

Abstract
Coronary collateral growth (CCG) is impaired in metabolic syndrome. microRNA-21 (miR-21) is a proproliferative and antiapoptotic miR, which we showed to be elevated in metabolic syndrome. Here we investigate whether impaired CCG in metabolic syndrome involved miR-21-mediated aberrant apoptosis. Normal Sprague-Dawley (SD) and metabolic syndrome [J. C. Russel (JCR)] rats underwent transient, repetitive coronary artery occlusion [repetitive ischemia (RI)]. Antiapoptotic Bcl-2, phospho-Bad, and Bcl-2/Bax dimers were increased on days 6 and 9 RI, and proapoptotic Bax and Bax/Bax dimers and cytochrome-c release concurrently decreased in JCR versus SD rats. Active caspases were decreased in JCR versus SD rats (∼50%). Neutrophils increased transiently on day 3 RI in the collateral-dependent zone of SD rats but remained elevated in JCR rats, paralleling miR-21 expression. miR-21 downregulation by anti-miR-21 induced neutrophil apoptosis and decreased Bcl-2 and Bcl-2/Bax dimers (∼75%) while increasing Bax/Bax dimers, cytochrome-c release, and caspase activation (∼70, 400, and 400%). Anti-miR-21 also improved CCG in JCR rats (∼60%). Preventing neutrophil infiltration with blocking antibodies resulted in equivalent CCG recovery, confirming a major role for deregulated neutrophil apoptosis in CCG impairment. Neutrophil and miR-21-dependent CCG inhibition was in significant part mediated by increased oxidative stress. We conclude that neutrophil apoptosis is integral to normal CCG and that inappropriate prolonged miR-21-mediated survival of neutrophils plays a major role in impaired CCG, in part via oxidative stress generation.
Keywords: apoptosis, collateral circulation, inflammation, microRNA, metabolic syndrome
studies in larger animals with epicardial coronary vessels have ascertained that collateral development progresses through two distinct stages. The early phase is characterized by endothelial progenitor cell and inflammatory cell recruitment and proliferation, degradation of the basement membrane, vascular smooth muscle cell (VSMC) switch to the synthetic, proliferative phenotype, and neointima formation (3). In the later phase, termed outward expansion and maturation, cell proliferation and inflammatory cell numbers are decreased, the neointima resolves, the basement membrane reforms, and VSMCs return to the contractile phenotype (3). Increased shear stress is proposed as the driving force for outward remodeling (luminal expansion). However, it is presently unclear what cellular processes underlie the resolution of the neointima as well as clearance of the inflammatory cells at the transition from inward to outward remodeling. Apoptosis, as a possible integral component of normal collateral development, has never been investigated and is presumed to be antithetical to growth processes. However, nitric oxide (NO), a principal factor upregulated by shear stress, has been shown to induce the mitochondrial apoptotic pathway in some cells (2) while simultaneously protecting endothelial cells (ECs). Whereas normal animals [Sprague-Dawley (SD), Wistar-Kyoto, and Zucker lean rats] have normal endothelial function (9) and develop collaterals (12, 14, 19), metabolic syndrome animals [Zucker obese (ZOF); J. C. Russel (JCR)] have impaired endothelial function (9) and do not develop collaterals (12, 14). This is at least in part a consequence of decreased NO. We have recently shown that the later stage of coronary collateral growth (CCG) is deregulated in metabolic syndrome. Specifically, we demonstrated that excessive and prolonged VSMC proliferation was associated with the inability of native collateral vessels to undergo outward expansion in a rat model of metabolic syndrome, the JCR rat (13). In this study we wanted to investigate a possible role apoptosis may play in CCG.
microRNA-21 (miR-21) is a major proproliferative and antiapoptotic miR, which promotes proliferation and cell survival via downregulating phosphatase and tensin homolog and consequently upregulating phosphatidylinositol 3-kinase and Akt signaling and by upregulating mitochondrial prosurvival signals, namely Bcl-2 (30). miR-21 is upregulated in the neointima following vascular injury, and its downregulation decreases neointima formation (30). miR-21 depletion increased apoptosis in a dose-dependent manner in cultured VSMCs (30). Its expression has been shown to be consistently elevated and associated with invasion in most solid cancers. miR-21 expression was significantly reduced in infarcted areas of the heart, and its overexpression reduced infarct size, suggesting that its prosurvival effects translate to the cardiovascular system. The role of miR-21 in angiogenesis, a process related to, but distinct from, collateral growth, is controversial. miR-21 induced tumor angiogenesis via Akt and ERK1/2 activation and HIF-1α expression. In contrast, miR-21 inhibited angiogenesis by decreasing RhoB expression and actin stress fiber formation (24). We hypothesized that these discrepancies are due to the fact that miR-21 is necessary for vascular growth, including angiogenesis and collateral growth but that the amount and especially timing of its expression must be strictly regulated. We have recently shown that miR-21 was indeed transiently and moderately upregulated in CCG in normal animals; this upregulation correlated with a transient increase in VSMC proliferation (13). However, in metabolic syndrome, miR-21 was highly upregulated and remained elevated through the late stages of CCG, correlating with excessive VSMC proliferation and failure of collaterals to undergo outward expansion (13). In this study we will investigate the role of miR-21 in the regulation apoptosis.
MATERIALS AND METHODS
Rat model of CCG/repetitive ischemia.
Male, 10–12-wk-old SD (300–350 g; Charles Rivers, Wilmington, MA) or JCR:LA-cp (650–700 g; JCR, J.C. Russell and S. Proctor, University of Alberta, Edmonton, Canada) rats were used for chronic (0–9 days) implantation of a pneumatic occluder over the left anterior descending coronary artery (LAD) as previously described (7, 14, 20). A suture was passed under the proximal portion of the LAD, and the occluder was sewn onto the surface of the heart. The occluder catheter was externalized between the scapulae. When the occluder is inflated, the suture is pulled toward the surface of the heart and the LAD is occluded. The LAD perfusion territory is termed the collateral-dependent zone (CZ) because perfusion in this area, while the LAD is occluded, depends on the development of coronary collaterals. The animals underwent the repetitive ischemia (RI) protocol consisting of eight 40-s occlusions, once every 20 min (2 h, 20 min total), followed by a rest period of 5 h, 40 min. This 8-h cycle was repeated 3 times/day for 0–9 days. Surgical procedures were performed in accordance with the Animal Welfare Act and are approved by the Institutional Animal Care and Use Committees of the University of South Alabama and New York Medical College. CZs are separated from normal zones (NZs) on the basis of visual assessment of blanching during LAD occlusion (occluder inflation), which is indicative of ischemia and hyperemia upon reperfusion in the CZ at the time of occluder implantation (initial surgery), before collaterals have formed. It is confirmed by measurements of baseline (day 0 RI) blood flows. Notes delineating the CZ and the NZ are made for each animal and referred to at terminal death, and the border zone is excluded from tissue samples.
