

Abstract
The pathophysiology of esophageal injury, repair, and inflammation in gastroesophageal reflux-disease (GERD) is complex. Whereas most studies have focused on the epithelial response to GERD injury, we are interested in the stromal response. We hypothesized that subepithelial esophageal myofibroblasts in GERD secrete proinflammatory cytokines in response to injurious agents encountered via epithelial barrier breaches or through dilated epithelial intercellular spaces. We determined the percentage of myofibroblasts [α-smooth muscle actin (α-SMA)+vimentin+CD31−] in the subepithelial GERD and normal esophageal stroma by immunomorphologic analysis. We performed α-SMA coimmunostaining with IL-6 and p65. We established and characterized primary cultures of α-SMA+vimentin+CD31−CD45− human esophageal myofibroblasts (HuEso MFs). We modeled GERD by treatment with pH 4.5-acidified media and Toll-like receptor 4 (TLR4) ligands, LPS and high-mobility group box 1 protein (HMGB1), and determined myofibroblast cytokine secretion in response to GERD injury. We demonstrate that spindle-shaped cell myofibroblasts are located near the basement membrane of stratified squamous epithelium in normal esophagus. We identify an increase in subepithelial myofibroblasts and activation of proinflammatory pathways in patients with GERD. Primary cultures of stromal cells obtained from normal esophagus retain myofibroblast morphology and express the acid receptor transient receptor potential channel vanilloid subfamily 1 (TRPV1) and TLR4. HuEso MFs stimulated with acid and TLR4 agonists LPS and HMGB1 increase IL-6 and IL-8 secretion via TRPV1 and NF-κB activation. Our work implicates a role for human subepithelial stromal cells in the pathogenesis of GERD-related esophageal injury. Findings of this study can be extended to the investigation of epithelial-stromal interactions in inflammatory esophageal mucosal disorders.
Keywords: gastroesophageal-reflux disease, acid-related disease, mucosal injury, inflammatory cells, inflammatory mechanisms
despite the profound morbidity and mortality associated with esophageal disorders, the basic mechanisms underlying esophageal injury and repair have not been fully elucidated. Gastroesophageal-reflux disease (GERD) is one of the most common problems encountered in clinical practice and is a recognized cause of esophageal injury, ranging from nonerosive to erosive esophagitis, strictures, intestinal metaplasia (35), and malignancy (1, 16). The pathophysiology of GERD-mediated injury is complex and incompletely understood. The stratified squamous epithelium of the human esophagus normally provides robust protection against reflux and potential luminal pathogens (18). In response to injury, damage to tight junctions, development of dilated intercellular spaces (DIS), and loss of squamous epithelium integrity are observed. Acid is one of the primary agents responsible for the initiation and perpetuation of esophageal injury in animal models (28). The traditional model of caustic damage to the squamous epithelium incompletely encapsulates the mechanism of injury, however. Pathologic exposure of the squamous epithelium to gastric and duodenal contents, including pepsin, trypsin, and bile acids, is involved (29). Clinical (25) and animal models (43) also suggest a more complex mechanism with an increasingly recognized role for inflammation in the perpetuation of injury (16). In addition, a pathogenic role for microbes has been suggested (18, 48). Esophageal biopsies from patients with GERD demonstrate an increase in inflammatory mediators IL-6, IL-8, and IL-1β (15). Cellular sources of these cytokines include squamous epithelial cells, immune cells, and a heterogeneous population of ill-defined stromal cells (29).
Rigorous evaluation of stromal cells in the GERD esophagus has been limited, in part, secondary to insufficient representation of the lamina propria in esophageal biopsies. There is a paucity of publications detailing the stromal response in GERD, although characterization of the stroma in Barrett's esophagus and esophageal adenocarcinoma (3) supports a potential role for the stroma beyond that of bystander. Because the esophageal stroma is incompletely characterized, and the capacity of stromal cells to respond to potential signals encountered in disease is undefined, mechanisms by which cell-to-cell signaling occurs between the esophageal epithelium and amongst stromal cells are unclear.
In the lower gastrointestinal (GI) tract, the importance of the stroma in maintaining epithelial integrity is established and well accepted. Myofibroblasts are a subpopulation of stromal cells, best characterized in the lower GI tract, at the interface between the epithelium and the lamina propria (32). They express α-smooth muscle actin (α-SMA) and vimentin and express desmin inconsistently or weakly. They participate in GI tissue injury and repair, inflammation (38), and carcinogenesis (40), predominantly via paracrine mechanisms (30, 31). Myofibroblast-derived mediators have known roles in limiting or promoting colitis and cancer (38, 40). The importance of this population of stromal cells and epithelial-stromal interactions is underscored further by their role in regulating epithelial cell proliferation and in contributing to the stem cell niche (6, 38–40, 46). It is reasonable to suppose that such interactions also play a critical role in the esophagus.
The esophageal stroma can be exposed to injurious luminal agents via barrier breach or dilated intracellular spaces. Stromal cells can also be stimulated by epithelial cellular debris or cytokines secreted from the epithelium or surrounding immune cells (2). We were interested in investigating the human esophageal stromal cell response in GERD. We have shown previously that α-SMA- and vimentin-expressing, myofibroblast-like cells in the murine esophagus secrete IL-6 in response to acid (36, 40). For this study, we hypothesized that subepithelial stromal cells with a myofibroblast phenotype respond to injurious luminal agents implicated in GERD pathogenesis with secretion of inflammatory cytokines.
To address this hypothesis, we examined the stroma of normal esophagus and identified those stromal cells with a myofibroblast phenotype. We then reviewed esophageal biopsies with histologic evidence of GERD and with sufficient stroma to determine if there is an expansion in myofibroblasts compared with normal esophagus. We evaluated expression of proinflammatory cytokine IL-6 and activation of NF-κB in these biopsies. An increase in IL-6 has been reported previously in GERD (15), although the stromal contribution to its expression has not been investigated. We modeled GERD by treating cells with acid and Toll-like receptor 4 (TLR4) ligand LPS and determined myofibroblast cytokine secretion in response to this injury.
We observed vimentin- and α-SMA-expressing nonimmune, nonendothelial, spindle-shaped cells located near the stratified squamous epithelium of normal esophagus, suggesting a possible role for crosstalk between these two populations in homeostasis and disease. We observed an increase in spindle-shaped α-SMA+ nonendothelial cells in GERD biopsies. An increase in extracellular stromal IL-6 expression and NF-κB activation in myofibroblasts in GERD implicated these cells in disease pathogenesis. To investigate further the role of these stromal cells in the pathogenesis of GERD, we established primary cultures of esophageal stromal cells using previously established techniques (8, 36). Primary cultures of esophageal stromal cells have myofibroblast morphology and express receptors for injurious agents encountered in GERD: the putative acid receptor transient receptor potential channel vanilloid subfamily 1 (TRPV1) and TLR4. These cells respond to acid and TLR4 ligands via an NF-κB-mediated mechanism with selective secretion of cytokines implicated in the pathogenesis of GERD-mediated injury. Our findings demonstrate a role for stromal cells in the pathogenesis of GERD and support further investigation of epithelial-stromal crosstalk in GERD.
METHODS
Esophageal Specimens
The protocol was approved by the Ethics Committee of Washington University School of Medicine in St. Louis and Keck School of Medicine of University of Southern California (USC). Sections of human esophagus (n = 8) were obtained from discarded esophagus during lung transplants and used for immunohistochemistry and immunofluorescence and establishment of primary cultures. These full-thickness sections along with esophageal biopsies (n = 2) without histopathologic evidence of GERD served as comparators to GERD biopsies.
De-identified, archived, formalin-fixed, paraffin-embedded generated slides of GERD biopsies were re-examined by a GI pathologist (exempt study HS-13-00648). Slide selection focused on histopathologic changes in squamous mucosa characteristic of GERD injury (n = 5). Further tissue selection required representative subepithelial stroma to be present for immunohistochemical analysis. All biopsies met accepted histopathologic criteria of GERD (11, 33), including varying degrees of basal intracellular edema; intraepithelial squamous infiltration by neutrophils, lymphocytes, and eosinophils; basal cell hyperplasia; and elongation of vascular papillae. Biopsies with intestinal metaplasia, consistent with Barrett's esophagus, were excluded.
Analysis of normal esophagus vs. GERD biopsies.
To characterize stromal changes in GERD, we used immunostains to identify the different cellular proliferations occurring with this type of injury, focusing on stromal fibroblasts, myofibroblasts, and endothelial cells. We then compared the same immunohistochemical battery in the subepithelial stroma of normal esophagus without histopathologic features of GERD. At least three fields of vision along the subepithelial region of each normal, full-thickness esophagus (n = 8) were viewed at 40× oil, with the epithelial layer comprising 50% of the field of vision and the stroma the remaining 50%. The same protocol was then applied to examination of GERD biopsies (n = 5). At this magnification, the stromal cells observed are those in the subepithelial region. This 50/50 distribution also standardizes the areas examined across all specimens and minimizes variability between biopsy samples. Immunofluorescent staining demonstrated fibroblasts (α-SMA negative/vimentin positive), myofibroblasts (α-SMA positive/vimentin positive and α-SMA positive/CD31 negative), and endothelial cells (α-SMA positive/vimentin positive and α-SMA positive/CD31 positive).
For quantification purposes, a nuclear counterstain [4′,6′-diamidino-2-phenylindole (DAPI)] was used to identify and obtain the total number of stromal cells present. DAPI-stained cells were then evaluated with immunohistochemical staining for α-SMA, vimentin, and CD31 to confirm cell types (myofibroblasts, fibroblasts, and endothelial cells). The total number of myofibroblasts, fibroblasts, and endothelial cells was divided by the total number of DAPI-stained nuclei to determine percentages.
In normal esophagus, it was straightforward to distinguish endothelial cells from myofibroblasts based on the circular configuration of blood vessels. To determine the percentage of cells that were α-SMA-positive/vimentin-positive myofibroblasts, therefore, DAPI-stained endothelial cells that were part of blood vessels were excluded initially in normal esophagus (n = 8). As described above, endothelial cells were also α-SMA positive/vimentin positive. The stroma of GERD biopsies was more cellular, however, and distinguishing endothelial cells from myofibroblasts based on morphology proved difficult. Evaluation of α-SMA and vimentin immunostaining in stromal cells was repeated in normal (n = 5) and GERD esophagus (n = 5) without exclusion of any DAPI-stained cells. Immunostaining was then performed on these sections for α-SMA and CD31 to distinguish myofibroblasts (α-SMA+CD31−) from endothelial cells (α-SMA+CD31+). Cell percentages were determined as described above. Immunostaining for IL-6 and p65 was also performed on normal and GERD esophagus (n = 5 each).
