq B, Sohl M, Sefland I, Carlsen H, Farnebo F, Blomhoff R, Valen G. Cardioprotection by hypoxia-inducible factor 1 alpha transfection in skeletal muscle is dependent on haem oxygenase activity in mice. Cardiovasc Res 82: 107–114, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med 6: 16–26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, Griendling KK. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ Res 82: 1094–1101, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Di Francesco L, Totani L, Dovizio M, Piccoli A, Di Francesco A, Salvatore T, Pandolfi A, Evangelista V, Dercho RA, Seta F, Patrignani P. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-alpha biosynthesis via heme oxygenase-1 in human endothelial cells. Circ Res 104: 506–513, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, Clinton Webb R, Lee ME, Nabel GJ, Nabel EG. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat Med 7: 693–698, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117: 231–241, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, Oh P, Schnitzer JE. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol 22: 985–992, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Farrugia G, Lei S, Lin X, Miller SM, Nath KA, Ferris CD, Levitt M, Szurszewski JH. A major role for carbon monoxide as an endogenous hyperpolarizing factor in the gastrointestinal tract. Proc Natl Acad Sci USA 100: 8567–8570, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, Ehlers MD, Zylka MJ, McCown TJ, Samulski RJ. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther 22: 1143–1153, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.High KA. The gene therapy journey for hemophilia: are we there yet? Hematology Am Soc Hematol Educ Program 2012: 375–381, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Hill-Kapturczak N, Agarwal A. Haem oxygenase-1–a culprit in vascular and renal damage? Nephrol Dial Transplant 22: 1495–1499, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Hillestad ML, Guenzel AJ, Nath KA, Barry MA. A vector-host system to fingerprint virus tropism. Hum Gene Ther 23: 1116–1126, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem 280: 27244–27250, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Hyvelin JM, Maurel B, Uzbekov R, Motterlini R, Lermusiaux P. Hemin prevents in-stent stenosis in rat and rabbit models by inducing heme-oxygenase-1. J Vasc Surg 51: 417–428, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Jais A, Einwallner E, Sharif O, Gossens K, Lu TT, Soyal SM, Medgyesi D, Neureiter D, Paier-Pourani J, Dalgaard K, Duvigneau JC, Lindroos-Christensen J, Zapf TC, Amann S, Saluzzo S, Jantscher F, Stiedl P, Todoric J, Martins R, Oberkofler H, Muller S, Hauser-Kronberger C, Kenner L, Casanova E, Sutterluty-Fall H, Bilban M, Miller K, Kozlov AV, Krempler F, Knapp S, Lumeng CN, Patsch W, Wagner O, Pospisilik JA, Esterbauer H. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 158: 25–40, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation 104: 1519–1525, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Juncos JP, Grande JP, Murali N, Croatt AJ, Juncos LA, Hebbel RP, Katusic ZS, Nath KA. Anomalous renal effects of tin protoporphyrin in a murine model of sickle cell disease. Am J Pathol 169: 21–31, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang L, Grande JP, Farrugia G, Croatt AJ, Katusic ZS, Nath KA. Functioning of an arteriovenous fistula requires heme oxygenase-2. Am J Physiol Renal Physiol 305: F545–F552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang L, Yamada S, Hernandez MC, Croatt AJ, Grande JP, Juncos JP, Vercellotti GM, Hebbel RP, Katusic ZS, Terzic A, Nath KA. Regional and systemic hemodynamic responses following the creation of a murine arteriovenous fistula. Am J Physiol Renal Physiol 301: F845–F851, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol 23: 2185–2191, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Korshunov VA, Mohan AM, Georger MA, Berk BC. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res 98: 1446–1452, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-κB and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA 91: 5987–5991, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Horke S, Forstermann U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol Sci 34: 313–319, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Lin HH, Chen YH, Yet SF, Chau LY. After vascular injury, heme oxygenase-1/carbon monoxide enhances re-endothelialization via promoting mobilization of circulating endothelial progenitor cells. J Thromb Haemost 7: 1401–1408, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Lin JP, Vitek L, Schwertner HA. Serum bilirubin and genes controlling bilirubin concentrations as biomarkers for cardiovascular disease. Clin Chem 56: 1535–1543, 2010. [DOI] [PubMed] [Google Scholar]