The JCR rat is a cross between the lean LA/N Zucker and the spontaneously hypertensive obese rat developed in the laboratory of Dr. Carl Hansen at the National Institutes of Health and sent to Dr. James C. Russell. By 8 wk of age, the JCR rats develop obesity with fatty liver, insulin resistance with glucose intolerance, complex dyslipidemia (low HDL, high LDL, and vLDL), and vasculopathy characterized by decreased endothelium-dependent and -independent vasorelaxation and intimal lesions morphologically identical to early atherosclerotic lesions in humans. By 12 wk, the rats exhibit widespread intimal hyperplasia, left ventricular (LV) hypertrophy, and myocardial and cerebral (micro)infarctions. At 16+ wk, the rats are prone to stroke and myocardial infarction, and at 18+ wk, they develop heart failure. Like the development of the metabolic syndrome and cardiovascular disease in humans, the apparent complexity of the cardiometabolic phenotype exhibited by the JCR rats is suspected to be multifactorial and polygenetic in etiology (23, 27).
Blocking antibodies.
JCR rats were treated with the blocking antibodies against the major monocyte/neutrophil adhesion receptor CD11b/CD18 (Mac-1) (MAb clones M1/70/M18/2, Abcam, Cambridge, MA) and with the blocking antibody against CD44 (MAb clone IM7, Abcam), the hyaluronan and osteopontin adhesion receptor, at the dose of 1 mg·kg−1·day−1, by direct LV injection on day 6 (12 h before animals were euthanized on day 6) through day 9 of RI.
Anti-miR-21 delivery.
Cultured VSMCs were treated with 60 nM locked nucleic acid (LNA)-modified anti-miR-21 (Exiqon, Woburn, MA) for 24 h before exposure to hypoxia-hyperoxia-normoxia. JCR rats were treated with the LNA-modified anti-miR-21 at 2 mg/kg in 100 μl of sterile saline via intracardiac injection directly into the LV cavity as previously described for anti-miR-145 (14), according to modification of previously used protocols for tail vein injection (18) on day 5 of RI. Scrambled LNA-anti-miR sequence was used as control.
miR quantitation.
miR quantitation was performed as previously described (13, 14). Total RNA was isolated from VSMCs or the heart (NZ and CZ) with QIAzol, followed by small RNA isolation with miRNeasy mini kits (Qiagen, Valencia, CA). Total and small RNA concentration and quality were determined by absorbance at 260/280 nm. The ratio of 18S and 28S ribosomal RNA and the degree of DNA contamination were assessed by agarose gel electrophoresis with SYBR Green II staining. cDNA synthesis and quantitative RT-PCR were performed with TaqMan miR assays using 250 ng RNA. Absolute quantities of miR-21 in CZ and NZ cardiac tissue were obtained by quantitative RT-PCR using standards constructed from a dilution series of the miR-21 standard (Integrated DNA Technologies, Coralville, IA). miR-21 was normalized to U6 RNA. Experiments were representative of n = 6 animals per time point (day of RI) and were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
Western blot analysis.
Unperfused hearts were excised, the LV was dissected, CZ was separated from the nonischemic NZ, and NZ and CZ were snap-frozen in liquid nitrogen before homogenization in lysis buffer containing 0.1% SDS and 1% Triton as previously described (15, 20). Cultured VSMCs were lysed in lysis buffer containing 0.1% SDS and 1% Triton. Equal amounts of protein (30 μg) were separated by SDS-PAGE and transferred to Hybond-ECL nitrocellulose membranes. Anti-Bcl-2, anti-Bax, anti-phospho-Ser155-Bad, anti-pro-caspase-3, antiactivated caspase-3, anti-pro-caspase-9, antiactivated caspase-9 antibodies (Cell Signaling, Boston, MA), and anti-osteopontin (Abcam) antibodies were used for Western blot analysis. Bands were visualized by enhanced chemiluminescence (GE Healthcare Biosciences, Pittsburg, PA) and quantified using Un-Scan-It Image software (Silk Scientific, Orem, UT). Data are normalized to β-tubulin (loading control), except phospho-Bad, which is also normalized to total Bad and expressed as CZ-to-NZ ratios, which is indicative of changes in response to RI in the CZ since protein expression in the NZ does not change for any protein. Experiments were representative of n = 6 animals per time point (day of RI) and were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
Immunoprecipitation.
Hearts were excised, the LV was dissected, CZ was separated from the nonischemic NZ, and NZ and CZ were snap-frozen in liquid nitrogen. Tissue lysates were prepared for immunoprecipitation by homogenization in lysis buffer containing 0.1% Triton X-100. Equal amounts of protein (500 μg) were immunoprecipitated with anti-Bcl-2 or anti-Bax polyclonal antibodies (Cell Signaling) overnight at 4°C, and immune complexes were collected by incubation with protein A-Sepharose beads for 2 h at 4°C as previously described (21, 22). Immunoprecipitates were separated by SDS-PAGE, and proteins were detected by Western blot analysis.
Cytochrome-c assay.
Cytochrome-c assay was performed using the cytochrome-c releasing apoptosis assay kit according to manufacturer's instructions (Abcam). Hearts were excised, the LV was dissected, CZ was separated from the nonischemic NZ, and NZ and CZ were homogenized in cytosol extraction buffer. Cytosolic fraction was collected and the pallet resuspended in mitochondria extraction buffer. Equal amounts of protein (30 μg) were separated by SDS-PAGE and transferred to Hybond-ECL nitrocellulose membranes. The anti-cytochrome-c antibody was used for Western blot analysis. Bands were visualized by enhanced chemiluminescence (GE Healthcare Biosciences) and quantified using Un-Scan-It Image software (Silk Scientific). Experiments were representative of n = 6 animals per time point (day of RI) and were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
Histology.
