

Abstract
Chronic ethanol consumption has been shown to significantly decrease hepatic glycogen content; however, the mechanisms responsible for this adverse metabolic effect are unknown. In this study, we examined the impact chronic ethanol consumption has on time-of-day-dependent oscillations (rhythms) in glycogen metabolism processes in the liver. For this, male C57BL/6J mice were fed either a control or ethanol-containing liquid diet for 5 wk, and livers were collected every 4 h for 24 h and analyzed for changes in various genes and proteins involved in hepatic glycogen metabolism. Glycogen displayed a robust diurnal rhythm in the livers of mice fed the control diet, with the peak occurring during the active (dark) period of the day. The diurnal glycogen rhythm was significantly altered in livers of ethanol-fed mice, with the glycogen peak shifted into the inactive (light) period and the overall content of glycogen decreased compared with controls. Chronic ethanol consumption further disrupted diurnal rhythms in gene expression (glycogen synthase 1 and 2, glycogenin, glucokinase, protein targeting to glycogen, and pyruvate kinase), total and phosphorylated glycogen synthase protein, and enzyme activities of glycogen synthase and glycogen phosphorylase, the rate-limiting enzymes of glycogen metabolism. In summary, these results show for the first time that chronic ethanol consumption disrupts diurnal rhythms in hepatic glycogen metabolism at the gene and protein level. Chronic ethanol-induced disruption in these daily rhythms likely contributes to glycogen depletion and disruption of hepatic energy homeostasis, a recognized risk factor in the etiology of alcoholic liver disease.
Keywords: liver, ethanol, diurnal, glycogen, glycogen synthase
chronic and excessive alcohol (ethanol) consumption causes a variety of serious liver diseases, including steatosis (i.e., fatty liver), steatohepatitis (i.e., steatosis with inflammation), fibrosis, cirrhosis, and hepatocellular carcinoma (6). The etiology of alcoholic liver disease (ALD) is complex and multifactorial, involving a combination of numerous molecular and cellular alterations, including lipid accumulation in hepatocytes, oxidative and nitrative stress, lipid peroxidation, acetaldehyde toxicity, inflammation, and fibrogenesis (40). Moreover, chronic ethanol consumption significantly impairs hepatic energy metabolism by decreasing mitochondrial and glycolytic ATP production, leading to hepatocyte necrosis (2, 46, 55). It is believed that these perturbations, as well as others, contribute to the initial development and progression of ALD in the chronic alcohol consumer (23).
Although the liver has regenerative capacity (25, 36), chronic ethanol-dependent decreases in hepatic energy levels will ultimately impede the liver's ability to repair, further increasing ethanol-induced liver damage (8, 55). Within this context, chronic ethanol-induced changes in liver glucose and glycogen metabolism will contribute to deficits in hepatic energy status and decreased hepatocyte viability (8). Notably, ethanol-induced decreases in hepatic glycolytic activity are paralleled by significant decreases in liver glycogen levels (47). Several studies have shown that both acute and chronic alcohol consumption reduce hepatic glycogen levels (28, 46, 47, 50, 52, 53). However, the mechanisms underlying these ethanol-mediated effects on liver glycogen metabolism remain poorly understood.
Studies by Van Horn et al. (47) suggested that chronic ethanol consumption lowered glycogen content in livers of male rats by decreasing the total protein level and activity of glycogen synthase (GS), the rate-limiting enzyme in glycogen synthesis. It is important to point out that these experimental results were measured in livers and hepatocytes collected at only a single time point during the 24-h day. Many metabolic processes, including hepatic glycogen metabolism, are dynamic and exhibit time-of-day rhythms (3). We therefore hypothesized that chronic ethanol consumption impairs time-of-day-dependent oscillations in the elements that regulate hepatic glycogen metabolism.
In the present study, mice were fed an ethanol-containing or control liquid diet for 5 wk to induce steatosis and glycogen depletion. Livers were collected every 4 h for a 24-h period to assess time-of-day and ethanol-dependent alterations in glycogen levels, as well as the expression and activities of enzymes involved in glycogen turnover. Our results show that chronic ethanol consumption significantly altered diurnal rhythms in liver glycogen content, and rhythms in key genes and proteins involved in glycogen turnover, glycogen particle composition, and glucose metabolism. Interestingly, the effect chronic ethanol consumption had on hepatic glycogen content was not readily explained by perturbations in assayable glycogen synthase and phosphorylase activities, suggesting primary influences on alternative mechanisms. In summary, these results demonstrate for the first time that chronic ethanol consumption significantly disrupts diurnal regulation of glycogen turnover in the liver. We propose that these perturbations render the liver unable to maintain its glycogen stores, an important energy source needed to preserve hepatocyte viability.
MATERIALS AND METHODS
Mice and feeding regimen.
Eight-week-old male C57BL/6J mice were purchased from Jackson Laboratories and allowed to acclimate for at least 1 wk before they were divided into control and ethanol feeding groups. For the feeding studies, each ethanol-fed mouse was weight-matched to a control-fed mouse partner to establish the pair-feeding regimen (13). Mice were singly housed in standard husbandry boxes with bedding and kept in a temperature- and humidity-controlled environment under a 12:12-h light-dark cycle. Mice were pair-fed isocaloric Lieber-DeCarli control and ethanol liquid diets (Bioserv) for 5 wk (13). Mice were acclimated to the ethanol-containing diet by gradually increasing the percentage of ethanol content from 0 to 4% (wt/vol) over an 8-day period, after which the concentration of ethanol in the diet was kept at 4% (wt/vol) until the end of the study. The ethanol diet (Bioserv, F1258SP) contained 15.1% calories from protein, 35.9% calories from fat, 20.6% calories from carbohydrate (maltose-dextrins), and 28.4% calories from ethanol. The pair-fed control mice received an identical diet (Bioserv, F1259SP) except ethanol calories were replaced isocalorically with maltose-dextrins. At the end of the 5-wk feeding period, livers were collected at Zeitgeber times (ZT) 3, 7, 11, 15, 19, and 23 (ZT 0 = lights on and ZT 12 = lights off). Liver tissue used for determining glycogen content, measuring enzyme activities, and performing Western blots was quickly frozen in liquid N2 and stored at −80°C until analyses were done. Liver tissue used for assessing gene expression was preserved in RNAlater (Life Technologies) to protect RNA from degradation and stored at −20°C until used for RNA isolation. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Alabama at Birmingham and were in compliance with National Institutes of Health animal use guidelines.
Hepatic glycogen content.
Homogenates (20 mg/ml in 1.0 M KOH) were prepared from frozen liver tissue and heated for 20 min at 70°C to solubilize tissues. The digested tissues (200 μl) were treated with glacial acetic acid (34 μl) to adjust pH and incubated overnight with amyloglucosidase (Roche) at 37°C to hydrolyze glycogen to free glucose (9). The concentration of free glucose generated in samples was determined using the hexokinase/glucose-6-phosphate dehydrogenase method, and glycogen standards were treated in the same manner as liver samples (4, 11). Results are expressed as milligram glycogen per gram liver weight.
Glycogen synthase and phosphorylase activity assays.
Glycogen synthase (GS) activity was assayed at 30°C based on the incorporation of radiolabeled glucose from UDP-[U-14C]glucose into glycogen (44). Liver homogenates were prepared as described in Ref. 47. Briefly, frozen livers were homogenized at a concentration of 100 mg/ml in buffer A [0.34 M sucrose, 1 mM EDTA, 5 mM Tris-HCl (pH 7.5), 40 μg/ml PMSF, 5 μl/ml leupeptin, 5 μg/ml aprotinin, and 7 μg/ml pepstatin]. Aliquots of homogenate (600 μl) were added to 1.0 ml of buffer B [50 mM glycylglycine (pH 7.4), 20 mM EDTA, 100 mM NaF, 5 mg/ml glycogen, 0.625 mM PMSF, and 0.125% 2-mercaptoethanol], and sonicated for 45 s. GS activity was measured by adding 30 μl of homogenate to 60 μl of assay buffer [50 mM Tris (pH 7.8), 20 mM EDTA, 87.5 mM KF, 10 mg/ml glycogen, 1 mM UDP-glucose, and 10 μl/ml radiolabeled UDP-glucose (American Radiolabelled Chemicals) in the presence or absence of 10 mM glucose-6-phosphate]. Enzyme activity is expressed as nanomoles glucosyl units incorporated into glycogen per minute per milligram liver protein.