Immunohistochemistry and Immunocytochemistry
Immunohistochemistry.
Esophageal resections were processed for histology. Diva Decloacker reagent was used for antigen retrieval on formalin-fixed, paraffin-embedded tissues. α-SMA and vimentin were detected with polyclonal rabbit anti-α-SMA (ab5694; Abcam, Cambridge, MA) 1:200 and anti-vimentin 1:200 (ab45939; Abcam), followed by goat anti-rabbit IgG, streptavidin-horseradish peroxidase (HRP), and diaminobenzidine.
Immunofluorescence.
Immunofluorescence staining was performed on paraffin-embedded sections of human esophagectomy specimens, on normal and diseased esophageal biopsies, on primary cultures of untreated esophageal stromal cells (2 × 104), and on esophageal myofibroblasts (5 × 104) grown in chamber slides (Nalge Nunc International, Rochester, NY). Rabbit polyclonal antibodies to vimentin (ab45939; Abcam) 1:500, vanilloid receptor 1 (VR1; ab74813; Abcam) 1:100, CD31 (ab28364; Abcam) 1:20, p65 (BioLegend, San Diego, CA) 1:250, and IL-6 (ab6672; Abcam) 1:400 were applied overnight, followed by goat anti-rabbit Cy2 (111-225-144; Jackson ImmunoResearch, West Grove, PA). Mouse MAb to α-SMA (ab7817; Abcam) 1:250, cytokeratin (ab7753; Abcam) 1:250, and TLR4 (ab22048; Abcam) 1:100 were applied overnight, followed by goat anti-mouse rhodamine (115-297-003; Jackson ImmunoResearch) or goat anti-mouse Cy5 (115-225-146; Jackson ImmunoResearch) at 1:1,000, 1:250, and 1:250, respectively. Nuclei were visualized by counterstaining with DAPI (blue). Cells were mounted with Fluoroshield (F6057; Sigma-Aldrich, St. Louis, MO) and examined with a fluorescence (TE 300 Nikon Eclipse; Nikon Instruments, Melville, NY) or confocal (LSM 510; Zeiss, Thornwood, NY) microscope.
Esophageal Stromal Cell Isolation and Cell Culture
Primary cultures of esophageal stromal cells were established from three freshly resected human esophagectomy specimens, as per Gargus et al. (8), using a modification of a technique described previously for establishment of murine esophageal stromal cells (36). Esophageal mucosa was separated from the muscle layers and adventitia by sharp dissection. Mucosa was cut into subcentimeter fragments and placed in HBSS (Sigma-Aldrich), transferred into fresh HBSS, and minced into 2- to 3-mm pieces. Tissue was washed eight times with HBSS, allowing time for esophageal fragments to sediment out between each wash. Tissue was incubated with 300 U/ml collagenase XI (Sigma-Aldrich) and 0.1 mg/ml dispase (GIBCO; Invitrogen, Carlsbad, CA) for up to 6 h with gentle shaking, further minced and centrifuged at 200 g for 10 min. Supernatant was discarded, and the pellet was transferred to a 5-ml tube and washed five times in DMEM (Sigma-Aldrich), supplemented with 2% sorbitol (Sigma-Aldrich) to eliminate nonviable cells and debris. Cells were seeded in six-well plates and cultured in DMEM with 10% FBS; 10 mg/ml insulin; and 10 μg/ml gentamicin, 2 ng/ml EGF (Sigma-Aldrich), and 10 μg/ml transferrin (Roche, Basel, Switzerland). This medium was used with murine esophageal myofibroblast-like stromal cells (36) and is called myofibroblast medium. For treatment studies, serum-free myofibroblast medium was used as serum interfered with cytokine analysis. Cells were grown at 37°C in a humidified 5% CO2 incubator.
At 80% confluence, adherent cells were passaged to T25 flasks using 0.05% trypsin/EDTA (Corning cellgro; Corning Life Sciences, Mediatech, Manassas, VA). Epithelial cells do not survive under these culture or passage conditions (36). Morphology was examined with an inverted microscope (Motic AE31), and images were digitally acquired using a Moticam 580 5.0 megapixel camera (Motic, Hong-Kong, China). Primary cultures at passages 5–15 were characterized by immunocytochemistry and flow cytometry before functional analyses.
Het-1A, a simian virus-40-immortalized human esophageal epithelial cell line (CRL-2692), was purchased from American Type Culture Collection (ATCC; Manassas, VA) and grown in flasks; precoated with 0.01 mg/ml fibronectin, 0.03 mg/ml bovine collagen type 1, and 0.01 mg/ml BSA; and dissolved in culture medium (BEGM; kit catalog no. CC-3170; Lonza/Clonetics, Walkersville, MD). 18Co cells were purchased from ATCC. These cells are derived from neonatal human colon and exhibit characteristics of subepithelial myofibroblasts (12).
Flow Cytometry
Antibodies to CD45 conjugated to FITC (11-0459-42), CD31 conjugated to eFluor 450 (48-0319-41), CD324 conjugated to peridinin chlorophyll protein-eFluor 710 (46-3249-80), and CD90 conjugated to allophycocyanin (17-0909-41) were purchased from eBioscience (San Diego, CA). Cells were blocked in BD PharMingen’s (San Diego, CA) mouse Fc block for CD16/CD32 (2136662) before being stained in PBS supplemented with 2% FBS (Sigma-Aldrich) and 1 mM EDTA. Viability was assayed using Annexin V and 7-aminoactinomycin D (559763; BD PharMingen). Cells were analyzed using the LSRII flow cytometer (BD Biosciences, San Jose, CA). All data were evaluated with FlowJo software (Tree Star, Ashland, OR). At least 6 × 104 esophageal stromal cells/sample were analyzed. An appropriate forward-scatter threshold was used to exclude debris. Commercially purchased peripheral blood mononuclear cells (HemaCare, Van Nuys, CA) were used as positive controls for CD31 and CD45 staining. The epithelial cell line Caco2 was used as a positive control for CD324 staining. Appropriate isotype controls were used to determine nonspecific antibody staining.
RNA Isolation and RT-PCR
RNA was harvested from primary cultures established from three different esophagi using the GeneElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich), per the manufacturer’s recommendations, and further cleaned with DNA-free (Ambion, Austin, TX). RNA was isolated from the human esophageal epithelial line Het-1A and colon myofibroblast 18Co for comparison. cDNA was synthesized, and mRNA expression of the following genes was evaluated: α-SMA (5′ CTG TTC CAG CCA TCC TTCAT 3′; 3′ CCG TGA TCT CCT TCT GCA TT 5′), vimentin (5′ TGT CCA AAT CGA TGT GGA TGT TTC 3′; 3′ TTG TAC CAT TCT TCT GCC TCC TG 5′), E-cadherin (5′ GAA GGT GAC AGA GCC TCT GGA T 3′; 3′ GAT CGG TTA CCG TGA TCA AAA TC 5′), TLR2 (5′ TGA TGC TGC CAT TCT CAT TC 3′; 3′ CGC AGC TCT CAG ATT TAC CC 5′), TLR4 (5′ CAG GGC TTT TCT GAG TCG TC 3′; 3′ TGA GCA GTC GTG CTG GTA TC 5′), TRPV1 (5′ GAC TTC AAG GCT GTC TTC ATC ATC C 3′; 3′ GAC TTC AAG GCT GTC TTC ATC ATC C 5′), CD45 (5′ CTGACATCATCACCTAGCAG 3′; 3′ TGCTGTAGTCAATCCAGTGG 5′), and cytokeratin (5′ CCA TGA AGA GGT GAG CGG GGA TTG 3′; 3′ CTG TGG GGA TGG GAA AGG AAG ATG T 5′). Human 18s (5′ CCA TGA AGA GGT GAG CGG GGA TTG 3′; 3′ ATT AAG TCC CTG CCC TTT GTA CAC 5′) was used as an endogenous control to normalize the samples using the ΔΔCT method of relative quantitation, where CT is the threshold cycle.
Treatment of Esophageal Stromal Cells with Acidified Media
Myofibroblast media were acidified with HCl, and pH 4.5 was used based on published literature (9, 17, 42) and our experience with murine myofibroblast-like stromal cells (36). Cells were treated with pH 4.5 serum-free myofibroblast media for 5 min–1 h. Trypan blue exclusion method and poor replating of cells confirmed cytotoxicity and death in 30- and 60-min-treated cells. In response to 15 min of acidified media, there was a <10% increase in cell death for each recovery time point. Replated cells grew normally. Cytotoxicity was also assessed by lactate dehydrogenase (LDH) release in response to pH 4.5-acidified media for 15 and 30 min using an LDH cytotoxicity assay kit (Thermo Scientific, Rockford, IL) in accordance with the manufacturer's instructions. Cytotoxicity was not observed with 15 min of treatment with acidified media. Based on the above, cells were grown in 12-well plates until 80% confluent and treated with acidified media for 15 min, followed by replacement with fresh, serum-free media and collection of conditioned media at 3, 6, and 24 h for further analysis. In separate experiments, conditioned media were collected from acid-treated myofibroblasts in the presence of TRPV1 inhibitor AMG9810 (A2731; Sigma-Aldrich; 1–10 μM) at 24 h.
Treatment of Esophageal Stromal Cells with TLR4 Ligands
Cells grown in 12-well plates (5.0 × 104 cells/well) were treated with LPS (Sigma-Aldrich) 10 μg/ml for 3, 6, and 24 h or with recombinant high-mobility group box 1 protein (HMGB1; R&D Systems, Minneapolis, MN) for 6 and 24 h. Conditioned media with appropriate controls were collected.
Luminex Multiplex Assay
Conditioned media of acid- and LPS-treated cells were submitted in triplicate for Luminex multiplex assay testing through the USC Beckman Center for Immune Monitoring Core Lab. Secreted cytokines and chemokines were analyzed with the Bio-Plex suspension array system (Bio-Rad Laboratories, Hercules, CA) using a Milliplex MAP human cytokine/chemokine 35-plex kit (EMD Millipore, Billerica, MA) for EGF, eotaxin, fractalkine, granulocyte colony-stimulating factor (CSF), granuloctye macrophage-CSF, growth-related oncogene (GRO), IFN-α, IFN-γ, IL-1α, IL-1β, IL-1rα, IL-2, IL-3, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p40, IL-15, IL-17, IFN-γ-induced protein 10 (IP-10), macrophage-derived chemokine, macrophage inflammatory protein (MIP)-1α, MIP-1β, monocyte chemoattractant protein 1 (MCP-1), soluble CD40 ligand, TGF-α, TNF-α, TNF-β, FGF-2, VEGF, PDGF-AA, PDGF-AB/BB, and regulated on activation, normal T cell expressed and secreted (RANTES).