Hematoxylin-and-eosin staining was performed according to standard protocols in perfused (isotonic saline followed by 4% paraformaldehyde) and unperfused hearts. Images (×100) are representative of n = 6 animals per time point (day of RI) from five consecutive cardiac cross sections per animal and five separate fields per slide. Immediately adjacent tissue sections were stained with anti-proliferating cell nuclear antigen (PCNA) (1:200, Millipore, San Diego, CA) to identify proliferating cells to differentiate actively remodeling arteries from preexisting arteries as in our recently published study (3). For larger arteries, arteries were distinguished from veins by assessment of vessel wall thickness, where wall thickness-to-lumen diameter ratio > 0.25 signified an artery and < 0.25, a vein (6).
Immunohistochemistry analysis.
Immunohistochemistry analysis was performed as previously described (14). Formalin-fixed, paraffin-imbedded cardiac tissue was cut into 10-μm sections. Primary anti-α-smooth muscle-specific actin (α-SMA; 1:1,000, Sigma, St. Louis, MO), anti-Gr-1 antibody (1:200, from Bethyl Laboratories, Montgomery, TX), anti-CD68/Mac-1 (1:500, MAb clone ED1/ED7, Bio-Rad, Hercules, CA), and anti-von Willebrand factor (1:100, Abcam) and secondary Alexa350-, Alexa488- Alexa568-conjugated (Invitrogen, Carlsbad, CA) antibodies were used. Blue (Alexa350), green (Alexa488), and red (Alexa568) fluorescence were visualized, and representative images were collected using a Nikon fluorescent microscope equipped with Nikon Elements software. Images are representative of n = 6 animals per time point (day of RI) from five consecutive cardiac cross sections per animal and five separate fields per slide.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling and Gr-1 immunohistochemistry.
Unperfused and perfused (isotonic saline followed by 4% paraformaldehyde), formalin-fixed, paraffin-imbedded, cardiac tissue was cut into 10-μm sections. Slides were deparaffinized and subjected to mild proteinase-K digestion, and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) was performed using the TUNEL kit (Roche, Indianapolis, IN). The kit contains an optimized terminal deoxynucleotide transferase and red fluorescent-labeled dUTP (tetramethylrhodamine-dUTP), which was visualized with a Nikon fluorescent microscope equipped with Nikon Elements software. Gr-1 antibody (1:200) was from Bethyl Laboratories. All slides were costained with anti-α-SMA (Sigma) to identify blood vessels, and immediately adjacent tissue sections were stained with anti-PCNA (1:200, Millipore) to identify proliferating cells, to differentiate actively remodeling arteries from preexisting arteries as in our published studies (13, 14). Arteries were distinguished from veins by assessment of vessel wall thickness, where wall thickness-to-lumen diameter ratio > 0.25 signified an artery and < 0.25, a vein (5). Images are representative of n = 6 animals per time point (day of RI) from five consecutive cardiac cross sections per animal and five separate fields per slide.
Flow cytometry.
Excised hearts were flushed with saline (1 ml, 3×) through the aortic root using a 5-ml syringe to create sufficient pressure to dislodge and separate adherent leukocytes without perturbing the integrity of the vascular wall, confirmed by high-power (×100) microscopy for 20–150-μM vessels in every heart. Flow cytometry was performed on the elute using a Gr-1 antibody (MAb clone Ly6G, Acris, San Diego, CA) to label peripheral neutrophils, CD115 (MAb clone 2E11, Acris) antibody to label monocytes, CD3 (delta chain MAb clone 1F4, Arcis) antibody to label T cells, CD23 (IgE receptor, MAb clone 2G8, Arcis) to label mast cells, and annexin V (Life Technologies, Molecular Probes, Grand Island, NY) to label apoptotic cells. Experiments were representative of n = 6 animals per time point (day of RI) and were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
Myocardial and collateral-dependent blood flow measurements.
Myocardial and collateral-dependent blood flow measurements were performed as in our published studies (7, 8, 13, 14). Color microspheres (5 × 105, 15 μM diameter) labeled with samarium (day 0 RI; initial surgery) or gold (day 10 RI) were injected into the LV during LAD occlusion. Arterial reference blood samples (carotid) and heart tissue from the NZ and the CZ were collected, weighed, and sent to BioPal (Worcester, MA) for analysis. Blood flows to the NZ and the CZ (in ml·min−1·g−1) were calculated from the formula: blood Flow = [(radioactive counts in myocardial tissue) × (blood reference withdrawal rate)/(radioactive count in blood reference)]/(weight of myocardial tissue). Blood flow was measured in the following groups of animals: SD RI, JCR RI, JCR RI + anti-miR-21, JCR + scrambled anti-miR, and JCR + CD11b/CD18 + CD44 blocking antibodies. Results were expressed as the CZ-to-NZ flow ratio on day 9 of RI. All experiments were representative of n = 8 animals per group. Results were analyzed by two-way ANOVA, followed by Bonferroni correction. P < 0.05 determined statistical significance.
RESULTS
Previous data from our laboratory demonstrate that RI induces maximal CCG in the normal (SD) rats but does not stimulate CCG in metabolic syndrome (JCR) rats (19). Nine days of RI stimulate maximal CCG in the SD rats; prolonging the duration of RI to 14, 21, or 28 days does not induce CCG in the JCR rats (8).
Mitochondrial apoptotic pathways and caspase activation favor cell survival in metabolic syndrome and apoptosis in the normal phenotype.
In our recent study, miR-21 downregulation by anti-miR-21 significantly improved CCG in JCR rats (∼60%) (13). miR-21 exerts its antiapoptotic actions by upregulating Bcl-2 expression; therefore, we first examined Bcl-2 expression along the time course of RI in SD versus JCR rats. Bcl-2 expression was increased ∼3-fold at day 6 and ∼2.5-fold at day 9 RI versus day 0 RI in JCR rats. In contrast, RI-induced expression of Bcl-2 in SD rats was early, transient (day 3 RI only) and smaller in magnitude (∼2-fold vs. day 0 RI) (Fig. 1A). Furthermore, Ser155(Bad) phosphorylation p(Ser155)Bad increased in an analogous manner (∼3-fold on days 6 and 9 RI) in response to RI in JCR but decreased (∼75% on days 3 and 6 RI) in SD rats (Fig. 1B). This is significant because when phosphorylated on Ser155, Bad cannot bind Bcl-2, thus leaving it free to bind to and sequester the proapoptotic Bax. Total Bad expression was not altered. Bax expression was decreased by 25% versus day 0 RI at days 6 and 9 RI in JCR but increased in SD rats (∼0.5-fold on day 3 RI and ∼2-fold on day 6 RI vs. day 0 RI) (Fig. 1C). Neither expression nor phosphorylation of any of these proteins changed in the NZ. Thus the balance of the relevant pro- versus antiapoptotic mitochondrial proteins of the Bcl-2 family overall favored cell survival in the metabolic syndrome phenotype and apoptosis on days 3 and 6 RI in the normal phenotype and resulted in a 50% decrease in formation of antiapoptotic Bcl-2/Bax dimers and a 2-fold increase in formation of proapoptotic Bax/Bax dimers on day 6 RI in SD rats versus an ∼6-fold increase in Bcl-2/Bax dimer and 50% decrease in Bax/Bax dimer formation at equivalent time points in JCR rats (Fig. 1D).