Glycogen phosphorylase (GP) activity was assayed at 30°C based on the incorporation of UDP-[U-14C]glucose-1-phosphate into glycogen as described previously (16). For this assay, frozen liver tissues were homogenized in buffer A (50 mM KF, 10 mM EDTA, 10% vol/vol glycerol; pH 7.0) at a concentration of 77 mg/ml. Liver homogenates were then centrifuged at 4°C for 10 min at 12,000 g. A 200-μl sample of the supernatant was added to 400 μl buffer B containing 50 mM MES, 50 mM KF, and 60 mM β-ME at pH 6.1. For measurement of GP activity, 30 μl of the diluted liver homogenate was added to 60 μl assay buffer C [200 mM KF, 80 mM cold glucose-1-phosphate, 1% glycogen, and 1 μCi/ml UDP-[U-14C]glucose-1-phosphate (American Radiolabelled Chemicals), pH 6.1] in the presence and absence of 3 mM 5′-AMP (54). The enzyme activity was expressed as micromoles glucosyl units incorporated into glycogen per minute per gram liver weight.
Western blot analyses.
Western blots were performed according to standard procedures (45) using mouse anti-phospho-GS (Ser641), anti-GS, anti-phospho-AKT (Ser473), anti-AKT, anti-phospho-GSK3β (Ser9), and anti-GSK3β (Cell Signaling Technology). Samples were prepared from frozen liver using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Cytosolic protein was separated on a 7.5% or 4–20% Criterion TGX Precast Gel (Bio-Rad Laboratories) via electrophoresis, transferred to nitrocellulose membranes, and incubated with primary antibodies overnight at 4°C. Protein detection was achieved by using appropriate secondary antibodies and enhanced chemiluminescence. Membranes were stripped and reprobed with appropriate loading control antibodies, anti-GAPDH (Cell Signaling Technology,) and anti-β-actin (Sigma-Aldrich). The intensities of immunoreactive protein bands were quantified using Quantity One software (Bio-Rad Laboratories) and expressed as total densitometry units.
RNA isolation and gene expression.
Total RNA was isolated from liver tissues preserved in RNAlater solution (Life Technologies) using Tri Reagent (Sigma-Aldrich), following the manufacturer's instructions, and DNase treated with DNA-free DNase treatment kit (Life Technologies). A NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific) was used to quantify RNA. cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and qPCR was performed with Taqman Gene Expression assays containing specific primers and probes using an Applied Biosystems 7900 HT instrument (Applied Biosystems). The relative levels of target gene PCR product were normalized to those of the housekeeping gene GAPDH. Relative gene expression was determined using the delta-delta Ct method (21). Gene expression data were expressed as fold change set relative to control trough.
Statistical analysis.
Two-factor analysis of variance (ANOVA) was performed to determine statistical significance of the main effects of time and diet, as well as an interaction (time × diet) on experimental measurements. In addition, cosinor analysis was performed to determine whether experimental measures where rhythmic during the course of the day, and to obtain the key chronobiological parameters of mesor, amplitude, and acrophase. For this, data were fitted to a cosine wave equation, f(t) = mesor + amplitude × cos[(2πt/T) + acrophase], in SPSS (IBM) using a nonlinear regression module (29), where the mesor (midline estimating statistic of rhythm) = mean of the oscillation; amplitude = 1/2 the distance between the peak and the trough; t = time point (ZT 3, 7, 11, 15, 19, or 23); T = the period (fixed to 24 h); and acrophase = the ZT time of the cosine maximum. Student's t-test was used to compare the parameter estimates between control and ethanol-fed mice. Data are expressed as means ± SE with n = 4–6 livers per diet group per time point. Statistical significance was set at P ≤ 0.05.
RESULTS
Ethanol consumption decreases liver glycogen content and alters its diurnal rhythm.
We investigated the effect of chronic ethanol consumption on time-of-day-dependent oscillations in hepatic glycogen levels. For these studies, male C57BL/6J mice were pair-fed control and ethanol diets for 5 wk and livers were collected every 4 h at ZT 3, 7, 11, 15, 19, and 23 (ZT 0–12 = lights on, and ZT 12–24 = lights off). Two-factor ANOVA showed a significant main effect of time (P = 0.016) and diet (P < 0.001) on hepatic glycogen levels, but no significant interaction (P = 0.092) (Fig. 1B). Glycogen content demonstrated a diurnal rhythm in livers of both control (R2 = 0.38, P = 0.006) and ethanol-fed (R2 = 0.36, P = 0.009) mice; however, rhythms were significantly different between the two groups (Fig. 1, A and C). For example, liver glycogen from control-fed mice displayed a strong diurnal oscillation that peaked at the end of the active (dark) phase (ZT 20.76 ± 0.22; Fig. 1, A and C). In contrast, the glycogen peak was significantly phase shifted to ZT 1.21 ± 0.23 (phase difference between groups, P = 0.008) in livers of ethanol-fed mice (Fig. 1, A and C). Chronic ethanol consumption also significantly decreased the mean (mesor) levels of hepatic glycogen by 53% (P < 0.001) such that glycogen levels remained significantly lower throughout the entire 24-h period in livers of ethanol-fed mice compared with livers of control-fed mice (Fig. 1, A and C).
Fig. 1.
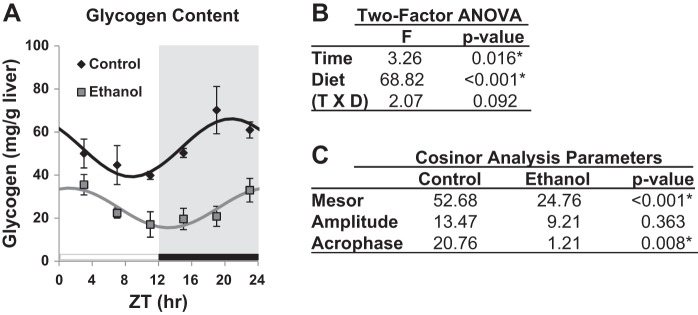
Diurnal oscillation in liver glycogen content. A: mice were fed a control (black symbols) or ethanol-containing (gray symbols) diet for 5 wk, and livers were collected every 4 h at ZT 3, 7, 11, 15, 19, and 23 to determine liver glycogen content. Mice were housed under a standard 12:12-h light-dark cycle where ZT 0 equals lights on (white bar) and ZT 12 equals lights off (black bar and gray shading). Data were fitted to a cosine curve and expressed as means ± SE for n = 4 mice per group per time point. Results are from 2-factor ANOVA (B) and cosinor analysis (C).
Ethanol consumption alters diurnal rhythms in liver glycogen synthase.