Immunoblot for TLR4 and TRPV1
Cell lysates were obtained from primary cultures of esophageal stromal cells, 18Co, HET-1A, HeLa, and Caco2, and protein was quantified by Bradford assay. An equivalent quantity (40 μg) of protein from each cell type was loaded, subjected to 4–20% SDS-PAGE gel, transferred to polyvinylidene difluoride membrane, blocked with 5% milk or BSA, and incubated overnight with mouse anti-TLR4 (76B357.1; ab22048; 1:250; Abcam) or TRPV1 [sc-12498; goat anti-VR1 antibody (P19); 1:200; Santa Cruz Biotechnology, Dallas, TX]. Membranes were washed with Tris-buffered saline with Tween 20 and incubated with donkey anti-mouse HRP or donkey anti-goat HRP (1:5,000; Santa Cruz Biotechnology) for 1 h at room temperature. Immunoreactive bands were detected by enhanced chemiluminescent substrates. Membranes were stripped and reprobed with β-actin (Sigma-Aldrich) as the loading control.
ELISA Cytokine Assay
Conditioned media from treated cells were collected for evaluation of the IL-6, IL-1β, and IL-8 at 3, 6, and 24 h using ELISA, according to the manufacturer's instructions. Cytokine levels in each sample were determined based on a standard curve generated by recombinant proteins provided with the kits. Results were expressed as the means ± SE in picograms/milliliter.
Evaluation of the NF-κB Pathway
Conditioned media were collected from LPS- or acid-treated cells in the presence of Bay 11-7082 (1–20 μM) at 6 and 24 h for IL-6 ELISA. For protein studies, cells were harvested at 30 min, 1 h, and 3 h, and immunoblot was performed as described above. Inhibitor of NF-κBα (IκBα) was detected by incubating membranes overnight with IκBα (1:1,000; Cell Signaling Technology, Danvers, MA), followed by donkey anti-mouse and/or donkey anti-rabbit IgG-HRP (Santa Cruz Biotechnology) for 1 h at room temperature. Immunoreactive bands were detected by enhanced chemiluminescent substrates (Santa Cruz Biotechnology). Membranes were stripped and reprobed with tubulin (Sigma-Aldrich) as the loading control. IκBα images were quantified using Image Studio Lite Software (LI-COR, Lincoln, NE) and normalized to tubulin-loading control.
Immunofluorescence staining for p65 was performed on acid- and LPS-treated esophageal myofibroblasts (5 × 104) grown in chamber slides (Nalge Nunc International). Rabbit polyclonal to p65 (BioLegend) 1:250 was applied overnight, followed by goat anti-rabbit Cy2 (111-225-144; Jackson ImmunoResearch) at 1:1,000. Nuclei were visualized by counterstaining with DAPI (blue). Cells were mounted with Fluoroshield (F6057; Sigma-Aldrich) and examined with a fluorescence microscope (TE 300 Nikon Eclipse; Nikon Instruments).
Statistics
All experiments were performed in triplicate and data presented as means ± SE. Data were analyzed using Student's two-tailed type 2 t-test (Excel; Microsoft, Redmond, WA) or ANOVA, followed by Bonferroni's multiple comparisons test as appropriate (Prism; GraphPad Software, La Jolla, CA).
RESULTS
Human Esophagus Contains Stromal Cells with Myofibroblast-Like Characteristics
Examination of hematoxylin and eosin (H&E) sections of normal esophagus demonstrated normal epithelium and a stroma characterized by a heterogenous population of spindle-shaped cells and blood vessels subjacent to the epithelial basement membrane (Fig. 1, A and B). Immunostaining demonstrated expression of α-SMA (Fig. 1, C and E) and vimentin (Fig. 1, D and F) in spindle-shaped cells adjacent to the epithelium. α-SMA and vimentin expression was also observed in endothelial cells forming blood vessels (Fig. 1, C–F). Confocal microscopy demonstrated spindle-shaped cells in the lamina propria that coexpressed myofibroblast markers α-SMA and vimentin (Fig. 2, A and B). Endothelial cells forming blood vessels were also α-SMA+vimentin+ and were distinguished in these sections from spindle-shaped cells by their circular configuration (Fig. 2, A and B). Throughout the lamina propria, vimentin-only-expressing, spindle-shaped cells, consistent with fibroblasts, were also observed (Fig. 2, A and B). Higher-power confocal images confirmed coexpression of α-SMA and vimentin in spindle-shaped cells adjacent to the squamous epithelium (Fig. 2C). Immunofluorescence of normal esophageal lamina propria similarly demonstrated the presence of vimentin-only-positive fibroblasts (Fig. 3, A and B), α-SMA+vimentin+ endothelial cells (Fig. 3A), and α-SMA+vimentin+ single, nonblood-vessel, spindle-shaped myofibroblasts (Fig. 3, A and B) near the squamous epithelium.
Fig. 1.

Immunostaining of normal human esophageal stroma. Hematoxylin and eosin staining was performed on full-thickness sections of normal esophagus. Examination of the stroma (n = 4; A and B) demonstrates a heterogeneous population with spindle-shaped cells near the basement membrane of the squamous epithelium (arrows). Blood vessels are delineated by arrowheads. Magnification, 400×. Immunostaining demonstrates α-smooth muscle actin (α-SMA; n = 2; C and E) expression in endothelial cells forming blood vessels (arrowheads) and in spindle-shaped cells adjacent to the squamous epithelium (arrows). Immunostaining for vimentin (n = 2; D and F) demonstrates expression in several stromal cells, including endothelial cells forming blood vessels (arrowheads) and in spindle-shaped cells near the squamous epithelium (arrows; A and B, 200×; C and D, 400×). The black boxes in C and D depict areas shown in E and F (630×).
Fig. 2.

Expression of α-SMA-positive, vimentin-positive cells in normal human esophagus. Representative confocal images of immunostaining for α-SMA [tetramethylrhodamine isothiocyanate (TRITC); red] and vimentin (FITC, green) in normal esophagus (n = 9) are shown (A and B). α-SMA and vimentin expression is observed in spindle-shaped cells adjacent to the squamous epithelium (sq. epi; solid arrows) and in endothelial cells forming blood vessels interspersed throughout the lamina propria and occasionally in the squamous epithelium (white arrowheads). Vimentin-expressing green fibroblasts (green, dashed arrows) were interspersed throughout the lamina propria (A and B; magnification, 40×). C: representative confocal image of immunostaining of normal esophagus at higher magnification (100×) demonstrates coexpression of α-SMA and vimentin in a spindle-shaped myofibroblast adjacent (yellow arrows) to the squamous epithelium. The 4 panels depict 4′,6′-diamidino-2-phenylindole (DAPI; blue), α-SMA (TRITC, red), vimentin (FITC, green), and α-SMA-vimentin coexpression in this cell. Scattered vimentin-only-expressing fibroblasts (green, dashed arrows) and endothelial cells (white arrowheads) are also seen.
Fig. 3.

Expression of α-SMA and vimentin in normal and gastroesophageal-reflux disease (GERD) esophagus. Immunostaining for α-SMA (TRITC, red) and vimentin (FITC, green) in normal esophagus (A and B) and GERD esophageal biopsies (C and D) was viewed with an immunofluorescent microscope. In the normal esophagus (A and B), endothelial cells forming blood vessels are organized in a circular configuration (white arrowheads) and coexpress α-SMA and vimentin. Scattered vimentin-only-expressing fibroblasts (green, dashed arrows) and single, spindle-shaped α-SMA- and vimentin-coexpressing cell myofibroblasts (white arrows) are also observed. GERD biopsies (C and D) demonstrate an increase in stromal cellularity. Two representative images are shown. C: blood vessels (white arrowheads) and α-SMA-positive, vimentin-positive, spindle-shaped cells (white, solid arrows) are observed. D: an increase in vimentin-expressing fibroblasts (green, dashed arrows) is observed as well.
An Increase in Esophageal Myofibroblasts in GERD Biopsies
We then aimed to investigate the stromal cell population in GERD esophagus. Our investigation was somewhat limited by the lack of standardization among esophageal biopsies acquired in GERD and by the predominance of samples that lack representative amounts of stroma in archived samples. Full-thickness esophageal specimens that are typically available by resections for malignancy are not typically available in GERD. To circumvent this limitation, we focused our investigation on reviewing biopsies that had representative stroma amenable to examination with H&E and immunostaining.
Three fields of vision at 40× magnification were evaluated for each normal esophagus. The number of DAPI-stained nuclei in the subepithelial region was counted and summed. There was little variability across each field of vision analyzed for normal esophagus. The average number of total DAPI-stained nuclei counted for each normal esophagus was 301 (n = 5; range 221–403). When blood vessels were excluded, the average number of total DAPI-stained nuclei was 174 (n = 8; range 153–192).
Compared with normal esophagus (Fig. 3, A and B), the stroma in diseased esophagus (Fig. 3, C and D) appeared more cellular. Because there was less available stroma in GERD biopsies vs. normal, full-thickness esophagus, all fields of vision that had stroma in GERD biopsies were analyzed (minimum of three and maximum of seven). In GERD biopsies, all DAPI-stained nuclei were counted, and blood vessels were not excluded. The average number of total DAPI-stained nuclei counted for GERD esophagus (n = 5) was 454 (range 313–519).
Each DAPI-stained nuclei in normal and GERD esophagus was evaluated individually for expression of α-SMA and vimentin. When all cells in the lamina propria were evaluated, a similar percentage of α-SMA- and vimentin-coexpressing cells was observed in normal vs. GERD esophagus [n = 5 each, 44 vs. 41%, P = not significant (NS)]. The remaining stromal cells (55–59%) were vimentin-positive fibroblasts. Of note, when blood was excluded from quantification in normal samples, the percentage of α-SMA- and vimentin-coexpressing stromal cells decreased to 22%. The remaining stromal cells (75%) were vimentin-positive fibroblasts. In both iterations, a few cells expressed neither marker, and all α-SMA-positive cells were vimentin positive.
