

Abstract
Reactive oxygen species (ROS) generated by Nox NADPH oxidases may play a critical role in the pathogenesis of diabetic nephropathy (DN). The efficacy of the Nox1/Nox4 inhibitor GKT137831 on the manifestations of DN was studied in OVE26 mice, a model of type 1 diabetes. Starting at 4–5 mo of age, OVE26 mice were treated with GKT137831 at 10 or 40 mg/kg, once-a-day for 4 wk. At both doses, GKT137831 inhibited NADPH oxidase activity, superoxide generation, and hydrogen peroxide production in the renal cortex from diabetic mice without affecting Nox1 or Nox4 protein expression. The increased expression of fibronectin and type IV collagen was reduced in the renal cortex, including glomeruli, of diabetic mice treated with GKT137831. GKT137831 significantly reduced glomerular hypertrophy, mesangial matrix expansion, urinary albumin excretion, and podocyte loss in OVE26 mice. GKT137831 also attenuated macrophage infiltration in glomeruli and tubulointerstitium. Collectively, our data indicate that pharmacological inhibition of Nox1/4 affords broad renoprotection in mice with preexisting diabetes and established kidney disease. This study validates the relevance of targeting Nox4 and identifies GKT137831 as a promising compound for the treatment of DN in type 1 diabetes.
Keywords: diabetic nephropathy, NADPH oxidase, Nox1/Nox4 inhibitor
oxidative stress with increased generation of reactive oxygen species (ROS) has emerged as a critical mechanism in the pathogenesis of diabetic complications including diabetic nephropathy (DN) (1, 22, 23, 30, 42). Although multiple pathways result in ROS generation, numerous studies identified the Nox family of NADPH oxidases as major sources of ROS in various nonphagocytic cells, including most kidney cells (4 5, 22, 23, 33). Evidence suggests that among the seven Nox homologs, it is the Nox1 and Nox4 homologs that are required for the damaging effects of high glucose in cultured cells and contributes to hyperglycemia-mediated microvascular and macrovascular complications of diabetes in the retina, vasculature, heart, and kidney (21–24, 27, 34).
Nox1, Nox2 (a.k.a. gp91phox), Nox4, and Nox5 are the NADPH oxidase homologs that are predominantly expressed in the kidney, including glomerular and tubulointerstitial cells (5, 22, 23). The calcium-dependent homolog Nox5 is found in higher species, including human kidney tissue and cells, but is not present in mice and rats. Nox2 and Nox1 require the membrane-bound regulatory subunit p22phox as well as multiple cytosolic factors for activation (5, 22, 23). While p22phox regulatory subunit enhances the activity of Nox4, the enzyme has constitutive activity, and consequently the overall ROS output of Nox4 may be directly governed by its expression level (22, 23). A role for Nox4 in DN is supported by previous studies by our group, which demonstrated that administration of antisense oligonucleotides for Nox4 to diabetic rats prevents renal hypertrophy and matrix expansion (18, 21). The orally available small-molecule Nox1/Nox4 inhibitors from the pyrazolo pyridine chemical series have recently drawn considerable attention. Preclinical studies performed with these inhibitors in experimental animal models indicate that they effectively attenuate the pathological changes observed in type 2 diabetes including accelerated atherosclerosis as well as ischemic retinopathy, liver fibrosis, and idiopathic pulmonary fibrosis (2, 10, 13, 25, 28, 32, 40, 44, 47). More recently, the first-generation Nox1/Nox4 inhibitor GKT136901 was found to ameliorate DN in db/db mice, a model of type 2 diabetes (40). A recent study using apolipoprotein E/Nox4 double knockout mice or Nox4 knockout mice on C57BL6/J background made type 1 diabetic with streptozotocin showed that systemic Nox4 deletion or pharmacological inhibition with the dual Nox4/Nox1 inhibitor GKT137831 reduces albuminuria and decreases matrix accumulation (27, 44).