In an attempt to determine the mechanism by which chronic ethanol consumption influences time-of-day-dependent oscillations in hepatic glycogen levels, we investigated glycogen synthase (GS), the rate-limiting enzyme involved in glycogen synthesis. We first measured 24 h expression patterns of several key glycogen metabolism genes. Mammals have two GS isoforms, the liver-specific isoform encoded by the GYS2 gene and the more ubiquitously expressed muscle isoform encoded by the GYS1 gene, which is also highly expressed in liver (38). Two-factor ANOVA of GYS1 gene expression showed significant main effects of time (P = 0.003), diet (P < 0.001), and interaction (P = 0.002) in the liver (Table 1). GYS1 displayed rhythmic expression peaking at ZT 3.73 ± 0.18 in livers of control mice. This rhythm was significantly decreased (45% and 58% decrease in mesor and amplitude, respectively) and phase-shifted (ZT 17.44 ± 0.29) by ethanol feeding (Fig. 2A and Table 2). GYS2 displayed a robust diurnal rhythm peaking at ZT 9.31 ± 0.08 in the liver of control-fed mice (Fig. 2B and Table 2). Chronic ethanol consumption significantly decreased the amplitude (50% decrease) and mesor (27% decrease) of GYS2 rhythmic expression (Table 2). Two-factor ANOVA indicated significant time (P < 0.001) and diet (P = 0.001) effects on GYS2 gene expression (Table 1).
Table 1.
Two-factor ANOVA of gene expression by time and diet
| Time |
Diet |
Interaction |
||||
|---|---|---|---|---|---|---|
| Gene | F | P value | F | P value | F | P value |
| GYS1 | 4.32 | 0.003* | 44.91 | <0.001* | 4.51 | 0.002* |
| GYS2 | 13.22 | <0.001* | 12.23 | 0.001* | 2.13 | 0.078 |
| GSK3β | 3.26 | 0.013* | 14.78 | <0.001* | 3.53 | 0.009* |
| AKT1 | 0.74 | 0.599 | 2.02 | 0.161 | 0.75 | 0.589 |
| AKT2 | 0.36 | 0.874 | 4.71 | 0.035* | 0.56 | 0.729 |
| PYGL | 0.52 | 0.756 | 4.74 | 0.034* | 0.32 | 0.899 |
| GYG | 5.98 | <0.001* | 0.18 | 0.673 | 2.54 | 0.041* |
| PPP1R3C | 23.14 | <0.001* | 4.33 | 0.043* | 0.26 | 0.933 |
| GLUT1 | 8.92 | <0.001* | 0.13 | 0.723 | 2.50 | 0.043* |
| GLUT2 | 5.88 | <0.001* | 1.11 | 0.298 | 0.75 | 0.589 |
| GCK | 7.44 | <0.001* | 16.68 | <0.001* | 3.83 | 0.005* |
| GCKR | 0.48 | 0.788 | 0.49 | 0.488 | 0.06 | 0.997 |
| PKLR | 4.62 | 0.002* | 0.53 | 0.470 | 1.21 | 0.320 |
Statistical significance, P ≤ 0.05.
Fig. 2.

Diurnal oscillation in liver glycogen synthase activity and related genes and proteins. Gene expression of glycogen synthase 1 (GYS1) (A), glycogen synthase 2 (GYS2) (B), and glycogen synthase kinase 3 beta (GSK3β) (C) was determined in livers from control (black symbols) and ethanol-fed (gray symbols) mice at ZT 3, 7, 11, 15, 19, and 23. Expression was normalized to GAPDH mRNA level and displayed as a fold change from control trough. Western blot analyses of GS1 (D), GS2 (E), phospho-GS1 (F), phospho-GS2 (G), phospho-AKT (H), and phospho-GSK3β (I) were examined. Data are from densitometry analysis with results fitted to a cosine curve. J: representative Western blots of samples from livers of control and ethanol-fed mice. Samples were loaded in duplicate across the gel. Blots are shown for total and phosphorylated proteins with loading controls (β-actin or GAPDH) provided for each. There were no significant effects of time for loading controls. There was a small effect of ethanol (a 14% decrease from control) for GAPDH on total AKT blots. Hepatic GS activity was measured in the absence (K) and presence (L) of 10 mM glucose-6-phosphate (G6P). Mice were housed under a standard 12:12-h light-dark cycle where ZT 0 equals lights on (white bar) and ZT 12 equals lights off (black bar and gray shading). Data are expressed as means ± SE for n = 4–6 mice per group per time point. Solid lines indicate a significant cosine curve fit, whereas dashed lines indicate a nonsignificant fit.
Table 2.
Cosinor analysis of glycogen and related metabolic gene expression in the livers of control and ethanol-fed mice
| Rhythmicity |
Cosinor Analysis Parameters |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Diet | R2 | P value | Mesor | P value | Amplitude | P value | Acrophase | P value |
| GYS1 | Control | 0.31 | 0.005* | 1.37 ± 0.02 | <0.001† | 0.45 ± 0.02 | 0.095 | 3.73 ± 0.18 | <0.001† |
| Ethanol | 0.22 | 0.049* | 0.75 ± 0.01 | 0.19 ± 0.01 | 17.44 ± 0.29 | ||||
| GYS2 | Control | 0.73 | <0.001* | 2.28 ± 0.02 | 0.002† | 1.38 ± 0.01 | 0.012† | 9.31 ± 0.08 | 0.536 |
| Ethanol | 0.31 | 0.010* | 1.66 ± 0.03 | 0.69 ± 0.04 | 8.58 ± 0.21 | ||||
| GSK3β | Control | 0.08 | 0.291 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.45 | 0.001* | 1.01 ± 0.01 | 0.18 ± 0.01 | 10.22 ± 0.16 | ||||
| AKT1 | Control | 0.04 | 0.527 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.02 | 0.795 | NA | NA | NA | ||||
| AKT2 | Control | 0.00 | 0.924 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.11 | 0.249 | NA | NA | NA | ||||
| PYGL | Control | 0.09 | 0.214 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.01 | 0.876 | NA | NA | NA | ||||
| GYG | Control | 0.54 | <0.001* | 1.35 ± 0.01 | NA | 0.41 ± 0.01 | NA | 4.42 ± 0.12 | NA |
| Ethanol | 0.11 | 0.225 | NA | NA | NA | ||||
| PPP1R3C | Control | 0.66 | <0.001* | 2.91 ± 0.03 | 0.049† | 2.28 ± 0.05 | 0.363 | 19.72 ± 0.08 | 0.831 |
| Ethanol | 0.71 | <0.001* | 2.35 ± 0.03 | 1.92 ± 0.05 | 19.87 ± 0.09 | ||||
| GLUT1 | Control | 0.15 | 0.082 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.66 | <0.001* | 1.28 ± 0.01 | 0.47 ± 0.01 | 15.73 ± 0.12 | ||||
| GLUT2 | Control | 0.46 | <0.001* | 1.60 ± 0.01 | 0.292 | 0.63 ± 0.02 | 0.066 | 9.05 ± 0.12 | 0.731 |
| Ethanol | 0.22 | 0.048* | 1.48 ± 0.02 | 0.31 ± 0.02 | 8.51 ± 0.27 | ||||
| GCK | Control | 0.33 | 0.001* | 1.61 ± 0.02 | <0.001† | 0.78 ± 0.03 | <0.001† | 17.38 ± 0.16 | <0.001† |
| Ethanol | 0.39 | 0.002* | 1.01 ± 0.01 | 0.43 ± 0.02 | 15.06 ± 0.19 | ||||
| GCKR | Control | 0.01 | 0.810 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.02 | 0.748 | NA | NA | NA | ||||
| PKLR | Control | 0.53 | <0.001* | 1.56 ± 0.01 | NA | 0.66 ± 0.02 | NA | 19.44 ± 0.11 | NA |
| Ethanol | 0.12 | 0.193 | NA | NA | NA | ||||
Values are means ± SE.
Statistical significance, P ≤ 0.05.
Statistically significant difference between control and ethanol, P ≤ 0.05. The R2 value represents the percent variance accounted for by a 24-h rhythm as determined by cosinor analysis.