Whereas spindle-shaped cell myofibroblasts were clearly distinct from endothelial cells in normal esophageal specimens, sectioning and biopsy orientation in GERD biopsies made it difficult to ascertain definitively whether the observed α-SMA+vimentin+ cells were endothelial cells or myofibroblasts. The similar percentage of α-SMA+vimentin+ cells in normal vs. GERD esophagus suggested an inability to distinguish myofibroblasts from endothelial cells using this combination of markers.
To determine whether the α-SMA+vimentin+ cells were myofibroblasts or endothelial cells, we performed immunostaining for α-SMA and CD31. Each DAPI-stained nuclei was evaluated for expression of α-SMA and CD31. In the normal esophageal stroma, α-SMA+CD31+ endothelial cells were readily identified (Fig. 4, A and B). We occasionally observed α-SMA+CD31− myofibroblasts adjacent to the squamous epithelium. In GERD biopsies (Fig. 4, C and D), there appeared to be a greater population of α-SMA+CD31− myofibroblasts compared with normal specimens. The number of α-SMA+CD31− myofibroblasts and α-SMA+CD31+ endothelial cells was determined and normalized to the total number of DAPI nuclei counted. Quantification demonstrated an increase in myofibroblasts (SMA+CD31−) in GERD vs. normal esophagus (7.8 vs. 2.3%, P < 0.05; Fig. 4E). The percentage of α-SMA+CD31+ endothelial cells was similar in GERD and normal esophagus (33.1 vs. 22.9%; P = NS).
Fig. 4.

Expression of α-SMA-positive, CD31-negative stromal cells in normal and GERD esophagus. α-SMA (TRITC, red) and CD31 (FITC, green) immunostaining was performed on normal esophagus (A and B) and GERD esophageal (C and D) biopsies to distinguish myofibroblasts (α-SMA+CD31−) from endothelial cells (α-SMA+CD31+). Images were viewed with an immunofluorescent microscope. In normal esophagus (A and B), coexpression of α-SMA and CD31 is readily observed in endothelial cells forming blood vessels (arrowheads). Occasional α-SMA+CD31− myofibroblasts (arrows) are also observed. In GERD biopsies (C and D), α-SMA+CD31− myofibroblasts are observed more frequently (arrows). Representative images of 2 GERD biopsies are shown; magnification, 400× oil. E: quantification of SMA+CD31− cells was performed. GERD esophagi (n = 5) had an increase in the percentage of subepithelial SMA+CD31− myofibroblasts compared with normal esophagus (mean 7.8 vs. mean 2.3%, P < 0.05).
Activation of Inflammatory Pathways in GERD Myofibroblasts
An increase in IL-6 expression has been reported previously in mucosal biopsies from GERD patients (15), although the cellular compartment responsible for this increase has not been identified. We evaluated IL-6 expression by immmunostaining of normal and GERD esophagus. In normal esophagus, IL-6 costaining was detected consistently in α-SMA+ blood-vessel walls (Fig. 5A). Spindle-shaped α-SMA+ myofibroblasts (Fig. 5A) did not express IL-6. In GERD biopsies, IL-6 was again detected in blood-vessel walls (Fig. 5B). Extracellular IL-6 was detected near α-SMA+ spindle-shaped cell myofibroblasts (Fig. 5B), α-SMA+ endothelial cells organized in a circular configuration (Fig. 5B), and α-SMA-negative fibroblasts (Fig. 5B). The faint ECM expression of IL-6 is consistent with its classification as a secreted cytokine.
Fig. 5.

Activation of inflammatory pathways in GERD myofibroblasts. Immunostaining for IL-6 and α-SMA was performed on paraffin-embedded, formalin-fixed esophageal biopsies from normal (n = 5) and GERD (n = 5) esophageal biopsies. Slides were viewed with an immunofluorescent microscope (A and B). Normal esophagus (A) demonstrates spindle-shaped α-SMA+ myofibroblasts (arrows) and endothelial cells forming blood vessels (arrowheads); IL-6 (FITC, green) costaining is observed in endothelial cells only. A representative GERD biopsy (B) demonstrates IL-6 expression (FITC, green) in the ECM (green, solid arrows). Spindle-shaped α-SMA+ myofibroblasts with surrounding IL-6 are observed (white arrows). Blood vessels again demonstrate IL-6 expression in endothelial cells (arrowheads). Magnification, 40×. Immunostaining for p65 and α-SMA was also performed in normal and GERD esophagus. Slides were viewed with a confocal microscope (C–E). In normal esophagus (C), cytoplasmic and nuclear p65 expression was observed in endothelial cells (arrowheads). Myofibroblasts (arrow) did not express nuclear p65. In a representative GERD biopsy (D), nuclear translocation of p65 was observed in myofibroblasts (white arrows). E: a magnification of the area demarcated by the box in D is shown.
IL-6 is an NF-κB transcription target, and in the setting of NF-κB activation, the p65 member of this complex translocates to the nucleus. In normal esophagus, cytoplasmic and nuclear p65 expression was observed in endothelial cells (Fig. 5C). In GERD biopsies, nuclear expression of p65 was observed in single, subepithelial, spindle-shaped, α-SMA-positive cells that were clearly not part of blood vessels (Fig. 5, D and E). Nuclear expression of p65 was also observed in scattered endothelial cells (Fig. 5, D and E).
Establishment and Characterization of Primary Cultures of Human Esophageal Stromal Cells
We aimed to investigate further the role of esophageal stromal cells by establishing and characterizing primary cultures and investigating their functional properties in response to GERD injury. Stromal cell cultures were prepared as per methods. Examination of primary cultures with an inverted microscope within 24 h of plating demonstrated a suspension of partially floating cells, loosely adherent to the plate bottom (Fig. 6A). The suspension of heterogeneous cells flattens out within 24–48 h of plating and adheres to the plate bottom, followed by an outgrowth of adherent cells with spindle-shaped morphology (Fig. 6B). Adherent esophageal stromal cells retained spindle-shaped morphology at low (Fig. 6C) and high (Fig. 6D) confluence.
Fig. 6.

Primary cultures of human esophageal stromal cells. Within hours of plating, a suspension of partially floating cells, loosely adherent to the plate bottom, is observed (A; arrow; magnification, 100×). Within 24–48 h, the mixed cell suspension flattens out and adheres to the plate bottom, followed by an outgrowth of spindle-shaped cells (B). Representative images of human stromal cell cultures at low (C) and high (D) confluence are shown at 100× magnification.
α-SMA and vimentin mRNA was detected readily across all esophageal primary stromal cultures, as well as in colonic myofibroblast 18Co cells (Fig. 7). The relative mRNA expression of vimentin was greatest, followed by expression of α-SMA across all cultures. mRNA expression of epithelial markers E-cadherin and cytokeratin was negligible in esophageal stromal cells. mRNA expression of hematopoietic and endothelial markers CD45 and CD31 was also undetectable.
Fig. 7.

mRNA expression of myofibroblast, epithelial, and hematopoietic markers in primary cultures of human esophageal stromal cells. RNA was harvested from esophageal stromal cell primary cultures [myofibroblasts 1, 2, and 3 (MF1, MF2, and MF3)], 18Co colon myofibroblasts, Het-1A esophageal epithelial cells, and commercially purchased peripheral blood mononuclear cells (PBMCs). cDNA was generated, and α-SMA, vimentin, E-cadherin, and CD45 mRNA expression was evaluated by quantitative RT-PCR (qRT-PCR). mRNA expression of α-SMA and vimentin was readily detected in esophageal stromal cells (MF1-3) and in colon myofibroblast line 18Co cells. mRNA expression of these myofibroblast markers was variable across primary cultures. E-Cadherin and CD45 mRNA expression was undetectable in esophageal stromal cells and 18Co cells. E-Cadherin and CD45 mRNA was readily detected in esophageal epithelial cell Het-1A and PBMCs, respectively. Results shown are representative of 3 individual experiments in primary cultures derived from 3 normal human esophagi.
Immunofluorescent staining for intracellular cytoskeletal proteins demonstrated that esophageal stromal cells in primary culture express α-SMA and vimentin (Fig. 8) and lacked expression of cytokeratin (data not shown). Flow cytometric analysis of cell-surface markers demonstrated that human esophageal stromal cells expressed stromal cell marker CD90. Esophageal stromal cells did not express hematopoietic, endothelial, or epithelial cell-surface markers CD45, CD31, or CD324 (E-cadherin), respectively (Fig. 9). These observations suggested that human esophageal stromal cells in primary culture exhibit a myofibroblast phenotype and will henceforth be referred to as esophageal myofibroblasts or as human esophageal myofibroblast (HuEso MF)1, MF2, and MF3, reflecting unique patient origins.
Fig. 8.

Primary cultures of human esophageal stromal cells express α-SMA and vimentin protein. Esophageal stromal cells at passages 5–15, grown on chamber slides, demonstrate expression of α-SMA (A and D, red rhodamine) and vimentin (B and E, green FITC). Primary cultures coexpress α-SMA and vimentin (C and F, yellow). Blue depicts DAPI nuclear stain.
Fig. 9.

Cell-surface marker characterization of primary cultures of human esophageal stromal cells. A: forward-scatter-area (FSC-A) and side-scatter-area (SSC-A) of cells grown in primary culture are shown. A total of 89.2% of events was gated. Stromal cell marker CD90 is present on the majority (97.7%) of human esophageal stromal cells in primary culture (B). Esophageal stromal cells in primary culture cells lack expression of hematopoietic CD45 (C), endothelial CD31 (D), and epithelial CD324 markers (E). Positive controls included commercially purchased human mononuclear cells (HemaCare). Isotype controls were used to determine nonspecific staining for each antibody.
HuEso MFs Increase in IL-6 and IL-8 Secretion in Response to Acid
Given our interest in GERD, we were interested in the myofibroblast cytokine secretory response to acid. We first conducted pilot studies to determine the duration of acid treatment. Treatment of esophageal myofibroblasts with pH 4.5-acidified media for 15 min did not result in cytotoxicity at 3, 6, or 24 h. Treatment with pH 4.5-acidified media for 30 min, however, did result in a slight increase in cytotoxicity at 3 and 6 h (P < 0.05 compared with no treatment) and a three-fold increase in toxicity at 24 h compared with no treatment (P < 0.05; Fig. 10A). Acid studies, therefore, were limited to 15 min of treatment.
Fig. 10.