- 38.Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31: 1565–1572, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9: 728–743, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 297: H1535–H1543, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Nath KA, Grande JP, Farrugia G, Croatt AJ, Belcher JD, Hebbel RP, Vercellotti GM, Katusic ZS. Age sensitizes the kidney to heme protein-induced acute kidney injury. Am J Physiol Renal Physiol 304: F317–F325, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nath KA, Grande JP, Kang L, Juncos JP, Ackerman AW, Croatt AJ, Katusic ZS. β-Catenin is markedly induced in a murine model of an arteriovenous fistula: the effect of metalloproteinase inhibition. Am J Physiol Renal Physiol 299: F1270–F1277, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol 156: 1527–1535, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nath KA, Hernandez MC, Croatt AJ, Katusic ZS, Juncos LA. Heme oxygenase activity as a determinant of the renal hemodynamic response to low-dose ANG II. Am J Physiol Regul Integr Comp Physiol 299: R1183–R1191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Negi SI, Nambi V. The role of carotid intimal thickness and plaque imaging in risk stratification for coronary heart disease. Curr Atheroscler Rep 14: 115–123, 2012. [DOI] [PubMed] [Google Scholar]
- 47.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 116: e66–73, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, Stachulak C, Bodyak N, Smith RN, Csizmadia E, Tyagi S, Akamatsu Y, Flavell RJ, Billiar TR, Tzeng E, Bach FH, Choi AM, Soares MP. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 9: 183–190, 2003. [DOI] [PubMed] [Google Scholar]
- 49.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: pathophysiologic correlates. Kidney Int 68: 611–622, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Platt JL, Nath KA. Heme oxygenase: protective gene or Trojan horse. Nat Med 4: 1364–1365, 1998. [DOI] [PubMed] [Google Scholar]
- 51.Riley WA. Cardiovascular risk assessment in individual patients from carotid intimal-medial thickness measurements. Curr Atheroscler Rep 6: 225–231, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez AI, Gangopadhyay A, Kelley EE, Pagano PJ, Zuckerbraun BS, Bauer PM. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler Thromb Vasc Biol 30: 98–104, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic Biol Med 35: 872–881, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000. [DOI] [PubMed] [Google Scholar]
- 56.Silacci P, Desgeorges A, Mazzolai L, Chambaz C, Hayoz D. Flow pulsatility is a critical determinant of oxidative stress in endothelial cells. Hypertension 38: 1162–1166, 2001. [DOI] [PubMed] [Google Scholar]
- 57.Sullivan CJ, Hoying JB. Flow-dependent remodeling in the carotid artery of fibroblast growth factor-2 knockout mice. Arterioscler Thromb Vasc Biol 22: 1100–1105, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Tinkel J, Hassanain H, Khouri SJ. Cardiovascular antioxidant therapy: a review of supplements, pharmacotherapies, and mechanisms. Cardiol Rev 20: 77–83, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Tulis DA, Durante W, Liu X, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 104: 2710–2715, 2001. [DOI] [PubMed] [Google Scholar]
- 60.Wang G, Hamid T, Keith RJ, Zhou G, Partridge CR, Xiang X, Kingery JR, Lewis RK, Li Q, Rokosh DG, Ford R, Spinale FG, Riggs DW, Srivastava S, Bhatnagar A, Bolli R, Prabhu SD. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 121: 1912–1925, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warabi E, Wada Y, Kajiwara H, Kobayashi M, Koshiba N, Hisada T, Shibata M, Ando J, Tsuchiya M, Kodama T, Noguchi N. Effect on endothelial cell gene expression of shear stress, oxygen concentration, and low-density lipoprotein as studied by a novel flow cell culture system. Free Radic Biol Med 37: 682–694, 2004. [DOI] [PubMed] [Google Scholar]