Glycogen synthase kinase 3 (GSK3) and AKT (i.e., protein kinase B) are well-studied signaling components in the glycogen synthesis pathway. Activated phospho-AKT phosphorylates and inhibits GSK3β activity, preventing the phosphorylation and inactivation of GS by GSK3β (39). Although GSK3β gene expression showed no significant diurnal rhythm in livers of control-fed mice, ethanol consumption induced a diurnal rhythm with a peak at ZT 10.22 ± 0.16 (Fig. 2C and Table 2). Two-factor ANOVA showed a significant main effect of diet, time, and interaction for GSK3β (Table 1). We saw no significant diurnal rhythmic expression for AKT1 and AKT2 in livers of mice fed either the ethanol or control diet (Table 2). However, two-factor ANOVA revealed a significant main effect of diet for AKT2 (Table 1) due to ethanol-dependent decrease in AKT2 expression. Together, these results show that ethanol consumption affects the diurnal gene expression patterns of key regulators of glycogen synthesis.
Western blots were performed to determine the effect chronic ethanol had on diurnal rhythms in GS1, GS2, AKT, and GSK3β protein abundance and phosphorylation status. There was no significant daily rhythm in total GS1 protein in livers of control or ethanol-fed mice (Fig. 2D; see Table 4). Two-factor ANOVA revealed no diet or time effect for GS1 (Table 3), whereas GS2 was significantly decreased in livers of ethanol-fed mice. Total GS2 protein displayed a diurnal rhythm that peaked at ZT 8.88 ± 0.22 in livers of control-fed mice (Fig. 2E and Table 4). Importantly, the GS2 rhythm was lost in livers from ethanol-fed mice (Fig. 2E and Table 4). Moreover, there were significant main effects of diet and time for GS2 (Table 3). To further explore the impact chronic ethanol consumption has on GS protein, we measured the diurnal phosphorylation status of GS. Both phospho-GS1 and phospho-GS2 displayed diurnal oscillations with peaks at ZT 6.69 ± 0.25 and ZT 6.64 ± 0.20, respectively, in the livers of control mice (Fig. 2, F and G, and Table 4). Ethanol consumption significantly decreased the mesor of phospho-GS1 and completely abolished the rhythmic abundance of phospho-GS2 (Fig. 2, F and G, and Table 4). A two-factor ANOVA indicated significant main effects of diet and time for both phospho-GS1 and phospho-GS2 (Table 3). Representative Western blots for total and phospho-GS1, GS2, and loading controls are provided in Fig. 2J.
Table 4.
Cosinor analysis of GS, AKT, and GSK3β protein abundance (total and phosphorylated) in the livers of control and ethanol-fed mice
| Rhythmicity |
Cosinor Analysis Parameters |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein | Diet | R2 | P value | Mesor | P value | Amplitude | P value | Acrophase | P value |
| GS1 | Control | 0.16 | 0.162 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.08 | 0.473 | NA | NA | NA | ||||
| GS2 | Control | 0.37 | 0.008* | 31.89 ± 0.49 | NA | 11.90 ± 0.70 | NA | 8.88 ± 0.22 | NA |
| Ethanol | 0.12 | 0.300 | NA | NA | NA | ||||
| phospho-GS1 | Control | 0.31 | 0.020* | 76.71 ± 0.82 | <0.001† | 17.49 ± 1.16 | 0.602 | 6.69 ± 0.25 | 0.248 |
| Ethanol | 0.44 | 0.004* | 39.04 ± 0.85 | 21.71 ± 1.19 | 4.79 ± 0.21 | ||||
| phospho-GS2 | Control | 0.42 | 0.003* | 36.52 ± 0.64 | NA | 17.42 ± 0.91 | NA | 6.64 ± 0.20 | NA |
| Ethanol | 0.22 | 0.094 | NA | NA | NA | ||||
| AKT | Control | 0.14 | 0.194 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.01 | 0.946 | NA | NA | NA | ||||
| phospho-AKT | Control | 0.05 | 0.591 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.03 | 0.705 | NA | NA | NA | ||||
| GSK3β | Control | 0.02 | 0.834 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.05 | 0.562 | NA | NA | NA | ||||
| phospho-GSK3β | Control | 0.03 | 0.749 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.13 | 0.218 | NA | NA | NA | ||||
Values are means ± SE.
Statistical significance, P ≤ 0.05.
Statistically significant difference between control and ethanol, P ≤ 0.05. The R2 value represents the percent variance accounted for by a 24-h rhythm as determined by cosinor analysis.
Table 3.
Two-factor ANOVA of protein densities by time and diet
| Time |
Diet |
Interaction |
||||
|---|---|---|---|---|---|---|
| Protein | F | P value | F | P value | F | P value |
| GS1 | 1.12 | 0.368 | 1.42 | 0.242 | 0.43 | 0.242 |
| GS2 | 3.59 | 0.010* | 28.29 | <0.001* | 1.40 | 0.251 |
| phospho-GS1 | 4.29 | 0.004* | 38.97 | <0.001* | 0.56 | 0.731 |
| phospho-GS2 | 3.53 | 0.011* | 48.93 | <0.001* | 1.38 | 0.258 |
| β-Actin (GS) | 0.04 | 0.999 | 2.09 | 0.157 | 0.34 | 0.885 |
| β-Actin (phospho-GS) | 0.12 | 0.988 | 0.02 | 0.880 | 0.08 | 0.995 |
| AKT | 0.29 | 0.942 | 0.09 | 0.762 | 0.39 | 0.855 |
| phospho-AKT | 0.49 | 0.782 | 16.83 | <0.001* | 0.45 | 0.814 |
| GAPDH (AKT) | 0.12 | 0.986 | 8.31 | 0.007* | 0.32 | 0.899 |
| GAPDH (phospho-AKT) | 0.04 | 0.999 | 1.33 | 0.257 | 0.15 | 0.980 |
| GSK3β | 0.20 | 0.962 | 0.04 | 0.846 | 0.26 | 0.934 |
| phospho-GSK3β | 0.40 | 0.846 | 0.20 | 0.660 | 1.20 | 0.328 |
| GAPDH (GSK3β) | 0.03 | 1.000 | 4.06 | 0.052 | 0.09 | 0.994 |
| GAPDH (phospho-GSK3β) | 0.02 | 1.000 | 1.44 | 0.238 | 0.19 | 0.964 |
Statistical significance, P ≤ 0.05.
We also measured total protein levels and the diurnal phosphorylation status of AKT and GSK3β as these proteins participate in the regulation of GS. Neither total protein nor phosphorylation status of AKT and GSK3β displayed significant rhythmicity in the livers of control and ethanol-fed mice (Fig. 2, H and I; Table 4). Further, two-factor ANOVA indicated no significant main effects for diet or time on total AKT protein, total GSK3β, or phospho-GSK3β (Table 3). However, chronic ethanol significantly decreased phospho-AKT levels in a non-time-dependent manner (P < 0.001 for diet) (Fig. 2H and Table 3).
Further, GS enzyme activity was measured in the absence and presence of glucose-6-phosphate (G6P), a key allosteric activator of GS. GS activity measured in the absence of G6P reflects the active (unphosphorylated) form of the enzyme, while GS activity measured in the presence of 10 mM G6P represents total enzyme activity (48). Two-factor ANOVA revealed significant main effects of time, but not diet, for GS activity with or without G6P, and a significant interaction for GS without G6P (Table 5). In the livers of control-fed mice, GS activity did not display a diurnal rhythm in the absence of the allosteric effector G6P (Fig. 2K and Table 6). In contrast, GS activity was rhythmic in livers of ethanol-fed mice (Fig. 2K and Table 6). During the inactive phase (ZT 0–12) GS activity was lower in the livers of mice fed the ethanol diet compared with the control mice (Fig. 2K). However, GS activity in livers of ethanol-fed mice was elevated during the active phase (ZT 12–24) relative to control-fed mice. In the presence of G6P total hepatic GS enzyme activity was rhythmic and comparable between the control and ethanol-fed mice (Fig. 2L and Table 6), with peak GS activity occurring in the active (dark) phase of the day.