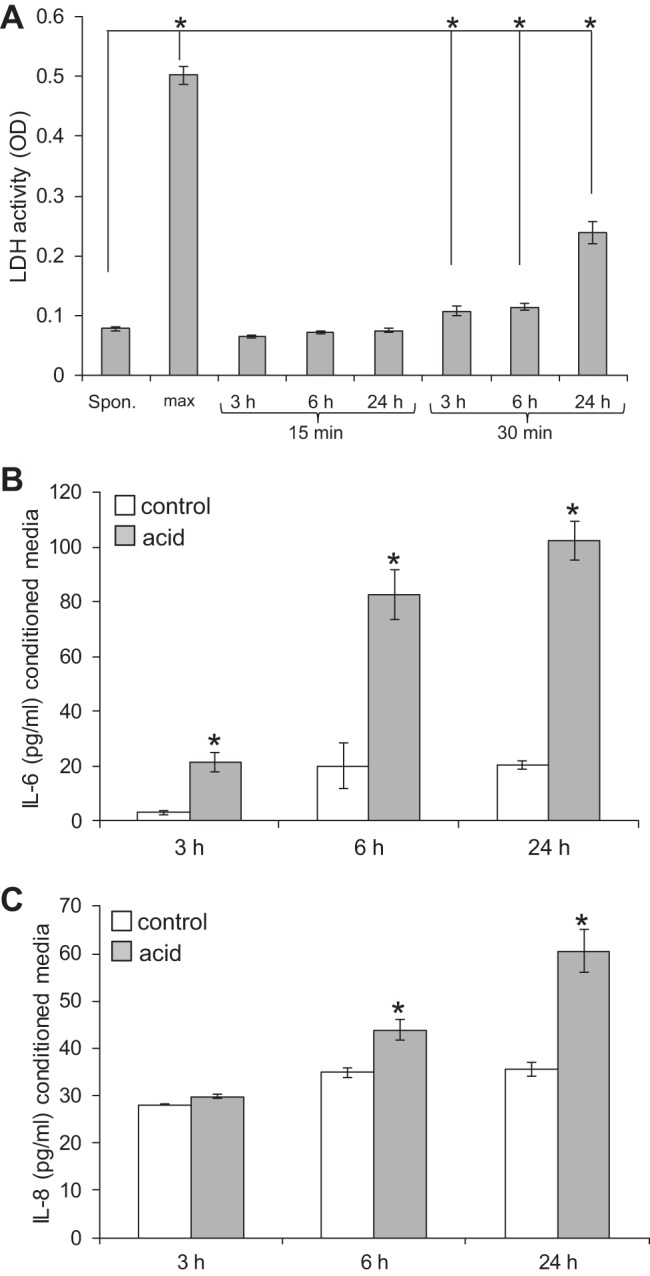
Human esophageal myofibroblasts (HuEso MFs) secrete of proinflammatory cytokines in response to noncytotoxic acid (A). Conditioned media of cells, 3, 6, and 24 h after treatment with pH 4.5-acidified media for 15 and 30 min, were evaluated for lactate dehydrogenase (LDH) cytotoxic activity and compared with spontaneous (Spon.) and maximum (max) LDH release. Treatment of cells for 15 min resulted in LDH activity similar to spontaneous release [optical density (OD) = 0.07] and did not result in cytotoxicity. Treatment of cells for 30 min resulted in a slight but significant increase of LDH activity at 3 and 6 h and a 3-fold increase in cytotoxicity at 24 h. *P < 0.05 compared with spontaneous LDH release. B: esophageal myofibroblasts treated with acidified media for 15 min increase IL-6 secretion at 3, 6, and 24 h. *P < 0.01 compared with recovery time in untreated cells. C: esophageal myofibroblasts treated with acidified media increase secretion of IL-8 at 6 and 24 h. *P < 0.05 compared with recovery time in untreated cells.
To focus our investigation on the most abundantly secreted cytokines in response to acid, we began with a preliminary Hu35-plex cytokine/chemokine screen. Esophageal myofibroblasts demonstrated a profound increase in secretion of IL-6 and IL-8. ELISA then performed on conditioned media demonstrated that esophageal myofibroblast secretion of IL-6 and IL-8 was enhanced in response to acidified media. Although absolute values of cytokine secretion differed across primary cultures, an increase in IL-6 secretion in response to treatment with acid was observed consistently across all cultures at 3, 6, and 24 h after treatment (Fig. 10B). Secretion of IL-8 increased after 6 and 24 h (Fig. 10C). IL-1β secretion was not detected by ELISA, consistent with the results of the screen.
HuEso MF-Like Stromal Cells Express TRPV1
The effects of acid on cells have long been considered to be a consequence of caustic injury or a nonspecific danger signal and survival response. TRPV1 is a nonselective ion channel activated by a variety of mediators, including hydrogen ions released from tissues during inflammation (44) or from gastric reflux (5). Expression of this receptor has been reported in human esophageal mucosal biopsies (10) and the human esophageal epithelial cell line Het-1A (22). We evaluated TRPV1 mRNA expression by quantitative RT-PCR (qRT-PCR) and observed that HuEso MFs express TRPV1 mRNA. mRNA expression of TRPV1 was greatest in Het-1A cells, followed by expression in esophageal myofibroblasts, and least in the colon myofibroblast line 18Co (Fig. 11A). Immunoblot demonstrated expression of a 97-kDa band in esophageal myofibroblasts (Fig. 11B), consistent with the size of TRPV1 reported in Het-1A cells (17, 22). TRPV1 protein could not be detected in 18Co cells by immunoblot. Immunostaining demonstrated predominantly cell-surface and cytoplasmic TRPV1 expression in esophageal myofibroblasts (Fig. 11C3) with some nuclear speckling in a pattern similar to that observed in Het-1A (Fig. 11, C1 and C2). Faint but detectable expression of TRPV1 was observed in 18Co cells when evaluated by immunocytochemistry, however. This discrepancy is likely a consequence of the limitations of the application of the available TRPV1 antibody in the detection of TRPV1 by immunoblot.
Fig. 11.

HuEso MFs express functional transient receptor potential channel vanilloid subfamily 1 (TRPV1), and (A) TRPV1 mRNA expression was determined in Het-1A, HuEso MFs (MF1-3), and 18Co by qRT-PCR. Fold change compared with expression in Het-1A cells is shown. Data represent the means ± SE of 3 independent experiments. B: immmunoblot demonstrates a 97-kDa band in cell lysates of HuEso MFs (MF1-3) consistent with the reported size of TRPV1. A band of similar size is observed in Het-1A cells. TRPV1 expression was not detected by immunoblot in 18Co cells. β-Actin was used as the loading control. C1–C3: immunostaining was performed for TRPV1, and images were examined with confocal microscopy. DAPI + FITC green TRPV1 images (top) and FITC green alone (bottom) are shown. C1: TRPV1 expression, reported previously in epithelial Het-1A, is demonstrated in the cuboidal cytoplasm of these cells. C2: cytoplasmic expression of TRPV1 (FITC, green) is observed in esophageal MFs. C3: TRPV1 expression in 18Co is faint compared with Het-1A and esophageal MF. The speckled nuclear staining is observed in all examined cells and is considered an artifact (magnification, 400× oil). D: after a pilot study was performed to determine nontoxic concentrations of TRPV1 antagonist AMG9810, HuEso MFs were treated with 1–10 μM AMG9810. At 1.0 μM AMG9810, an increase in constitutive and acid-stimulated IL-6 secretion is observed. At 2.5, 5, and 7.5 μM AMG9810, constitutive secretion of IL-6 remains unaffected, and acid-stimulated IL-6 secretion is suppressed progressively. Maximal suppression of acid-stimulated IL-6 secretion occurs at 7.5 μM AMG9810. At doses of AMG9810 >7.5 μM, constitutive and stimulated IL-6 secretion is suppressed. IL-6 secretion was determined in the presence of AMG9810 in acid- and no acid-treated cells. ∧P < 0.001 in the acid vs. no acid treatment for each dose of AMG9810; *P < 0.001 compared with IL-6 secretion in the no acid, no Bay 11-7082 control; #P < 0.001 compared with acid-treated cell without Bay 11-7082. Results represent the means ± SE of 3 individual experiments in primary cultures derived from 3 normal human esophagi. P values were determined with ANOVA, followed by Bonferroni's multiple comparisons test.
The mRNA and protein detection of this putative acid receptor suggested a potential role for this receptor in signaling. To determine whether cytokine secretion that we observed in response to acid was mediated via TRPV1 activation, we treated cells with acidified media in the presence of a TRPV1 inhibitor AMG9810 (Fig. 11D). Based on the findings of a pilot study conducted to determine nontoxic concentrations of AMG9810 that would inhibit TRPV1 activation, we cultured HuEso MFs with 1–10 μM of AMG9810. At 1.0 μM AMG9810, an increase in constitutive and acid-stimulated IL-6 secretion was observed. Between 2.5 and 7.5 μM AMG9810, constitutive secretion of IL-6 remained unaffected, whereas stimulated secretion was progressively suppressed. Maximal suppression of acid-stimulated IL-6 secretion occurred with 7.5 μM AMG9810 (P < 0.001). At >7.5 μM AMG9810, constitutive and stimulated IL-6 secretion is inhibited.
Esophageal Myofibroblasts Express Functional TLR4
A potential role of TLRs and TLR signaling has been described recently in the esophageal epithelium (18, 19). We were curious whether HuEso MFs expressed TLRs and evaluated TLR4 and TLR2 mRNA expression by qRT-PCR. Although TLR2 and TLR4 mRNA were detected, TLR4 mRNA was expressed at least 30-fold more abundantly than TLR2 in all cultures (Fig. 12A). mRNA expression of TLR4 was similar across all esophageal myofibroblasts. Immunoblot demonstrated a protein band at 94 kDa, consistent with the reported size of TLR4 in all esophageal primary cultures (Fig. 12B). A protein band of similar size was observed in 18Co colon myofibroblasts, HeLa cervical epithelial cells, and Caco2 colon epithelial cells (Fig. 12B), as reported previously (13, 47). TLR4 expression was detected in the membrane and cytoplasm of esophageal myofibroblasts (Fig. 12C), consistent with the pattern observed in human aortic valve interstitial cells (23) and similar to TLR4 expression in 18Co colon myofibroblasts. Negligible TLR4 protein was detected by immunocytochemistry in Het-1A cells.
Fig. 12.