Fig. 1.

J. C. Russel (JCR) rats were treated with anti-microRNA-21 (miR-21) or scrambled (nontargeting) anti-miR on day 5 repetitive ischemia (RI) where indicated, and Sprague-Dawley (SD) and JCR rats were euthanized on days 0, 3, 6, or 9 of RI (n = 6). Tissue samples were collected from normal zone (NZ) and collateral-dependent zone (CZ). Top: cumulative data (n = 6). Bottom: representative Western blots from CZ. A: anti-Bcl-2. B: anti-phospho (p)-(Ser155)-Bad. C: anti-Bax. D: anti-Bcl-2 or anti-Bax antibodies were used for immunoprecipitation (IP) as indicated. Samples were then subject to SDS-PAGE, followed by Western blot analysis with anti-Bax antibodies. The same blot was stripped and reprobed for Bcl-2, p(Ser115)Bad, total Bad, Bax, and β-tubulin. E: tissue samples were collected from CZ and NZ, and mitochondria were separated from the cytosol. Cytochrome c was detected by Western blot analysis. The cytoplasmic sample was stripped and reprobed with β-tubulin. *P < 0.05 vs. sham; #P < 0.05 vs. Wistar-Kyoto (WKY) RI; $P < 0.05 vs. JCR RI.
Formation of Bax/Bax dimers leads to loss of mitochondrial outer membrane integrity and cytochrome-c release into the cytoplasm where it interacts with the apoptotic protease-activating factor to form a macromolecular complex, apoptosome, which recruits and activates pro-caspase 9 to initiate and commit the cell to the apoptotic death cascade (29). Cytochrome c in the cytoplasm decreased ∼75% in response to RI in JCR rats but increased ∼4-fold in SD rats on day 6 RI, clearly indicating progression toward apoptosis in normal but not metabolic syndrome animals (Fig. 1E). Neither cytochrome-c distribution nor expression changed in the NZ.
Caspase 3 (∼2-fold day 3 RI, ∼3-fold day 6 RI vs. day 0) and caspase 9 (∼2.5-fold day 6 RI vs. day 0 RI) were activated by RI in SD rats (Fig. 2, A and B). In JCR rats, neither caspase was activated in response to RI (Fig. 2, A and B). RI did not alter expression of active caspase 3 or 9 in the NZ. Pro-caspase expression was likewise not altered in either rat phenotype on any day of the protocol (data not shown). In the aggregate, patterns of mitochondrial apoptotic pathway and caspase expression on day 6 RI favor cell survival in metabolic syndrome and apoptosis in the normal phenotype.
Fig. 2.

JCR rats were treated with anti-miR-21 or scrambled (nontargeting) anti-miR on day 5 RI where indicated, and SD and JCR rats were euthanized on days 0, 3, 6, or 9 of RI (n = 6). Tissue samples were collected from the NZ and the CZ. Top: cumulative data (n = 6). Bottom: representative Western blots from the CZ. The same blot was stripped and reprobed for caspase 9, caspase 3, and β-tubulin. A: antiactivated caspase 3. B: antiactivated caspase 9. *P < 0.05 vs. sham; #P < 0.05 vs. WKY RI; $P < 0.05 vs. JCR RI.
miR-21 promotes cell survival in the metabolic syndrome by upregulating Bcl-2 expression.
Successful downregulation of native miR-21 by anti-miR-21 has been demonstrated in our recent study (13). In this study, anti-miR-21, delivered on day 5 RI, blocked the RI-induced increase in Bcl-2 expression in JCR rats without effecting p(Ser155)Bad or Bax expression (Fig. 1); however, this was sufficient to block the relative amount of antiapoptotic Bax/Bcl-2 dimers, increase the relative amount of proapoptotic Bax/Bax dimers (∼70%) (Fig. 1D), increase cytochrome-c release from the mitochondria ∼5-fold (Fig. 1E), and increase caspase-9 and -3 activation (Fig. 2), the final steps toward the cell's commitment to apoptosis. These results suggest that miR-21 promotes cell survival in the metabolic syndrome by upregulation of Bcl-2 expression.
Apoptosis is a key feature of normal CCG, which is absent in the metabolic syndrome.
To determine the localization of apoptotic cells in the heart, myocardial cross sections were costained with α-SMA and TUNEL to identify coronary arteries and arterioles (vessels > 20 μM diameter with wall thickness-to-lumen diameter ratio > 0.25) and apoptotic cells, respectively. Adjacent sections were stained with PCNA (not shown) to identify proliferating cells to distinguish actively remodeling collaterals from preexisting, nonremodeling arteries. Results in Fig. 3, A and B, show that in normal animals, apoptosis was restricted to the neointima and lumen of remodeling collaterals, began on day 3 RI, and was maximal on day 6 RI, immediately preceding the beginning of noticeable increase in collateral-dependent blood flow on day 7 RI (Rocic, unpublished observations). In contrast, in metabolic syndrome animals, apoptosis did not occur in the remodeling immature collaterals but in the ECs of the large preexisting arteries. The fact that increased EC apoptosis in the preexisting coronary vasculature of JCR rats is not explained by global myocardial changes in pro- and antiapoptotic markers (cytochrome-c release, caspase activation; Figs. 1 and 2) is not surprising since ECs comprise a very small percentage of total myocardial cells.
Fig. 3.

JCR rats were treated with anti-miR-21 on day 5 RI where indicated (C–F), and SD or JCR rats were euthanized on days 0, 3, 6, or 9 of RI as indicated. Representative images of cardiac cross sections for unperfused (A, C, and E) and perfused (B, D, and F) hearts stained with terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL, red), anti-α-smooth muscle-specific actin (α-SMA, green), anti-Gr-1 or anti-von Willebrand factor (vWF) or anti-Mac-1 (blue, as indicated on individual images in C–F) are shown. Note differences in size bars.
miR-21 downregulation induces neutrophil apoptosis in metabolic syndrome.