Table 5.
Two-factor ANOVA of enzyme activities by time and diet
| Time |
Diet |
Interaction |
||||
|---|---|---|---|---|---|---|
| Enzyme (+ Allosteric Activator) | F | P value | F | P value | F | P value |
| GS | 5.26 | 0.001* | 0.00 | 0.971 | 2.68 | 0.036* |
| GS (+ G6P) | 8.81 | <0.001* | 1.20 | 0.279 | 0.55 | 0.739 |
| GP | 7.55 | <0.001* | 80.05 | <0.001* | 1.67 | 0.170 |
| GP (+ AMP) | 5.65 | 0.001* | 65.96 | <0.001* | 1.90 | 0.117 |
Statistical significance, P ≤ 0.05.
Table 6.
Cosinor analysis of glycogen synthase and glycogen phosphorylase activities in the livers of control and ethanol-fed mice
| Rhythmicity |
Cosinor Analysis Parameters |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Enzyme (+ Allosteric activator) | Diet | R2 | P value | Mesor | P value | Amplitude | P value | Acrophase | P value |
| GS | Control | 0.06 | 0.503 | NA | NA | NA | NA | NA | NA |
| Ethanol | 0.45 | 0.001* | 2.11 ± 0.05 | 1.58 ± 0.07 | 17.53 ± 0.19 | ||||
| GS (+ G6P) | Control | 0.55 | <0.001* | 29.27 ± 0.26 | 0.269 | 9.14 ± 0.35 | 0.458 | 18.20 ± 0.16 | 0.460 |
| Ethanol | 0.49 | 0.001* | 31.75 ± 0.35 | 11.42 ± 0.49 | 17.33 ± 0.17 | ||||
| GP | Control | 0.42 | 0.003* | 13.81 ± 0.12 | <0.001† | 3.25 ± 0.16 | 0.269 | 8.10 ± 0.19 | 0.035† |
| Ethanol | 0.44 | <0.001* | 8.04 ± 0.07 | 2.19 ± 0.09 | 5.22 ± 0.17 | ||||
| GP (+ AMP) | Control | 0.42 | 0.003* | 17.80 ± 0.11 | <0.001† | 3.08 ± 0.16 | 0.073 | 9.76 ± 0.18 | 0.334 |
| Ethanol | 0.51 | 0.262 | 12.46 ± 0.10 | 1.13 ± 0.13 | 7.32 ± 0.46 | ||||
Values are means ± SE.
Statistical significance, P ≤ 0.05.
Statistically significant difference between control and ethanol, P ≤ 0.05. The R2 value represents the percent variance accounted for by a 24-h rhythm as determined by cosinor analysis.
Ethanol consumption alters diurnal rhythms in liver glycogen phosphorylase.
To determine whether the ethanol-induced alteration in liver glycogen content could be linked to changes in the expression and/or activity of the rate-limiting enzyme for glycogen degradation, we next measured glycogen phosphorylase (GP) transcript levels and activity. We are unaware of a commercially available antibody for GP that works well in the mouse, and as such, were unable to determine protein levels and/or phosphorylation status of GP. Gene expression of the liver form of GP (PYGL) did not display rhythmic expression in livers of control or ethanol-fed mice (Fig. 3A and Table 2). However, a significant main effect of diet (P = 0.034) was shown by two-factor ANOVA (Table 1), due to ethanol-dependent decrease in PYGL expression. GP activity was also measured in livers from control and ethanol-fed mice over the course of the day. GP activity measured in the absence and presence of 3 mM AMP represents activity of the active form of GP (phosphorylated) and total GP activity, respectively (34). Chronic ethanol consumption caused a significant decrease in liver GP activity (Fig. 3B and Table 5). GP activity in the absence of the allosteric effector AMP was significantly rhythmic in livers of both control and ethanol-fed mice (Fig. 3B and Table 6). Ethanol consumption significantly decreased mean expression (mesor) by 42% and caused a 2.88 h phase advance in peak GP activity (Table 6). Addition of AMP stimulated an increase in GP activity in both control and ethanol-fed mice; however, GP activity remained significantly lower in the ethanol-fed group compared with the control-fed mice (Fig. 3C and Table 6). In addition, the diurnal rhythm of total GP activity (+ AMP) was lost in the ethanol-fed group (Fig. 3C and Table 6). Significant main effects of time and diet were observed in GP activity by a two-factor ANOVA with no significant interaction (Table 5). Together, our findings indicate that chronic ethanol decreased mRNA expression and activity of GP in the liver.
Fig. 3.
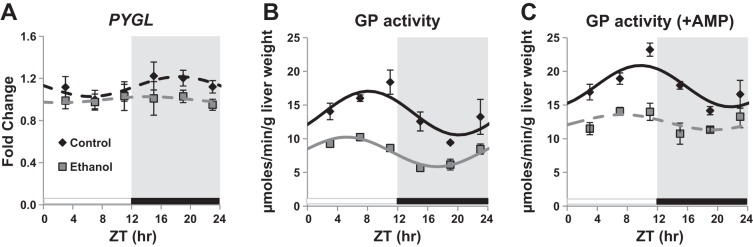
Diurnal oscillation in liver glycogen phosphorylase gene expression and activity. Livers of mice fed a control (black symbols) or ethanol-containing (gray symbols) diet for 5 wk were collected at ZT 3, 7, 11, 15, 19, and 23 for determination of glycogen phosphorylase gene expression (PYGL) (A) and enzyme activities (GP) measured in the absence (B) and presence of 3 mM 5′-AMP (C). Data presented in A–C were fitted to a cosine curve and expressed as means ± SE for n = 4–6 mice per group per time point. Gene expression levels were normalized to GAPDH mRNA levels and displayed as a fold change from control trough. Mice were housed under a standard 12:12-h light-dark cycle where ZT 0 equals lights on (white bar) and ZT 12 equals lights off (black bar and gray shading). Solid lines indicate a significant cosine curve fit, whereas dashed lines indicate a nonsignificant fit.
Ethanol consumption alters diurnal rhythms in glycogen particle components in the liver.
Given that the effects of ethanol feeding on assayable GS and GP activities did not readily explain attenuated hepatic glycogen content, we also investigated ethanol-mediated effects on other regulatory components present in the glycogen particle, namely glycogenin (GYG) and protein phosphatase 1 regulatory subunit 3C (PPP1R3C). We found that GYG, the primer for glycogen synthesis, displayed diurnal rhythmic expression that peaked at ZT 4.42 ± 0.12 in livers of control-fed mice (Fig. 4A and Table 2), whereas GYG expression was not rhythmic in livers of ethanol-fed mice (Fig. 4A and Table 2). Further, we observed a main effect of time (P < 0.001) and an interaction (P = 0.041) on GYG expression (Table 1). We also examined expression of PPP1R3C, also known as PTG (protein targeting to glycogen). PPP1R3C is a molecular scaffold protein that participates in the regulation of glycogen synthesis by targeting protein phosphatase-1 to glycogen particles to facilitate dephosphorylation of GS and GP (38). We observed a robust diurnal rhythm in PPP1R3C gene expression peaking around ZT 20 in livers of both control and ethanol-fed mice (Table 2 and Fig. 4B). Ethanol consumption caused a significant decrease in the mesor of PPP1R3C, but had no effect on the acrophase or amplitude (Table 2). Two-factor ANOVA revealed significant main effects of diet (P = 0.043) and time (P < 0.001) for PPP1R3C (Table 1).
Fig. 4.