HuEso MFs express Toll-like receptor 4 (TLR4) and secrete proinflammatory cytokines in response to TLR4 ligands. A: TLR2 and TLR4 mRNA expression was evaluated across 3 primary cultures by qRT-PCR. TLR4 mRNA was expressed more profoundly than TLR2. Results shown are representative of 3 individual experiments in primary cultures derived from 3 normal human esophagi. B: immunoblot was performed to evaluate expression of the TLR4 protein (94 kDa) in esophageal myofibroblasts MF1-3, colon myofibroblast 18Co, human cervical cancer cell line HeLa, and human colorectal cancer Caco2 cells as positive controls. β-Actin was used as the loading control. C: immunofluorescent staining for TLR4 was performed in esophageal myofibroblasts (MF1), 18Co (positive control), and Het-1A (negative control). Images of the TLR4 protein, obtained by fluorescence microscopy, demonstrate TLR4 expression in esophageal myofibroblasts (red TLR4; blue DAPI; magnification, 200×), similar to that observed in 18Co. D: HuEso MFs treated with LPS increase IL-8 secretion at 3, 6, and 24 h. *P < 0.05 vs. no treatment. E: esophageal myofibroblasts treated with TLR4 ligands LPS and high-mobility group box 1 protein (HMGB1) increase secretion of IL-6 after 6 h (*P < 0.005 vs. 6 h control) and 24 h (#P < 0.005 vs. 24 h control). Results shown represent the means ± SE of 3 independent experiments conducted with primary cultures derived from 3 normal human esophagi.
Esophageal Myofibroblasts Increase IL-6 and IL-8 Secretion in Response to TLR4 Ligands
To determine if TLR4 was functional in esophageal myofibroblasts, we treated esophageal myofibroblasts with the TLR4 ligand bacterial LPS and determined cytokine and chemokine secretion. Hu35-plex cytokine/chemokine screen demonstrated an increase in secretion of IL-8, MCP-1, IL-6, GRO, and IP-10 in response to LPS (data not shown). Hu35-plex findings were first confirmed with ELISAs for IL-8, MCP-1, and IL-6. We observed an increase in esophageal myofibroblast secretion of IL-8 that was most profound at 24 h (Fig. 12D) and a nearly five-fold increase in MCP-1 secretion at 24 h (not shown). To model inflammatory stress further, HuEso MFs were also treated with another TLR4 ligand, HMGB1. We observed a several-fold increase in IL-6 at 6 and 24 h in response to LPS and HMGB1 (Fig. 12E).
HuEso MFs Increase Cytokine Secretion in Response to Treatment with Acid and TLR4 Ligands via an NF-κB-Dependent Mechanism
Activation of the NF-κB pathway in esophageal epithelium and impairment of barrier function have been demonstrated in murine models of GERD (7), and implicated cytokines IL-6 and IL-8 are NF-κB target genes (7, 27). Given an increase in IL-6 and IL-8 secretion in response to acid and TLR4 ligands, we suspected activation of NF-κB in acid- and TLR4 ligand-treated HuEso MFs. Under resting conditions, p65 is one of the cytoplasmic components of the NF-κB complex, bound to the IκB protein family. With activation of the pathway, IκB phosphorylation results in release of the NF-κB complex, followed by nuclear translocation and transcription of NF-κB target genes (49). We first observed nuclear p65 expression, 10 min after acid treatment was complete. Nuclear translocation of p65 was most profound in esophageal myofibroblasts, 30 min after acid treatment (Fig. 13B). Three hours after acid treatment, p65 was, once again, cytoplasmic.
Fig. 13.

Activation of the NF-κB pathway in acid- and LPS-treated esophageal myofibroblasts. Primary cultures of esophageal myofibroblasts were grown in chamber slides in serum-free myofibroblast media and fixed at 10 min, 30 min, 1 h, and 3 h. These cells served as controls (A). Esophageal myofibroblasts treated with pH 4.5-acidified media for 15 min were then recovered in serum-free myofibroblast media for 10 min, 30 min, 1 h, and 3 h and fixed (B). Cells cultured in LPS were fixed at similar time points (C). Cells were immunostained for p65 with DAPI counterstain. Untreated cells cultured in serum-free myofibroblast media demonstrate cytoplasmic p65 expression at all times (A, green cytoplasm, blue DAPI-stained nucleus). Cells treated with acidified media (B) demonstrate green specks in the nucleus, consistent with nuclear translocation beginning at 10 min (arrows), with more intense nuclear staining of p65 observed at 30 min and 1 h (arrows) compared with untreated cells. At 3 h, esophageal myofibroblasts treated with acidified media demonstrate predominantly cytoplasmic p65 staining. Magnification, 400×. Cells treated with LPS (C) demonstrate green nuclear specks with minimal cytoplasmic staining, consistent with near-complete p65 nuclear translocation, beginning at 30 min and persisting at 3 h. D: cell lysates were harvested from primary cultures grown in serum-free myofibroblast media (SF) and from cultures treated with LPS and harvested at indicated time points after treatment (30 min, 1 h, and 3 h). Immunoblots for inhibitor of NF-κBα (IκBα) were performed on protein harvested from untreated cells and cells treated with LPS. Blots were stripped and reprobed for tubulin as loading control. At each time point, a decrease in IκBα expression is observed after treatment with LPS (top). Relative quantification of IκBα bands was achieved by normalizing the densitometry of the band to the tubulin-loading control using ImageJ software.
Nuclear translocation of p65 in esophageal myofibroblasts was also evaluated in LPS treatment. Nuclear translocation of p65 persists for the duration of the treatment with LPS and appears maximal at 30 min into treatment (Fig. 13C). We further evaluated activation of the NF-κB pathway in LPS-treated cells by immunoblot for IκBα expression. We observed a decrease in IκBα expression at 30 min, 1 h, and 3 h (Fig. 13D), which was confirmed with relative quantification of band densitometry normalized to tubulin.
To confirm further the contribution of the NF-κB pathway in cytokine secretion in acid- and LPS-treated myofibroblasts, cytokine secretion was evaluated in the presence of the NF-κB inhibitor Bay 11-7082, which prevents phosphorylation and degradation of IκBα (7, 49). A dose-response study was performed to determine the optimal concentration of this NF-κB inhibitor in acid-treated cells at the 6-h recovery time point. In the presence of increasing concentrations of Bay 11-7082, stimulated secretion of IL-6 decreased progressively. At concentrations >5 μM, however, constitutive secretion of IL-6 (treated with nonacidified media) was also decreased. Maximal suppression of IL-6 secretion in response to acidified media was achieved in the presence of 5 μM Bay 11-7082 (Fig. 14A).
Fig. 14.
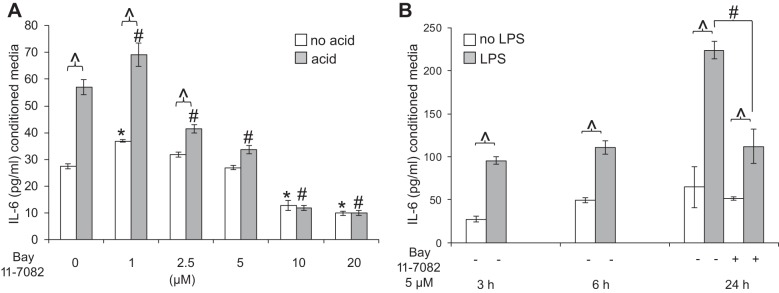
HuEso MFs secrete IL-6 in response to acid and TLR4 ligands via activation of the NF-κB pathway. A: IL-6 secretion was determined in the presence of increasing concentrations of Bay 11-7082 in acid- and no acid-treated cells at 6 h. Bay 11-7082 at 5 μM resulted in a profound decrease in IL-6 secretion without affecting constitutive secretion. ∧P < 0.001 in the acid vs. no acid treatment for each dose of Bay 11-7082; *P < 0.001 compared with IL-6 secretion in the no acid, no Bay 11-7082 control; #P < 0.001 compared with the acid-treated cell without Bay 11-7082. Results represent the means ± SE of 3 individual experiments in primary cultures derived from 3 normal human esophagi. P values were determined with ANOVA, followed by Bonferroni's multiple comparisons test. B: HuEso MFs increase IL-6 secretion at 3, 6, and 24 h in response to LPS. Based on pilot studies, 5 μM Bay 11-7082 was used to determine the effect of NF-κB inhibition on LPS-stimulated IL-6 secretion, which at 24 h, was inhibited by 5 μM Bay 11-7082. Bay 11-7082 treatment was not evaluated at 3 and 6 h. ∧P < 0.001 in LPS vs. no LPS treatment for each time point; #P < 0.01 in IL-6 secretion in LPS-treated cells without vs. with Bay 11-7082. Results represent the means ± SE of 3 individual experiments in primary cultures derived from 3 normal human esophagi. P values were determined with paired Student's t-test.
We also investigated the contribution of the NF-κB pathway in mediating esophageal myofibroblast IL-6 secretion in response to TLR4 ligands. Based on our observation that 5 μM Bay 11-7082 achieved inhibition of acid-stimulated IL-6 secretion without affecting constitutive secretion, we treated esophageal myofibroblasts with the TLR4 ligand in the presence of 5 μM Bay 11-7082. We observed that IL-6 secretion of IL-6 in response to LPS was suppressed in the presence of Bay 11-7082 at 24 h (Fig. 14B).
DISCUSSION
Mechanisms mediating esophageal injury, repair, and inflammation are complex and incompletely understood (2). We can postulate that luminal agents gain access to the mesenchyme through DIS or directly through a barrier breach. DIS, observed in patients with nonerosive and erosive GERD, allow for the infiltration/penetration of deeper epithelial layers and stromal cells with luminal agents (45). Acid-mediated epithelial cell necrosis (28, 29) and immune and epithelial cell-derived cytokines contribute to mucosal injury (43). Cellular debris (20) and the microbial contents of the esophagus (48) may further promote the inflammatory response. Investigation of the sources and effect of inflammatory mediators have been limited predominantly to study of epithelial cells. Although esophageal mesenchymal cells have been reported as sources of inflammatory cytokines (34), studies of the normal human esophageal lamina propria are frequently descriptive (2). Mechanistic studies frequently use fibroblast lines (26), and the use of well-characterized primary cultures that more accurately reflect the in vivo environment is limited.
Our aims were to characterize the stromal cell population in the normal human esophagus and to begin to define the stroma in GERD. We began our approach by examination of the stroma in normal esophagus and in GERD biopsies. We performed immunostaining for myofibroblast markers and for activation of inflammatory pathways implicated in GERD injury. We then used well-characterized primary cultures of esophageal stromal cells treated with injurious agents encountered in GERD to establish an in vitro model of GERD injury and examined stromal cell cytokine secretion.