Treatment of JCR rats with anti-miR-21 on day 5 RI induced apoptosis in JCR collaterals on day 6 RI and was accompanied by slight lumen expansion similar to that seen in normal SD rats (Fig. 3, C and D). To determine which cell types undergo apoptosis in response to anti-miR-21 treatment, we costained consecutive cardiac cross sections with α-SMA and TUNEL (to label VSMCs) (Fig. 3, C and D) or von Willebrand factor (to label ECs) (Fig. 3, E and F) or Gr-1 (to label neutrophils) (Fig. 3, C and D) or Mac-1 (CD11b/CD18) (to label monocytes) (Fig. 3, E and F) or F4/80 (to label macrophages) (data not shown) or CD3 (to label T cells) (data not shown) or CD23 (IgE receptor, to label mast cells) (data not shown). Whereas Gr-1-positive cells (neutrophils) are present in SD, JCR, and anti-miR-21-treated JCR rats on day 6 of RI, apoptosis of Gr-1-positive cells (Gr-1-positive and TUNEL-positive cells) is evident only in SD and anti-miR-21-treated JCR rats but not in untreated JCR animals (Fig. 3, C and D). Consequently, on day 9 of RI, Gr-1-positive cells persist in unremodeled collaterals of untreated JCR animals (and are still not apoptotic), but not in enlarged collaterals of SD and anti-miR-21-treated JCR rats (Fig. 3, C and D). The remaining TUNEL-positive, but Gr-1-negative, cells present in SD animals on day 6 RI are synthetic, proliferative VSMCs, as evident by α-SMA-positive staining (Fig. 3, C and D). We have recently demonstrated excessive miR-21-dependent accumulation of these synthetic VSMCs in the lumen and vascular wall of JCR collaterals (by nonmuscle myosin heavy chain-B-positive staining) (13). The current data suggest that apoptosis is also a mechanism partially responsible for VSMC clearance from the lumen of forming collaterals in normal but not in metabolic syndrome animals. Neither ECs nor monocytes underwent apoptosis in response to RI in the collaterals of either SD or JCR rats, nor was their apoptosis induced by anti-miR-21 treatment (Fig. 3, E and F).
Thus, in summary, TUNEL-positive staining mainly colocalized with Gr-1-positive staining, indicating that cells undergoing apoptosis on day 6 RI in normal animals are predominantly neutrophils (Fig. 3). Neutrophil apoptosis or apoptosis of any other cell types examined was absent in the collaterals of metabolic syndrome animals. This increased neutrophil survival in metabolic syndrome is miR-21 dependent since anti-miR-21 induced neutrophil apoptosis (to the exclusion of all other cell types) in JCR rats (Fig. 3). Flow cytometry results using Annexin V-labeling support this conclusion (Fig. 4, A and B).
Fig. 4.


SD and JCR rats were treated with anti-miR-21 or scrambled anti-miR on day 5 RI or with anti-CD11b/CD18 and CD44 blocking antibodies on days 6–9 RI as indicated and euthanized on days 6 (12 h post-blocking antibody administration) or 9 RI, or untreated and euthanized on days 0, 3, 6, and 9 of RI. A–D: flow cytometry was performed on CZ and NZ perfusates using Gr-1 (Ly6G) antibodies to label neutrophils, CD115 antibodies to label monocytes, and annexin V antibodies to label apoptotic cells. Representative analysis (A and B) and cumulative data (C and D) are shown. *P < 0.05 vs. sham; #P < 0.05 vs. WKY RI; $P < 0.05 vs. JCR RI. E and F: representative hematoxylin-and-eosin images (×100) of remodeling collateral arteries (left) and large preexisting arteries (right) in cardiac cross sections from unperfused (E) and perfused (F) JCR rat hearts on day 9 RI. White arrows point to neutrophils. Note differences in size bars.
miR-21-dependent CCG recovery is mainly due to miR-21-induced neutrophil apoptosis.
If neutrophil apoptosis was a major component of miR-21-dependent recovery of CCG in JCR rats, then prevention of neutrophil infiltration into the CZ should have an equivalent effect on CCG to that seen with anti-miR-21. Our results demonstrate that both anti-miR-21 and neutralizing antibodies against the major neutrophil/monocyte EC adhesion receptor CD11b/CD18 and CD44, an axillary adhesion receptor that binds to a variety of ligands, resulted in collateral vessel enlargement equivalent to that achieved with anti-miR-21 treatment (Fig. 4E) and equal restoration of CCG in JCR rats (∼50%); collateral-dependent to NZ flow ratio was 0.56 ± 0.05 (JCR RI + anti-miR-21) and 0.49 ± 0.05 (JCR + anti-CD11b/CD18 + anti-CD44) versus 0.16 ± 0.03 (JCR RI) versus 0.85 ± 0.05 (SD RI) (Fig. 5B). In response to either treatment, the increase in CZ blood flow was due to collateral growth (arteriogenesis) and not angiogenesis since the number of arteries per area increased in the CZ, but capillary number did not change (Fig. 5A). Although the anti-CD11b/CD18 and CD44 antibodies target both neutrophil and monocyte adhesion, at the time when they were delivered, monocyte numbers are already very low (Fig. 4, B–D); therefore, we trust that their effect on CCG is primarily due to inhibition of neutrophil adhesion. Blockade of the CD44, in addition to the major neutrophil and monocyte adhesion receptor CD11b/CD18, was necessary to sufficiently decrease neutrophil numbers in JCR rats [CD11b/CD18 blockade alone resulted in an ∼70% decrease in neutrophil numbers (data not shown)] (Fig. 4, B and D).
Fig. 5.

JCR rats were treated with anti-miR-21 or scrambled (scram) anti-miR on day 5 RI or anti-CD11b/CD18 and CD44 blocking antibodies on days 6–9 RI where indicated, and SD or JCR rats underwent 9 days of RI (n = 6). A: arteriolar [smooth muscle-myosin heavy chain (SM-MHC)- and proliferating cell nuclear antigen-positive vessels > 20 μM] and capillary (vessels < 20 μM) densities were determined in cardiac (CZ) cross sections on day 9 RI. B: coronary blood flow was measured in CZ and NZ using microspheres during LAD occlusion and is expressed as the ratio between CZ and NZ flows on day 9 of RI. *P < 0.05 vs. sham; #P < 0.05 vs. WKY RI; §P < 0.05 vs. JCR RI.