Because Nox-derived ROS have been implicated in diabetic complications, these enzymes are attractive therapeutic targets. This is particularly relevant since treatment with antioxidants, such as vitamins E and C, failed to ameliorate the complications related to cardiovascular disease or diabetes (11, 12, 14, 38). Consequently, inhibiting the sources of ROS in the setting of diabetes may be a superior approach compared with nonselective scavengers.
Building on these encouraging results, it is important to determine whether pharmacological Nox inhibition attenuates disease severity when therapy is initiated once diabetic kidney disease is already established. It is also important to evaluate novel therapies in a murine model that closely reproduces the human manifestations of DN, such as the OVE26 mouse model. Accordingly, the present study was designed to assess whether the therapeutic administration of GKT137831 in the OVE26 type 1 diabetic mouse model attenuates the severity of established diabetic nephropathy.
MATERIALS AND METHODS
Pharmacokinetics of Nox1/Nox4 inhibitor.
Pharmacokinetic (PK) studies were performed in mice by Genkyotex to characterize the PK properties of GKT137831 and its main active metabolite GKT138184. Male C57BL/6 mice were dosed daily for 8 days at 5, 20, and 60 mg·−1·kg−1·day−1 (Genkyotex study GSN000168). GKT138184 is a N-desmethylated phase 1 metabolite with a nearly identical potency and selectivity profile against Nox isoforms as its parent compound GKT137831. The vehicle used was 1.2% methyl cellulose/0.1% polysorbate 80 in water. Levels of GKT137831 and GKT138184 were measured in plasma at 0.17, 0.33, 0.67, 1.5, 4, 8, and 24 h (n = 3 animals/time point) on days 0 and on 7, and PK parameters were calculated. There was no notable difference in exposure to either analyte upon repeated dosing, and thus plasma concentrations on day 7 (i.e., after the 8th daily dose) were deemed to be representative of PK steady state.
Cmax (maximal concentration), AUC0-t (area under the curve from time 0 to time t), Clast (last concentration), and Tlast (last time point) values on day 7 at each dose level for each analyte are presented in Tables 1 and 2.
Table 1.
Cmax, AUC0-t, Clast, and Tlast values on day 7 at each dose level for GKT137831
| GKT137831 |
|||||
|---|---|---|---|---|---|
| Dose, mg·kg−1·day−1 | Day | Cmax, ng/ml | AUC0-t, ng·h−1·ml−1 | Clast, ng/ml | Tlast, h |
| 5 | 7 | 2,495 | 1,578 | 241 | 1.5 |
| 20 | 7 | 8,675 | 9,657 | 148 | 8 |
| 60 | 7 | 28,616 | 34,481 | 1,479 | 8 |
Cmax, maximal concentration; AUC0-t, area under the curve from time 0 to time t; Clast, last concentration; Tlast, last time point.
Table 2.
Cmax, AUC0-t, Clast, and Tlast values on day 7 at each dose level for GKT138184
| GKT138184 |
|||||
|---|---|---|---|---|---|
| Dose, mg·kg−1·day−1 | Day | Cmax, ng/ml | AUC0-t, ng·h−1·ml−1 | Clast, ng/ml | Tlast, h |
| 5 | 7 | 1,873 | 1,473 | 294 | 1.5 |
| 20 | 7 | 5,677 | 8,130 | 175 | 8 |
| 60 | 7 | 15,506 | 25,366 | 1,134 | 8 |
Combined GKT137831 and GKT138184 steady-state plasma concentrations were maintained above 200 ng/ml (i.e., 5 times the IC50) for 1.5 h at a dose of 5 mg·−1·kg−1·day−1, and for 8 h at doses of 20 and 60 mg·−1·kg−1·day−1. It is therefore reasonable to assume that, at doses of 10 and 40 mg·−1·kg−1·day−1, combined GKT137831 and GKT138184 plasma concentrations would be maintained above 200 ng/ml for >1.5 and >8 h, respectively. Collectively, the PK data indicate that the Genkyotex compounds exhibit a good exposure and absorption, as evidenced by the values of the Cmax and AUC and a good distribution volume allowing a good distribution in the various tissues.