Diurnal oscillation in glycogen particle and glucose metabolism genes. Mice were a fed control (black symbols) or ethanol-containing (gray symbols) diet for 5 wk, and liver gene expression was examined at ZT 3, 7, 11, 15, 19, and 23 h for glycogenin (GYG) (A), protein phosphatase 1 regulatory subunit 3C (PPP1R3C) (B), glucose transporter 1 (GLUT1) (C), glucose transporter 2 (GLUT2) (D), glucokinase (GCK) (E), and pyruvate kinase (PKLR) (F). Data were fitted to a cosine curve and expressed as means ± SE for n = 4–6 mice per group per time point. Gene expression levels were normalized to GAPDH mRNA levels and displayed as fold change from control trough. Mice were housed under a standard 12:12-h light-dark cycle where ZT 0 equals lights on (white bar) and ZT 12 equals lights off (black bar and gray shading). Solid lines indicate a significant cosine curve fit, whereas dashed lines indicate a nonsignificant fit.
Ethanol consumption alters diurnal rhythms in other glucose metabolism components in the liver.
To gain greater insight regarding alternative mechanisms of ethanol-induced glycogen depletion, we also examined diurnal expression of genes involved in glucose uptake and glucose metabolism, including glucose transporters (GLUT1, GLUT2), glucokinase (GCK), glucokinase regulatory protein (GCKR), and pyruvate kinase (PKLR). We observed significant diurnal variations in gene expression for GLUT2, GCK, and PKLR in livers of mice fed the control diet (Fig. 4, D–F, and Table 2). These rhythms were decreased (GLUT2, GCK) or abolished (PKLR) in livers of ethanol-fed mice (Table 2). In addition, chronic ethanol consumption significantly phase advanced the peak of GCK by 2.32 h (Table 2). Interestingly, while GLUT1 gene expression was not rhythmic in livers of control-fed mice, chronic ethanol consumption induced a significant diurnal rhythm with peak GLUT1 expression occurring at the beginning of the active phase (Fig. 4C and Table 2). GCKR was not rhythmic in livers of either control or ethanol-fed mice (Table 2). Additionally, there was a significant main effect of time on GLUT1, GLUT2, GCK, and PKLR, a significant main effect of diet on GCK, and a significant interaction (time × diet) on GCK and GLUT1, as determined by a two-factor ANOVA (Table 1). These results indicate that ethanol consumption alters diurnal gene expression profiles of several components involved in glucose uptake and glucose metabolism, which, in turn, could influence glycogen metabolism.
DISCUSSION
Previous studies report that acute and chronic ethanol consumption reduce liver glycogen content (28, 46, 50, 52); however, the mechanisms responsible for this adverse metabolic outcome are not known. Herein, we evaluated the impact chronic ethanol consumption has on diurnal oscillations in various glycogen metabolic processes using a mouse model. We show that hepatic glycogen was significantly lower in livers of ethanol-fed mice throughout the entire 24-h day, and the peak of glycogen content was shifted to a different time (phase) of the day in livers of ethanol-fed mice compared with control-fed mice. We also observed that chronic ethanol consumption altered time-of-day-dependent fluctuations in the enzymatic activities of GS and GP, the rate-limiting enzymes of glycogen metabolism. Similar to previous studies (47), we observed lower GS activity during the inactive (light) phase in livers from ethanol-fed mice. However, higher GS activities were observed in livers of ethanol-fed mice during the active (dark) phase. GP activity was lower in the livers of mice fed the ethanol diet throughout the entire 24-h day. We originally predicted that the ethanol-mediated decrease in hepatic glycogen content would have been associated with decreased GS and/or increased GP activities in the livers of ethanol-fed mice; however, this was not the case, suggesting other mechanisms. It is important to point out that the assays for GS and GP activities were measured in vitro under nonphysiological conditions with saturating substrate concentrations. Accordingly, in vitro enzymatic measurements do not necessarily represent the in situ activity state of these enzymes. For example, substrate levels, allosteric effector concentrations, and glycogen binding regulatory subunit levels are likely different within hepatocytes of control and ethanol-fed mice. It is likely that chronic ethanol consumption disrupted the availability of substrates and allosteric effectors of GS and GP in vivo, thereby reducing liver glycogen. In support of this, we observed an ethanol-induced phase shift and dampening of GCK expression, an enzyme that facilitates the phosphorylation of glucose to G6P, an important allosteric activator of GS (17, 48). It is also well established that phosphorylation activates GP and inactivates GS (17, 39). For example, in these studies we observed that the phospho-GS2 peak (Fig. 2G) coincided with higher GP enzyme activity (Fig. 3B) and the trough of glycogen content (Fig. 1) in livers of control-fed mice at the end of the inactive (light) phase of the day. Similarly, the ethanol-mediated decrease in phosphorylated GS1 and GS2 (Fig. 2, F and G) may partially explain elevated levels of GS activity in the livers of ethanol-fed mice during the active (dark) phase of the day (Fig. 2K). Our findings in control livers strongly support the concept that temporal maintenance (synchrony) of these metabolic rhythms plays an important role in glycogen metabolism. In contrast, chronic temporal dysregulation in these pathways likely contributed, in part, to decreased glycogen in the livers of ethanol-fed mice.
GS is phosphorylated by multiple upstream kinases. For example, AKT phosphorylates and inactivates GSK3β. Consequently, phospho-GSK3β is unable to phosphorylate and inactivate GS, thus leading to an increase in glycogen synthesis (39). In the present study, levels of phospho-AKT-Ser473 were significantly lower in livers of mice fed the ethanol diet. This finding is in agreement with other studies showing that acute (19) and chronic (5) ethanol consumption decreased phospho-AKT-Ser473, which could account for impaired glycogen synthesis in the ethanol-exposed liver. However, we observed no effect of chronic ethanol on phospho-GSK3β. These results suggest a lack of key involvement of GSK3β in glycogen metabolism. In support of this, Wan et al. showed that GSK3 double mutant knockin mice (i.e., mice expressing GSK3α/β mutants that cannot be phosphorylated and inhibited by AKT) had normal liver glycogen levels despite GSK3 being constitutively active (51). We also found that the diurnal oscillation and content of glycogen was the same in livers from GSK3 double mutant knockin mice and wild-type littermates (data not shown), suggesting that other kinases may be more important in regulating GS. In addition to GSK3, GS activity is regulated by protein kinase A and C, phosphorylase kinase, casein kinase l and ll, and calmodulin-dependent protein kinase (49). Future studies are needed that are directed towards determining the effect chronic ethanol has on these regulatory kinases.
We also observed that chronic ethanol abolished the diurnal oscillation of glycogenin (GYG), a self-glucosylating protein that plays a crucial role in initiating glycogen synthesis (22, 37). This ethanol-induced alteration in GYG expression pattern likely contributes to the lower level and lack of a rhythm in glycogen in the liver of ethanol-fed mice. Ethanol consumption also decreased mean expression of PPP1R3C, a member of the glycogen-targeting protein family (37). Recent studies indicate that these proteins participate in the regulation of GS and other glycogen metabolism enzymes by targeting protein phosphatase 1 to glycogen particles (30). For example, adenovirus-mediated PTG overexpression in rat livers increased glycogen levels (32), whereas mice possessing heterozygous deletion of the PTG gene had reduced glycogen levels in liver (7). As such, the ethanol-mediated decrease in PPP1R3C chronically may contribute, in part, to reduced glycogen in livers of ethanol-fed mice.