Among other ligands, CD44 binds a proinflammatory extracellular matrix protein osteopontin, which has been shown to prolong the life span of neutrophils in the periphery (1). Results in Fig. 6 demonstrate that osteopontin is increased in JCR rats at baseline (∼2-fold) and in response to RI (∼2-fold vs. day 0 RI), which might explain the necessity for additional blockade of the CD44 receptor in this study.
Fig. 6.
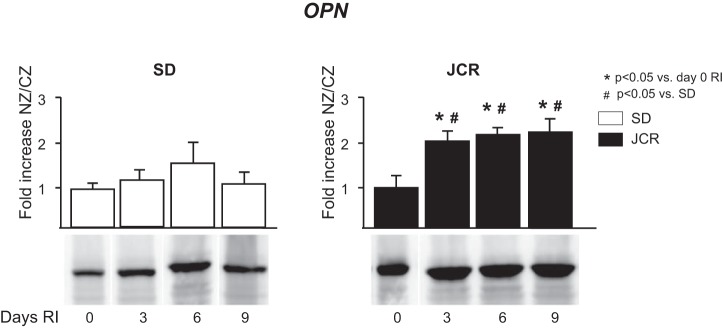
SD and JCR rats were euthanized on days 0, 3, 6, or 9 of RI (n = 6). Tissue samples were collected from NZ and CZ. Top: cumulative data, (n = 6). Bottom: representative Western blots from the CZ using anti-osteopontin (OPN) antibodies.
Effect of anti-miR-21 and blocking antibody treatment on inflammatory cell numbers in the CZ and the NZ was evaluated by flow cytometry. The anti-miR and blocking antibodies were administered to reduce neutrophil numbers to those observed at equivalent time points in normal (SD) animals, where their increase was early and transient (∼2-fold on day 3 RI) and in JCR rats at baseline (day 0 RI) (Fig. 4, A–D). Absence of neutrophils is also evident on large magnification (×100) hematoxylin-and-eosin images of anti-miR-21 and anti-CD11b/CD18 and anti-CD44-treated JCR collateral vessels on day 9 of RI versus untreated JCR rats (Fig. 4, E and F). Taken together, these results demonstrate that miR-21-dependent CCG recovery is mainly due to miR-21-induced neutrophil apoptosis since it equals the effect achieved by inhibition of neutrophil infiltration. Although the combination of blocking antibodies used prevents both neutrophil and monocyte adhesion, this is not a concern in this study because monocyte numbers at the time points at which the neutralizing antibodies were used (days 6–9 RI) are already low and were not further reduced by the blocking antibodies (Fig. 4).
Neutrophils inhibit CCG in metabolic syndrome by excessive reactive oxygen species production.
We have previously shown that excessive oxidative stress in the metabolic syndrome animals (ZOF and JCR rats) was detrimental to collateral development; reduction in reactive oxygen species (ROS) significantly improved CCG (12, 19, 20). However, the question of the cellular origin of those ROS remained unanswered. Neutrophils are capable of production of high amounts of ROS via their NAD(P)H oxidase. In this study, we conclusively demonstrate that neutrophils are a significant source of ROS during CCG in metabolic syndrome, since prevention of neutrophil adhesion with anti-CD11b/CD18 and anti-CD44 blocking antibodies significantly decreased ROS (superoxide) production in the CZ (Fig. 7). No significant changes were observed in the NZ in any treatment group at any time point. Treatment with anti-miR-21 also reduced superoxide production in metabolic syndrome animals to levels similar to those achieved by anti-CD11b/CD18 and anti-CD44 blocking antibodies (Fig. 7), which is not surprising since anti-miR-21 inhibited the prolonged inappropriate neutrophil survival in the late stages of CCG in metabolic syndrome (Fig. 4).
Fig. 7.
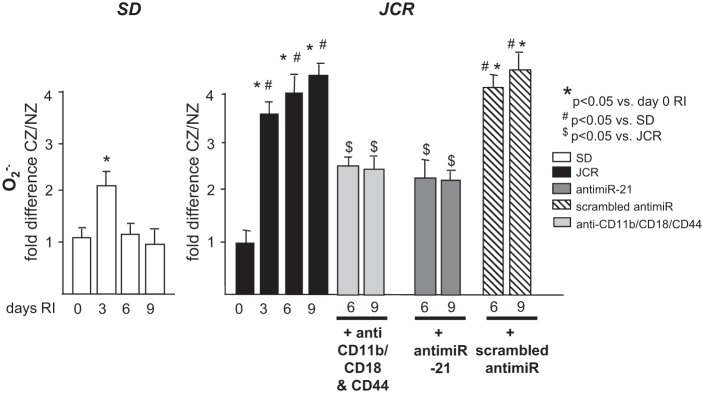
SD and JCR rats were treated with anti-miR-21 or scrambled anti-miR on day 5 RI or with anti-CD11b/CD18 and CD44 blocking antibodies on days 6–9 RI as indicated and euthanized on days 6 (12 h post-blocking antibody administration) or 9 RI, or untreated and euthanized on days 0, 3, 6, and 9 of RI. Superoxide (O2·−) levels were measured by X-band EPR in CZ and NZ and are shown as fold difference CZ/NZ. *P < 0.05 vs. sham; #P < 0.05 vs. WKY RI; $P < 0.05 vs. JCR RI.
DISCUSSION
The most important finding of this study is that neutrophil apoptosis is an integral component of normal collateral growth, which like collateral growth itself, is severely compromised in the metabolic syndrome. Maximal cytochrome-c release and caspases-3 and -9 activation observed in normal animals on day 6 RI are in concordance with our hypothesis that day 6 of RI represents the transition from inward to outward remodeling in the rat and that apoptosis of certain cells is a fundamental component of this transition. Further evidence from this study identifies the majority of neointimal apoptotic cells at the transition point from inward to outward remodeling in normal animals to be neutrophils. We also demonstrate that whereas in the normal animals both monocytes and neutrophils comprise the inflammatory cell population, which increases early and transiently (day 3 RI) during CCG, in agreement with multiple studies [reviewed in Meisner and Price (17)], in metabolic syndrome the infiltrating inflammatory cells are predominantly neutrophils with very little monocytes. Significantly, it is the neutrophils that accumulate to levels much higher than in normal animals and remain in the area long past the window observed for normal animals, which prevent normal collateral remodeling (expansion) in the late stages of growth.