We have demonstrated that the human esophageal stroma contains a small percentage of spindle-shaped, subepithelial myofibroblasts that increase in GERD. In addition, we observed an increase in stromal expression of IL-6, an NF-κB transcription target and cytokine implicated in GERD pathogenesis. Activation of the NF-κB pathway in esophageal myofibroblasts suggests that these cells may be contributing to the cytokine milieu observed in GERD. Overall, these observations support a role for myofibroblasts in injury, repair, and inflammation in the esophagus.
To investigate further the role of these cells, we established primary cultures of esophageal stromal cells. These cells have myofibroblast morphology and gene expression, lack endothelial and immune-cell markers, and express CD90, consistent with a myofibroblast phenotype (32). Myofibroblasts are potentially involved in numerous signaling pathways relevant to esophageal epithelial injury, repair, and inflammation. For this paper, we focused on their role in inflammation. We have shown that esophageal myofibroblasts respond to acid and TLR4 ligands with secretion of IL-6 and IL-8. The esophageal myofibroblast response to acid appears to be, at least partially, via the putative acid receptor TRPV1, as inhibition of this receptor markedly suppressed IL-6 secretion. Furthermore, pharmacological inhibition of the NF-κB pathway markedly suppressed acid and TLR4 ligand-stimulated IL-6 secretion, suggesting at least partial involvement of this pathway in mediating myofibroblast cytokine secretion. Our work suggests that HuEso MFs in GERD are not mere bystanders but rather, active participants in inflammation via selective secretion of cytokines in response to epithelial or luminal sources of stimulation.
Delineation of the role of stromal cells, such as esophageal myofibroblasts, is critical, as much of the study of esophageal disorders has been limited to the epithelium. We focused on IL-6 secretion, given its increased expression in mucosal biopsies from patients with GERD-related esophagitis (15, 27) and in stimulated esophageal epithelial cell lines (18). Our observations that esophageal myofibroblasts secrete IL-6 and the chemoattractants IL-8 and MCP-1 are in line with our hypothesis that these cells contribute to the cytokine milieu. Esophageal biopsies in GERD are often limited to the epithelium with at best little stromal component. Therefore, studies reporting an increase in cytokine expression in GERD biopsies have often overlooked the stromal contribution. Whereas structural alterations, such as an increased shedding of microvesicles from the surface of stromal cells, have been described in the stroma of Barrett's esophagus along the metaplasia-dysplasia-adenocarcinoma sequence (3), this level of scrutiny has not been achieved in GERD esophagitis, the condition that precedes the development of Barrett's. Whether the observed increase in myofibroblasts is a consequence of an increase in proliferation of existing cells or a consequence of infiltration of bone marrow-derived cells was not investigated in this study. Although the role of bone marrow-derived cells in GERD has not been formally investigated, a possible contribution to epithelial and stromal components has been suggested in complications of long-standing GERD, such as Barrett's esophagus and esophageal adenocarcinoma (14). An improved understanding of the stroma before the development of preneoplastic or neoplastic states, i.e., an understanding of stroma in normal and GERD esophagus, would allow for timely prevention and perhaps more targeted therapy of more advanced pathology.
This study adds to the existing relative paucity of literature on esophageal mesenchymal cells (9, 34) in several ways. To begin to determine mechanisms by which these cells contribute to injury, repair, and inflammation, we attempted to model the GERD environment by treating esophageal myofibroblasts with potentially encountered, injurious agents. Our work delineates two possible mechanisms by which myofibroblasts contribute to esophageal inflammatory disorders. Our findings suggest that esophageal myofibroblasts can respond to luminal- and epithelial-derived noxious stimuli via putative acid receptors and TLR4 and that on an intracellular level, secretion of cytokines in response to both stimuli is mediated, at least partially, via the NF-κB pathway.
The effects of acid on esophageal tissue are reportedly mediated potentially via a TRPV1 (5), which is a member of the transient receptor potential family of ion channels that includes seven subfamilies that participate in cellular activation and signaling. TRPV1 expression has not been reported previously in esophageal stromal cells or myofibroblasts. We have begun to address the definitive role of the TRPV1 receptor in regulating the response to acid in esophageal myofibroblasts by using the selective TRPV1 inhibitor AMG9810. Interestingly, there have been recent publications detailing the interaction of TRPV1 with the EGF receptor (4). Although the delineation of this interaction in our cells is beyond the scope of this study, it is plausible that this interaction may partially account for the increase in constitutive IL-6 secretion observed at lower concentrations (1 μM) of AMG9810.
Our findings also describe a novel role for TLRs in esophageal myofibroblasts, in addition to their described role in the epithelium (18). As the conduit from the oropharynx to the distal GI tract, the esophagus is exposed to oropharyngeal and food-borne pathogens (18). In the setting of injury, there is also potential release of necrotic cell debris, which can be engaged by TLR4. We have demonstrated that esophageal myofibroblasts express pattern receptors for and respond to microbial components with cytokine secretion. It has been postulated that by favoring an inflammatory environment, acid reflux might shift the esophageal flora from a type 1 to type 2 flora that consists of gram-negative bacteria (48). We can postulate that esophageal myofibroblasts have the potential to engage with altered flora via TLR4.
We have also demonstrated that esophageal myofibroblasts augment IL-6 secretion in response to the TLR4 ligand HMGB1, a chromatin protein that serves as an alarmin when released from necrotic cells (20). It can be envisaged that in the setting of erosive disease, myofibroblasts engage bacterial components or necrotic cell debris (20) or potentially other endogenous TLR4 ligands, such as heat-shock proteins, hyaluronic acid, and β-defensin 2 (21). Our work suggests that esophageal myofibroblasts are potential sensors of danger signals.
Activation of the NF-κB pathway and upregulation of proinflammatory genes in acid-treated esophageal epithelial cells have been reported previously (1). Reports of activation of this pathway in the stromal compartment are more limited. Activation of the NF-κB pathway in human esophageal fibroblasts in response to direct treatment with acid and to conditioned media of acid-treated human esophageal squamous epithelium has been described previously (9). Our study demonstrates similar activation of the NF-κB in esophageal myofibroblasts in response to acid and TLR4 ligands. Of note, stromal cell generation and characterization in this prior study differed from the present study. In addition, consequences of cytokine secretion from esophageal stromal cells in the setting of NF-κB inhibition have not been studied previously. We have demonstrated that the NF-κB-mediated increase in esophageal myofibroblast cytokine secretion in response to acid and TLR4 ligands is suppressed with inhibition of this pathway. The present study does not exclude the role of other potentially involved pathways, such as STAT1 or p38 MAPK.
Immunostaining for IL-6 and p65 in GERD biopsies supports the contention that esophageal myofibroblasts are one of the stromal constituents that contribute to the inflammatory cytokine milieu in GERD. Endothelial cells and fibroblasts are other likely contributors, as demonstrated by our observation of IL-6 and nuclear p65 in these cells. Although these stromal constituents were not the focus of our current work, their contribution to pathogenesis certainly merits investigation in future studies that more fully recapitulate the complexity of the esophageal mucosa. Our work is an important first step in investigating the importance and contribution of these cells to inflammatory esophageal disorders.
Our study has some limitations. The majority of normal samples used in this study was full-thickness specimens, whereas GERD samples were derived from biopsies. As such, we may have unintentionally masked potential differences in the stroma of these two groups and skewed the results toward acceptance of the null hypothesis (no difference between normal and GERD stroma). We minimized this limitation by limiting examination of the stroma to the subepithelial region in normal and GERD esophagus. The lack of a difference in quantification of α-SMA-positive-, vimentin-positive-expressing cells is likely a consequence of difficulties differentiating blood vessels from myofibroblasts and prompted CD31 immunostaining. We were only then able to observe a difference in the α-SMA+CD31− myofibroblast population in normal vs. GERD esophagus.
Another limitation is the small sample size for GERD specimens and the lack of injury stratification between samples. Our work was not meant to be a full histopathologic study, however, but rather, a first step in recognizing the contribution of the stroma in GERD. Differences in biopsy technique between archived GERD specimens made it difficult to perform quantitation of stromal cellularity rigorously. There is remarkably little published information on what happens to the lamina propria in nonmalignant esophageal disorders. We were, however, able to appreciate a subjective increase in stromal cellularity in GERD vs. normal esophagus. Our experience in reviewing GERD biopsies certainly suggests an increase in stromal cellularity in GERD, and our study has taken the next step of defining and characterizing these stromal cells.
Our work also adds to the existing literature of studies using esophageal stromal cell cultures. Previous studies established cultures using normal-appearing tissue adjacent to diseased esophagus (9). The microenvironment of such tissue may not reflect normal esophagus. Prior studies do not indicate whether the muscularis propria (9) was stripped before subjection of remaining tissue to mechanical and enzymatic digestion, and the reported enzymes (34) did not include both collagenase and dispase, as outlined in our protocol. Such differences could result in isolation of different populations of mesenchymal cells. mRNA, protein, and cell-surface marker characterization of the human esophageal primary stromal cell cultures used in these studies demonstrates their similarity to myofibroblasts in the lower GI tract.
Finally, primary cultures have inherent limitations, including source-derived variability that is best offset by the use of multiple cultures, as we have done in this study. Well-characterized primary cultures derived from three normal esophagi were used to study the mechanisms of acid and TLR4 stimulation. It is possible that our protocol isolated predominantly vimentin-expressing fibroblasts that became activated and thereby demonstrated α-SMA expression in culture. This possibility would explain the low percentage of α-SMA-positive, vimentin-positive cells in vivo and the uniform coexpression of these markers in our primary cultures. Such activation is inherent to the isolation procedure, however, and is unavoidable, as even low-passage cells expressed both α-SMA and vimentin. Another possibility is that these cells are pericytes or are derived from muscularis mucosa, although the absence of hematopoietic markers and coexpression of α-SMA and vimentin differentiate myofibroblasts from the former and latter cells, respectively (24). Future studies will investigate the potential origin of esophageal myofibroblasts in the normal and GERD esophagus.
In conclusion, esophageal myofibroblasts appear to be increased in the subepithelial stroma in GERD. Our work suggests that these cells can respond to injurious agents encountered in GERD via TRPV1 and TLR4 and that they contribute to the cytokine milieu via NF-κB-mediated proinflammatory cytokine secretion. The secretory capacity of HuEso MFs and their physical proximity to the basal layer of the squamous epithelium suggest a role in paracrine signaling. They may function primarily as immune-cell modulators and are likely under-recognized participants in the immune-mediated injury responses of the human esophagus. The interaction of esophageal myofibroblasts with surrounding cells and the epithelium, therefore, merits future investigation. Our work contributes to the foundation of knowledge in the study of epithelial-mesenchymal interactions and the development of targeted therapeutics in esophageal disorders.