Another possible explanation for the ethanol-mediated decrease in glycogen levels is alteration in glucose uptake. As such, we examined diurnal expression of the glucose transporters. We observed that GLUT2 was rhythmic in livers of control-fed mice with peak expression occurring in the inactive (light) phase. Ethanol consumption decreased the amplitude of the GLUT2 rhythm by 50%; however, this difference just missed being statistically significant (P = 0.066, Table 2). In contrast, GLUT1 expression was rhythmic in livers of ethanol-fed mice with expression peaking during the active (dark) phase, whereas GLUT1 expression was not rhythmic in livers of control mice. Interestingly, our results are similar to those of Nanji et al. (27), who demonstrated that chronic ethanol feeding decreased GLUT2 and increased GLUT1 protein levels in livers of rats, whereas Van Horn et al. (47) showed decreased GLUT1 and no change in GLUT2 protein levels in livers of ethanol-fed rats (47). These differing results indicate that ethanol-mediated effects on hepatic glucose metabolism are complex and likely influenced by multiple factors, including the experimental animal model used (rats vs. mice), the time of day of tissue collection, the nutritional state of the animal at the time of tissue collection, and/or composition of the experimental diets. It is important to point out that the glucose transporters exhibit different locations (i.e., zones) of expression in the liver. For example, GLUT2 transporters are more predominantly distributed in periportal (zone 1) hepatocytes (33), whereas GLUT1 is localized to a special subset of centrilobular hepatocytes just surrounding the terminal hepatic venules (43). Importantly, the centrilobular region of the liver is the primary site of ethanol-induced liver injury and hypoxic stress (14). Consistent with our findings, other metabolic stressors (e.g., inhibition of mitochondrial respiration and endotoxemia) have also been shown to increase GLUT1 expression (18, 42). It has been proposed that this may lead to increased glucose transport into this select population of centrilobular hepatocytes for energy production (27), thus serving as an adaptive response mitigating hepatocyte necrosis in ethanol-exposed livers.
While it is well-accepted that postprandial glucose is taken up by the liver and converted to glycogen, recent studies suggest that glycogen is also formed from gluconeogenic substrates (1, 24). Moreover, gluconeogenesis and glycogenesis may form an interconnected metabolic circuit, instead of being separate metabolic pathways (17). For example, isotopic studies in rats (31) and humans (35) reported that a significant fraction of glycogen synthesized after glucose ingestion is from gluconeogenic precursors. Thus ethanol-mediated glycogen depletion may be attributed, in part, to ethanol-induced disruption in gluconeogenesis. In support of this, ethanol has been shown to significantly reduce hepatic gluconeogenesis in rats (26). Importantly, we found that the diurnal oscillation of phosphoenolpyruvate carboxykinase 1 (PCK1), the rate-limiting enzyme of gluconeogenesis, was abolished and dampened in livers of ethanol-fed mice (13). Thus an ethanol-mediated decrease in PCK1 and consequently gluconeogenesis may also contribute, in part, to decreased glycogen synthesis.
Taken together, this present study shows that hepatic glycogen content, along with several regulatory enzymes of glycogen metabolism, display diurnal rhythms in gene expression, protein abundance, protein phosphorylation status, and activities in livers of control (i.e., ethanol-naive) mice. Importantly, chronic ethanol consumption significantly altered and disrupted diurnal patterns in many of these critical metabolic rhythms. These rhythms in glycogen metabolism, along with rhythms in physiology and behavior, are regulated, in part, by a molecular circadian clock that allows organisms/tissues/cells to anticipate and adapt to changes in their environment (3, 12). Accordingly, hepatic glycogen levels display diurnal rhythms that persist during fasting (15, 20, 41). A recent study by Doi et al. (10) reported that the diurnal expression of GYS2 is transcriptionally regulated by the core clock component CLOCK (circadian locomotor output cycles kaput). Further, CLOCK mutant mice (10) and mice lacking a functional Period 2 (PER2) protein (a clock component) (56) display altered and dampened hepatic glycogen rhythms. In line with this, we found that chronic ethanol consumption altered the function of the liver circadian clock and dampened the diurnal rhythms of several clock genes including CLOCK and PER2 (13). Therefore, it is possible that ethanol-induced disruption of the liver molecular clock function may underpin alterations in the daily rhythms of hepatic glycogen metabolism, which in turn leads to glycogen depletion. Futures studies are needed to examine the role of the liver circadian clock in ethanol-induced changes in hepatic glycogen metabolism.
Conclusions.
In summary, hepatic glycogen metabolism is a tightly regulated and complex dynamic process that helps higher organisms store and use glycogen for energy and maintenance of blood glucose. The data presented in this new study show that chronic ethanol consumption dysregulates key metabolic processes in the liver, leading to decreased glycogen. Importantly, chronic ethanol-mediated alteration in the timing of glycogen metabolism genes in the liver creates a situation where gene and/or protein expression are occurring at times least optimal for liver and whole body metabolism. While further studies are needed to determine the role of other glycogen metabolism modulators (e.g., protein kinases involved in GS regulation) and the liver clock in ethanol-induced reduction of liver glycogen, the results presented here show for the first time that chronic ethanol consumption significantly alters several dynamic (time-of-day) processes of glycogen metabolism.
GRANTS
This work was supported in part by National Institutes of Health Grants R01-NS-082413 to K. L. Gamble, R01-HL-123574 and R01-HL-122975 to M. E. Young, and R01-AA-018841 and R21-AA-020199 to S. M. Bailey.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: U.S.U., K.L.G., M.E.Y., and S.M.B. conception and design of research; U.S.U., T.M.S., and A.N.F. performed experiments; U.S.U. and S.M.B. analyzed data; U.S.U., K.L.G., M.E.Y., and S.M.B. interpreted results of experiments; U.S.U. prepared figures; U.S.U. and S.M.B. drafted manuscript; U.S.U., K.L.G., M.E.Y., and S.M.B. edited and revised manuscript; U.S.U., T.M.S., A.N.F., K.L.G., M.E.Y., and S.M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for A. N. Filiano: Bridgewater College, 402 E. College St, Box 65, Bridgewater, VA 22812.