The metabolic syndrome is a chronic proinflammatory state characterized by persistent endothelial dysfunction and upregulation of adhesion molecules, including P-selectin and ICAM-1, which promote neutrophil adhesion (6, 11, 31). Our results also demonstrate that osteopontin expression is increased in the JCR rats. Osteopontin is a glycoprotein secreted predominantly by synthetic VSMCs and activated macrophages. In JCR rats, its elevated expression correlates with persistent synthetic VSMC phenotype (14) and VSMC proliferation (13), which we have recently demonstrated as another important contributor to disrupted CCG in metabolic syndrome. The population of synthetic VSMCs, the proliferation of which was also decreased by anti-miR-21 (7), are a likely source of osteopontin and may thus be an indirect regulator of inappropriate miR-21-dependent neutrophil survival in the metabolic syndrome. Osteopontin binds to several integrins, including the α4β1-integrin (1), expressed on monocytes/macrophages, neutrophils, T cells, and mast cells (17). Binding to osteopontin prolongs inflammatory cell, especially neutrophil, survival from hours to days and induces neutrophil adhesion and migration (26). We were not able to improve CCG in the JCR rats by blocking neutrophil adhesion to endothelial ICAM-1 through the Mac-1 (CD11b/CD18) receptor (10) alone. Instead, joint inhibition of the Mac-1 receptor and the CD44 receptor was required to significantly improve CCG in JCR rats, indicating the importance of increased osteopontin deposition in CCG impairment in the metabolic syndrome.
Osteopontin was also associated with very high levels of ROS in the metabolic syndrome and similar pathologies (25, 28). Neutrophils are major producers of ROS in the vasculature via their NAD(P)H oxidases. We have previously established that high ROS levels in the metabolic syndrome play a major role in inhibiting CCG, since lowering ROS significantly improved CCG (19, 20). The source of this excessive ROS generation in the metabolic syndrome has never been established. Results in the present study indicate that these long-lived neutrophils contribute very significantly (∼50%) to ROS generated by RI.
Whereas neutrophils and other neointimal cells, which block the lumen, fail to undergo apoptosis in metabolic syndrome, ECs of large, preexisting vessels undergo apoptosis at increasing rates. This is consistent with recent studies that report increased EC and endothelial progenitor cell apoptosis in type II diabetes and metabolic syndrome (Chen CH, 2012 No. 125; Kuliszewski MA, 2013 No. 126). Taken together, these findings suggest a scenario in which aberrant prolonged survival of neutrophils in combination with excessive VSMC proliferation (13) create an environment that is not conducive to collateral expansion. It is currently thought that as collateral remodeling progresses and HIF-1α levels decline as ischemia diminishes, increasing NO induced by increasing shear stress carries on continuing remodeling (4). Under certain conditions, NO has been shown to stimulate the mitochondrial apoptotic pathway by directly disrupting the mitochondrial membrane potential, leading to cytochrome-c release and subsequent caspase-9 activation (2) through unknown mechanisms. NO also prevents VSMC proliferation, migration, and conversion to the synthetic phenotype via the protein kinase G pathway. On the other hand, NO promotes EC survival during angiogenesis (16). Endothelial dysfunction and decreased NO levels are a hallmark of metabolic syndrome and are in part due to elevated ROS. Combined with absence of increased shear stress because of luminal occlusion in this scenario, this could provide the low NO environment, which favors apoptosis of ECs and proliferation of VSMCs and survival of inflammatory cells. In our study, similar luminal accumulation of cells was observed in both perfused and unperfused hearts. We believe that this supports the initial accumulation of synthetic VSMCs, mediated by their excessive proliferation, and subsequent production of proinflammatory extracellular matrix, including osteopontin, results in accumulation of leukocytes, specifically neutrophils. Together, these processes result in decreased flow that further exacerbates neutrophil accumulation and prevents collateral enlargement.
In our recent study, we have demonstrated that downregulation of miR-21 significantly improves CCG in JCR rats (13). This correlated with decreased VSMC proliferation (7); however, it was clear that decreased VSMC proliferation alone could not account for the extent of CCG recovery. Results in this study demonstrate that blockade of neutrophil infiltration improves CCG to the same extent as miR-21 downregulation, indicating that abnormal neutrophil survival at the transition from early to late phase of collateral remodeling is the principal mechanism by which miR-21 inhibits CCG in the metabolic syndrome. miR-21 is now well accepted as a major prosurvival and proproliferative miR. Specifically related to this study, miR-21 has been shown to upregulate Bcl-2 expression (30). Our results clearly demonstrate upregulation of Bcl-2 expression during the late days of RI in JCR rats, which was blocked by miR-21 downregulation (anti-miR-21). This resulted in tilting the balance during the late days of RI in JCR rats in favor of forming antiapoptotic Bcl-2/Bax dimers and preventing cytochrome-c release from the mitochondria and caspase-9 activation, which was also corrected by miR-21 downregulation.
miR-21 is ubiquitously produced by all cell types in the heart and the blood vessels, and our anti-miR-21 treatment was not cell specific; therefore, the reason for its apparently specific effect on neutrophil apoptosis is not known. It may have been preferentially taken up by neointimal cells. While this is certainly a direction for future investigation, from the perspective of potential therapeutic potential for anti-miR-21 and/or induction of neutrophil apoptosis to promote CCG in metabolic syndrome, this point may be less important. Another limitation inherent to the rodent models of CCG is that we were not able to distinguish collateral arteries from total coronary arteries within the CZ and NZ, respectively, by flow cytometry. Thus the collateral-specific analysis of luminal cell content in this study is limited to qualitative (vs. quantitative) analysis achieved via immunohistochemistry.
In conclusion, this study establishes a completely novel concept with respect to regulation of collateral growth. Contrary to the assumption that impaired collateral development in the metabolic syndrome is a consequence of global impairment in cell survival, our data demonstrate that compromised CCG in the metabolic syndrome is in large part a consequence of inadequate neutrophil apoptosis. Furthermore, our findings point to fundamentally different inflammatory mechanisms governing the response to RI in normal versus metabolic syndrome myocardium. Finally, this study emphasizes the critical importance of understanding the temporal regulation of miR expression (miR-21) to design of successful therapeutic approaches.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-093052.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.H., R.T., B.H., R.J., J.C., E.S., and P.R. performed experiments; R.H., R.T., B.H., R.J., E.S., and P.R. analyzed data; R.H., R.T., and P.R. interpreted results of experiments; R.H., R.T., B.H., R.J., J.C., E.S., R.B., S.D.P., and P.R. approved final version of manuscript; R.T., R.B., S.D.P., and P.R. conception and design of research; R.T., R.J., and P.R. prepared figures; R.T. and P.R. drafted manuscript; P.R. edited and revised manuscript.