GRANTS
Support for this work was provided by U.S. National Institutes of Health (NIH) Grants P30 DK048522 and S10 RR022508; National Cancer Institute (NCI) Cancer Center Shared Grant Awards P30CA014089 and P30CA014089 and USC Office of the Provost, Dean's Development Funds, Keck School of Medicine of USC; American Gastroenterological Association-General Mills Bell Institute of Health and Nutrition Research Scholar Award in Gut Physiology and Health (to A. Shaker); NIH/NCI K08CA153-01A1 (to A. Shaker); and Wright Foundation Pilot Award (to A. Shaker).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.C.R. and A.S. conception and design of research; M.G., C.N., J.B., and A.S. performed experiments; M.G., C.N., J.G.V., and A.S. analyzed data; M.G., C.N., J.G.V., and A.S. interpreted results of experiments; M.G., C.N., and A.S. prepared figures; M.G., C.N., and A.S. drafted manuscript; D.C.R, J.G.V., and A.S. edited and revised manuscript; A.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Microscopy and histology services were provided by the Cell and Tissue Imaging Core of USC Research Center for Liver Diseases. Multiplex cytokine assays and statistical analysis were performed by Diane Da Silva in the USC Immune Monitoring Core Facility. Flow cytometry was performed in the USC Flow Cytometry Core Facility.
REFERENCES
- 1.Abdel-Latif MM, Duggan S, Reynolds JV, Kelleher D. Inflammation and esophageal carcinogenesis. Curr Opin Pharmacol 9: 396–404, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Appelman HD, Streutker C, Vieth M, Neumann H, Neurath MF, Upton MP, Sagaert X, Wang HH, El-Zimaity H, Abraham SC, Bellizzi AM. The esophageal mucosa and submucosa: immunohistology in GERD and Barrett's esophagus. Ann N Y Acad Sci 1300: 144–165, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bobryshev YV, Killingsworth MC, Lord RV. Structural alterations of the mucosa stroma in the Barrett's esophagus metaplasia-dysplasia-adenocarcinoma sequence. J Gastroenterol Hepatol 27: 1498–1504, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Bode AM, Cho YY, Zheng D, Zhu F, Ericson ME, Ma WY, Yao K, Dong Z. Transient receptor potential type vanilloid 1 suppresses skin carcinogenesis. Cancer Res 69: 905–913, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng L, de la Monte S, Ma J, Hong J, Tong M, Cao W, Behar J, Biancani P, Harnett KM. HCl-activated neural and epithelial vanilloid receptors (TRPV1) in cat esophageal mucosa. Am J Physiol Gastrointest Liver Physiol 297: G135–G143, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chivukula RR, Shi G, Acharya A, Mills EW, Zeitels LR, Anandam JL, Abdelnaby AA, Balch GC, Mansour JC, Yopp AC, Maitra A, Mendell JT. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell 157: 1104–1116, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang Y, Chen H, Hu Y, Djukic Z, Tevebaugh W, Shaheen NJ, Orlando RC, Hu J, Chen X. Gastroesophageal reflux activates the NF-kappaB pathway and impairs esophageal barrier function in mice. Am J Physiol Gastrointest Liver Physiol 305: G58–G65, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gargus M, Niu C, Shaker A. Isolation of myofibroblasts from mouse and human esophagus. J Vis Exp 95: 52215, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green NH, Huang Q, Corfe BM, Bury JP, MacNeil S. NF-kappaB is activated in oesophageal fibroblasts in response to a paracrine signal generated by acid-exposed primary oesophageal squamous cells. Int J Exp Pathol 92: 345–356, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guarino MP, Cheng L, Ma J, Harnett K, Biancani P, Altomare A, Panzera F, Behar J, Cicala M. Increased TRPV1 gene expression in esophageal mucosa of patients with non-erosive and erosive reflux disease. Neurogastroenterol Motil 22: 746–751, e219, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Haggitt RC. Histopathology of reflux-induced esophageal and supraesophageal injuries. Am J Med 108, Suppl 4a: 109S–111S, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Hinterleitner TA, Saada JI, Berschneider HM, Powell DW, Valentich JD. IL-1 stimulates intestinal myofibroblast COX gene expression and augments activation of Cl− secretion in T84 cells. Am J Physiol 271: C1262–C1268, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, Ferri LE. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res 71: 1989–1998, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Hutchinson L, Stenstrom B, Chen D, Piperdi B, Levey S, Lyle S, Wang TC, Houghton J. Human Barrett's adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev 20: 11–17, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kandulski A, Malfertheiner P. Gastroesophageal reflux disease—from reflux episodes to mucosal inflammation. Nat Rev Gastroenterol Hepatol 9: 15–22, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Kavanagh ME, O'Sullivan KE, O'Hanlon C, O'Sullivan JN, Lysaght J, Reynolds JV. The esophagitis to adenocarcinoma sequence; the role of inflammation. Cancer Lett 345: 182–189, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Kishimoto E, Naito Y, Handa O, Okada H, Mizushima K, Hirai Y, Nakabe N, Uchiyama K, Ishikawa T, Takagi T, Yagi N, Kokura S, Yoshida N, Yoshikawa T. Oxidative stress-induced posttranslational modification of TRPV1 expressed in esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol 301: G230–G238, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Lim DM, Narasimhan S, Michaylira CZ, Wang ML. TLR3-mediated NF-{kappa}B signaling in human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol 297: G1172–G1180, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim DM, Wang ML. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic cells. Am J Physiol Gastrointest Liver Physiol 301: G91–G99, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331–342, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 42: 145–151, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Ma J, Harnett KM, Behar J, Biancani P, Cao W. Signaling in TRPV1-induced platelet activating factor (PAF) in human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol 298: G233–G240, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC Jr, Fullerton DA. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol 294: C29–C35, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Metz CN. Fibrocytes: a unique cell population implicated in wound healing. Cell Mol Life Sci 60: 1342–1350, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miwa H, Takubo K, Shimatani T, Furuta T, Oshima T, Tanaka J, Aida J, Ito M, Kurosawa S, Joh T, Wada T, Habu Y, Watanabe Y, Hongo M, Chiba T, Kinoshita Y, Aid-Related Symptom Research Group. Histology of symptomatic gastroesophageal reflux disease: is it predictive of response to proton pump inhibitors? J Gastroenterol Hepatol 28: 479–487, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Muir AB, Lim DM, Benitez AJ, Modayur Chandramouleeswaran P, Lee AJ, Ruchelli ED, Spergel JM, Wang ML. Esophageal epithelial and mesenchymal cross-talk leads to features of epithelial to mesenchymal transition in vitro. Exp Cell Res 319: 850–859, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Riordan JM, Abdel-latif MM, Ravi N, McNamara D, Byrne PJ, McDonald GS, Keeling PW, Kelleher D, Reynolds JV. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol 100: 1257–1264, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Orlando RC. Mechanisms of reflux-induced epithelial injuries in the esophagus. Am J Med 108, Suppl 4a: 104S–108S, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Orlando RC, Paterson WG, Harnett KM, Ma J, Behar J, Biancani P, Guarino MP, Altomare A, Cicala M, Cao W. Esophageal disease: updated information on inflammation. Ann N Y Acad Sci 1232: 369–375, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol 277: C1–C9, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am J Physiol 277: C183–C201, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol 73: 213–237, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riddell RH. The biopsy diagnosis of gastroesophageal reflux disease, “carditis,” and Barrett's esophagus, and sequelae of therapy. Am J Surg Pathol 20 Suppl 1: S31–S50, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Rieder F, Cheng L, Harnett KM, Chak A, Cooper GS, Isenberg G, Ray M, Katz JA, Catanzaro A, O'Shea R, Post AB, Wong R, Sivak MV, McCormick T, Phillips M, West GA, Willis JE, Biancani P, Fiocchi C. Gastroesophageal reflux disease-associated esophagitis induces endogenous cytokine production leading to motor abnormalities. Gastroenterology 132: 154–165, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Ronkainen J, Talley NJ, Storskrubb T, Johansson SE, Lind T, Vieth M, Agreus L, Aro P. Erosive esophagitis is a risk factor for Barrett's esophagus: a community-based endoscopic follow-up study. Am J Gastroenterol 106: 1946–1952, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Shaker A, Binkley J, Darwech I, Swietlicki E, McDonald K, Newberry R, Rubin DC. Stromal cells participate in the murine esophageal mucosal injury response. Am J Physiol Gastrointest Liver Physiol 304: G662–G672, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaker A, Gargus M, Fink J, Binkley J, Darwech I, Swietlicki E, Levin MS, Rubin DC. Epimorphin(−/−) mice are protected, in part, from acute colitis via decreased interleukin 6 signaling. Transl Res 164: 70–83, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaker A, Rubin DC. Intestinal stem cells and epithelial-mesenchymal interactions in the crypt and stem cell niche. Transl Res 156: 180–187, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaker A, Swietlicki EA, Wang L, Jiang S, Onal B, Bala S, DeSchryver K, Newberry R, Levin MS, Rubin DC. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J Clin Invest 120: 2081–2093, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shan J, Oshima T, Chen X, Fukui H, Watari J, Miwa H. Trypsin impaired epithelial barrier function and induced IL-8 secretion through basolateral PAR-2: a lesson from a stratified squamous epithelial model. Am J Physiol Gastrointest Liver Physiol 303: G1105–G1112, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Souza RF, Huo X, Mittal V, Schuler CM, Carmack SW, Zhang HY, Zhang X, Yu C, Hormi-Carver K, Genta RM, Spechler SJ. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 137: 1776–1784, 2009. [DOI] [PubMed] [Google Scholar]
- 44.Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev 51: 159–212, 1999. [PubMed] [Google Scholar]
- 45.Tobey NA, Hosseini SS, Argote CM, Dobrucali AM, Awayda MS, Orlando RC. Dilated intercellular spaces and shunt permeability in nonerosive acid-damaged esophageal epithelium. Am J Gastroenterol 99: 13–22, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, Medema JP. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 12: 468–476, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Weng Y, Shi Y, Xia X, Wang S, Duan H. Expression and functional analysis of Toll-like receptor 4 in human cervical carcinoma. J Membr Biol 247: 591–599, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 137: 588–597, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Eppenberger-Castori S, Eppenberger U, Benz CC. The NFkappaB pathway and endocrine-resistant breast cancer. Endocr Relat Cancer 12 Suppl 1: S37–S46, 2005. [DOI] [PubMed] [Google Scholar]