REFERENCES
- 1.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J 414: 1–18, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Bailey SM, Cunningham CC. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcohol Clin Exp Res 23: 1210–1218, 1999. [PubMed] [Google Scholar]
- 3.Bailey SM, Udoh US, Young ME. Circadian regulation of metabolism. J Endocrinol 222: R75–R96, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmeyer HU, Bernt E, Schmidt F, Stork H. d-Glucose: determination with hexokinase and glucose-6-phosphate dehydrogenase. In: Methods of Enzymatic Analysis, edited by Bergmeyer HU. New York: Academic, 1974, p. 1196–1201. [Google Scholar]
- 5.Carr RM, Dhir R, Yin X, Agarwal B, Ahima RS. Temporal effects of ethanol consumption on energy homeostasis, hepatic steatosis, and insulin sensitivity in mice. Alcohol Clin Exp Res 37: 1091–1099, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol 20: 17756–17772, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosson SM, Khan A, Printen J, Pessin JE, Saltiel AR. PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance. J Clin Invest 111: 1423–1432, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunningham CC, Van Horn CG. Energy availability and alcohol-related liver pathology. Alcohol Res Health 27: 291–299, 2003. [PMC free article] [PubMed] [Google Scholar]
- 9.Dietrich Keppler KD. Glycogen determination with amyloglucosidase. In: Methods of Enzymatic Analysis, edited by Bergmeyer HU. New York: Academic, 1974, p. 1127–1131. [Google Scholar]
- 10.Doi R, Oishi K, Ishida N. CLOCK regulates circadian rhythms of hepatic glycogen synthesis through transcriptional activation of Gys2. J Biol Chem 285: 22114–22121, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durgan DJ, Moore MW, Ha NP, Egbejimi O, Fields A, Mbawuike U, Egbejimi A, Shaw CA, Bray MS, Nannegari V, Hickson-Bick DL, Heird WC, Dyck JR, Chandler MP, Young ME. Circadian rhythms in myocardial metabolism and contractile function: influence of workload and oleate. Am J Physiol Heart Circ Physiol 293: H2385–H2393, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Edery I. Circadian rhythms in a nutshell. Physiol Genomics 3: 59–74, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Filiano AN, Millender-Swain T, Johnson R Jr, Young ME, Gamble KL, Bailey SM. Chronic ethanol consumption disrupts the core molecular clock and diurnal rhythms of metabolic genes in the liver without affecting the suprachiasmatic nucleus. PLoS One 8: e71684, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.French SW, Benson NC, Sun PS. Centrilobular liver necrosis induced by hypoxia in chronic ethanol-fed rats. Hepatology 4: 912–917, 1984. [DOI] [PubMed] [Google Scholar]
- 15.Fuller RW, Diller ER. Diurnal variation of liver glycogen and plasma free fatty acids in rats fed ad libitum or single daily meal. Metabolism 19: 226–229, 1970. [DOI] [PubMed] [Google Scholar]
- 16.Gilboe DP, Larson KL, Nuttall FQ. Radioactive method for the assay of glycogen phosphorylases. Anal Biochem 47: 20–27, 1972. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg CC, Jurczak MJ, Danos AM, Brady MJ. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab 291: E1–E8, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Hamrahian AH, Zhang JZ, Elkhairi FS, Prasad R, Ismail-Beigi F. Activation of Glut1 glucose transporter in response to inhibition of oxidative phosphorylation. Arch Biochem Biophys 368: 375–379, 1999. [DOI] [PubMed] [Google Scholar]
- 19.He J, de la Monte S, Wands JR. Acute ethanol exposure inhibits insulin signaling in the liver. Hepatology 46: 1791–1800, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa K, Shimazu T. Circadian rhythm of liver glycogen metabolism in rats: effects of hypothalamic lesions. Am J Physiol Endocrinol Metab 238: E21–E25, 1980. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−DeltaDeltaC(T) method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Lomako J, Lomako WM, Whelan WJ. Glycogenin: the primer for mammalian and yeast glycogen synthesis. Biochim Biophys Acta 1673: 45–55, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med 44: 1259–1272, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGarry JD, Kuwajima M, Newgard CB, Foster DW, Katz J. From dietary glucose to liver glycogen: the full circle round. Annu Rev Nutr 7: 51–73, 1987. [DOI] [PubMed] [Google Scholar]
- 25.Michalopoulos GK. Liver regeneration. J Cell Physiol 213: 286–300, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mokuda O, Tanaka H, Hayashi T, Ooka H, Okazaki R, Sakamoto Y. Ethanol stimulates glycogenolysis and inhibits both glycogenesis via gluconeogenesis and from exogenous glucose in perfused rat liver. Ann Nutr Metab 48: 276–280, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Nanji AA, Fogt F, Griniuviene B. Alterations in glucose transporter proteins in alcoholic liver disease in the rat. Am J Pathol 146: 329–334, 1995. [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson P, Wagle SR, Ashmore J. Ethanol effects on hepatic oxidations and gluconeogenesis. Proc Soc Exp Biol Med 131: 707–710, 1969. [DOI] [PubMed] [Google Scholar]
- 29.Nelson W, Tong YL, Lee JK, Halberg F. Methods for cosinor-rhythmometry. Chronobiologia 6: 305–323, 1979. [PubMed] [Google Scholar]
- 30.Newgard CB, Brady MJ, O'Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes 49: 1967–1977, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Newgard CB, Hirsch LJ, Foster DW, McGarry JD. Studies on the mechanism by which exogenous glucose is converted into liver glycogen in the rat. A direct or an indirect pathway? J Biol Chem 258: 8046–8052, 1983. [PubMed] [Google Scholar]
- 32.O'Doherty RM, Jensen PB, Anderson P, Jones JG, Berman HK, Kearney D, Newgard CB. Activation of direct and indirect pathways of glycogen synthesis by hepatic overexpression of protein targeting to glycogen. J Clin Invest 105: 479–488, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogawa A, Kurita K, Ikezawa Y, Igarashi M, Kuzumaki T, Daimon M, Kato T, Yamatani K, Sasaki H. Functional localization of glucose transporter 2 in rat liver. J Histochem Cytochem 44: 1231–1236, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Ortmeyer HK, Bodkin NL, Hansen BC. Insulin regulates liver glycogen synthase and glycogen phosphorylase activity reciprocally in rhesus monkeys. Am J Physiol Endocrinol Metab 272: E133–E138, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Radziuk J. Sources of carbon in hepatic glycogen synthesis during absorption of an oral glucose load in humans. Fed Proc 41: 110–116, 1982. [PubMed] [Google Scholar]
- 36.Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. J Gastroenterol Hepatol 26, Suppl 1: 203–212, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roach PJ. Glycogen and its metabolism. Curr Mol Med 2: 101–120, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J 441: 763–787, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rui L. Energy metabolism in the liver. Compr Physiol 4: 177–197, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. J Gastroenterol Hepatol 26: 1089–1105, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Sollberger A. The control of circadian glycogen rhythms. Ann NY Acad Sci 117: 519–554, 1964. [DOI] [PubMed] [Google Scholar]
- 42.Spolarics Z, Pekala PH, Bagby GJ, Spitzer JJ. Brief endotoxemia markedly increases expression of GLUT1 glucose transporter in Kupffer, hepatic endothelial and parenchymal cells. Biochem Biophys Res Commun 193: 1211–1215, 1993. [DOI] [PubMed] [Google Scholar]
- 43.Tal M, Schneider DL, Thorens B, Lodish HF. Restricted expression of the erythroid/brain glucose transporter isoform to perivenous hepatocytes in rats. Modulation by glucose. J Clin Invest 86: 986–992, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas JA, Schlender KK, Larner J. A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-[14C]glucose. Anal Biochem 25: 486–499, 1968. [DOI] [PubMed] [Google Scholar]
- 45.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Horn CG, Cunningham CC. Contributions of dietary carbohydrate and ethanol to alterations in liver glycogen levels and glycolytic activity. Alcohol 19: 139–144, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Van Horn CG, Ivester P, Cunningham CC. Chronic ethanol consumption and liver glycogen synthesis. Arch Biochem Biophys 392: 145–152, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Villar-Palasi C, Guinovart JJ. The role of glucose 6-phosphate in the control of glycogen synthase. FASEB J 11: 544–558, 1997. [PubMed] [Google Scholar]
- 49.Voet DV, Judith G. Biochemistry. New York: Wiley, 2010, p. 1428. [Google Scholar]
- 50.Walker JE, Gordon ER. Biochemical aspects associated with an ethanol-induced fatty liver. Biochem J 119: 511–516, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wan M, Leavens KF, Hunter RW, Koren S, von Wilamowitz-Moellendorff A, Lu M, Satapati S, Chu Q, Sakamoto K, Burgess SC, Birnbaum MJ. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab 18: 99–105, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winston GW, Reitz RC. Effects of chronic ethanol ingestion on glucose homeostasis in males and females. Life Sci 26: 201–209, 1980. [DOI] [PubMed] [Google Scholar]
- 53.Winston GW, Reitz RC. Effects of chronic ethanol ingestion on liver glycogen phosphorylase in male and female rats. Am J Clin Nutr 34: 2499–2507, 1981. [DOI] [PubMed] [Google Scholar]
- 54.Young ME, Radda GK, Leighton B. Activation of glycogen phosphorylase and glycogenolysis in rat skeletal muscle by AICAR—an activator of AMP-activated protein kinase. FEBS Lett 382: 43–47, 1996. [DOI] [PubMed] [Google Scholar]
- 55.Young TA, Bailey SM, Van Horn CG, Cunningham CC. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol 41: 254–260, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Zani F, Breasson L, Becattini B, Vukolic A, Montani JP, Albrecht U, Provenzani A, Ripperger JA, Solinas G. PER2 promotes glucose storage to liver glycogen during feeding and acute fasting by inducing Gys2 PTG and G L expression. Mol Metab 2: 292–305, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]