REFERENCES
- 1.Barry ST, Ludbrook SB, Murrison E. Analysis of the a4b1 integrin-osteopontin interaction. Exp Cell Res 258: 2–51, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Boyd CS, Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol Chem 383: 411–423, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Cai W, Schaper W. Mechanisms of arteriogenesis. Acta Biochim Biophys Sin (Shanghai) 40: 681–692, 2008. [PubMed] [Google Scholar]
- 4.Chilian WM, Penn MS, Pung YF, Dong F, Mayorga M, Ohanyan V, Logan S, Yin L. Coronary collateral growth—back to the future. J Mol Cell Cardiol 52: 905–911, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choy JS, Kassab GS. Wall thickness of coronary vessels varies transmurally in the LV but not the RV: implications for local stress distribution. Am J Physiol Heart Circ Physiol 297: H750–H758, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag 1: 55–71, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodd T, Jadhav R, Wiggins L, Stewart J, Smith E, Russell JC, Rocic P. MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J Mol Cell Cardiol 51: 1015–1025, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dodd T, Wiggins L, Hutcheson R, Smith E, Musiyenko A, Hysell B, Russell JC, Rocic P. Impaired coronary collateral growth in the metabolic syndrome is in part mediated by matrix metalloproteinase 12-dependent production of endostatin and angiostatin. Arterioscler Thromb Vasc Biol 33: 1339–1349, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Focardi M, Dick GM, Picchi A, Zhang C, Chilian WM. Restoration of coronary endothelial function in obese Zucker rats by a low-carbohydrate diet. Am J Physiol Heart Circ Physiol 292: H2093–H2099, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Futosi K, Fodor S, MA. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17: 638–650, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanzu FA, Palomo M, Kalko SG, Parrizas M, Garaulet M, Escolar G, Gomis R, Diaz-Ricart M. Translational evidence of endothelial damage in obese individuals: inflammatory and prothrombotic responses. J Thromb Haemost 9: 1236–1245, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Hattan N, Chilian WM, Park F, Rocic P. Restoration of coronary collateral growth in the Zucker obese rat: impact of VEGF and ecSOD. Basic Res Cardiol 102: 217–223, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Hutcheson R, Chaplin J, Hutcheson B, Borthwick F, Proctor S, Gebb S, Jadhav R, Smith E, Jadhav R, Rocic P. Mir-21 normalizes vascular smooth muscle proliferation and improves coronary collateral growth in metabolic syndrome. FASEB J Jun 5. pii: fj.14-251223, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutcheson R, Terry R, Chaplin J, Smith E, Musiyenko A, Russell JC, Lincoln T, Rocic P. MicroRNA-145 restores contractile vascular smooth muscle phenotype and coronary collateral growth in the metabolic syndrome. Arterioscler Thromb Vasc Biol 33: 727–736, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jadhav R, Dodd T, Smith E, Bailey E, Delucia AL, Russell JC, Madison R, Potter B, Walsh K, Jo H, Rocic P. Angiotensin type I receptor blockade in conjunction with enhanced Akt activation restores coronary collateral growth in the metabolic syndrome. Am J Physiol Heart Circ Physiol 300: H1938–H1949, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci 120: 492–501, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Meisner JK, Price RJ. Spatial and temporal coordination of bone marrow-derived cell activity during arteriogenesis: regulation of the endogenous response and therapeutic implications. Microcirculation 17: 583–599, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 43: 371–378, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed R, Kolz C, Potter B, Rocic P. The mechanistic basis for the disparate effects of angiotensin II on coronary collateral growth. Arterioscler Thromb Vasc Biol 28: 61–67, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Reed R, Potter B, Smith E, Jadhav R, Villalta P, Jo H, Rocic P. Redox-sensitive Akt and Src regulate coronary collateral growth in metabolic syndrome. Am J Physiol Heart Circ Physiol 296: H1811–H1821, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocic P, Griffin TM, McRae CN, Lucchesi PA. Altered PYK2 phosphorylation by ANG II in hypertensive vascular smooth muscle. Am J Physiol Heart Circ Physiol 282: H457–H465, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Rocic P, Lucchesi PA. Down-regulation by antisense oligonucleotides establishes a role for the proline-rich tyrosine kinase PYK2 in angiotensin II-induced signaling in vascular smooth muscle. J Biol Chem 276: 21902–21906, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Russell JC, Graham SE, Richardson M. Cardiovascular disease in the JCR:LA-cp rat. Mol Cell Biochem 188: 113–126, 1998. [PubMed] [Google Scholar]
- 24.Sabatel C, Malvaux L, Bovy N, Deroanne C, Lambert V, Gonzalez ML, Colige A, Rakic JM, Noël A, Martial JA, Struman I. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One 6: e16979, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.San Martín A, Du P, Dikalova A, Lassègue B, Aleman M, Góngora MC, Brown K, Joseph G, Harrison DG, Taylor WR, Jo H, Griendling KK. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in type 2 diabetes. Am J Physiol Heart Circ Physiol 292: H2073–H2082, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol 27: 2302–2309, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Schneider D, Absher P, Neimane D, Russell JC, Sobel B. Fibrinolysis and atherogenesis in the jcr:La-cp rat in relation to insulin and triglyceride concentrations in blood. Diabetologia 41: 141–147, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Shannahan JH, Alzate O, Winnik WM, Andrews D, Schladweiler MC, Ghio AJ, Gavett SH, Kodavanti UP. Acute phase response, inflammation and metabolic syndrome biomarkers of libby asbestos exposure. Toxicol Appl Pharmacol 260: 105–114, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Shroff EH, Snyder C, Chandel NS. Bcl-2 family members regulate anoxia-induced cell death. Antiox Redox Signal 9: 1405–1409, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Silvestri P, Di Russo C, Rigattieri S, Fedele S, Todaro D, Ferraiuolo G, Altamura G, Loschiavo P. Micrornas and ischemic heart disease: towards a better comprehension of pathogenesis, new diagnostic tools and new therapeutic targets. Recent Pat Cardiovasc Drug Discov 4: 109–118, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Park Y, Zhang H, Gao X, Wilson E, Zimmer W, Abbott L, Zhang C. Role of MCP-1 in tumor necrosis factor-α-induced endothelial dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297: H1208–H1216, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]