Note that the PK properties of single- and multiple-dose GKT137831 were also assessed in healthy human subjects (46).
Animals and treatment.
OVE26 mice (FVB background, The Jackson Laboratory, Bar Harbor, ME) were used as the experimental model of type 1 diabetes, and 24-wk-old FVB mice served as controls. At ∼20 wk (4–5 mo) of age, OVE26 mice were treated with dual Nox1/4 inhibitor GKT137831 (Genkyotex) for 4 wk and then euthanized. Mice were divided into the following groups: group 1, FVB control mice receiving drug vehicle (1.2% methylcellulose+0.1% polysorbate 80) by oral gavage once a day (n = 7); group 2, OVE26 mice receiving drug vehicle by gavage once a day (n = 6); group 3, OVE26 mice receiving 10 mg/kg of the GKT137831 inhibitor by oral gavage once a day (n = 6); and group 4, OVE26 mice receiving 40 mg/kg of GKT137831 by gavage once a day (n = 5). Three of the six OVE26 mice not on treatment died toward the end of the study. The analysis included two OVE26 mice of the same batch and of the same age that were not part of this study. One of the six OVE26 mice treated with the higher dose of GKT137831 died toward the end of the study. The dose of 40 mg/kg was selected to provide a maximally effective dose, and the 10 mg/kg dose was at least a half log smaller. Since the inception of the present study, several in vivo studies have been conducted with GKT137831. Overall, the maximally effective dose was generally ∼20 mg/kg. Blood glucose was determined at 3- to 4-wk intervals in all groups by glucometer on blood samples obtained from the tail vein. Before euthanasia, mice were placed in metabolic cages and 24-h urine was collected for albumin measurements. Urine albumin was measured by a mouse albumin ELISA (Bethyl Laboratories) and expressed as micrograms of albumin per 24 hours (15–17). Animals were euthanized by exsanguination under anesthesia, and both kidneys were removed and weighed. A slice of kidney cortex at the pole was embedded in paraffin or flash-frozen in liquid nitrogen for microscopy and protein analyses. All experiments were approved by the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee.
NADPH oxidase assay.
NADPH oxidase activity was measured by the lucigenin-enhanced chemiluminescence method as described previously (6, 15–18, 21). Superoxide production was expressed as relative chemiluminescence (light) units (RLU)/mg protein.
Measurement of hydrogen peroxide production in the kidney cortex.
Hydrogen peroxide was measured using an Amplex Red Assay Kit (Invitrogen/Molecular Probes) as described previously (6, 39). Hydrogen peroxide generation was expressed as fluorescence units per 50 micrograms protein.
In situ detection of intracellular ROS production in the kidney cortex using dihydroethidium and confocal microscopy.
Frozen cortical sections (6 μm thick) were placed on cover glasses (no. 1, 25 mm diameter) and set into a Attofluor cell chamber (A7816, Invitrogen/Molecular Probes). The frozen sections were air-dried and incubated with 10 μM dihydroethidium (DHE; D11347, Invitrogen Molecular Probes) for 10 min at room temperature in the dark. The fluorescence images by DHE staining were taken at the wavelength of 515 nm (excitation)/580 nm (emission) by a Olympus confocal microscope (FV 1000).
Detection of intracellular superoxide production in the kidney cortex using DHE and HPLC.
Cellular superoxide production in the kidney cortex was assessed by HPLC analysis of DHE-derived oxidation products, as described previously (27, 31). The HPLC-based assay allowed the separation of the superoxide-specific 2-hydroxyethidium (EOH) from the nonspecific ethidium. Superoxide was measured in finely minced kidney cortex from the experimental animals. The minced tissue was washed three times with PBS/diethylenetriaminepentaacetic acid (DTPA) followed by a 30-min incubation with 100 μM DHE (Sigma-Aldrich) in 500 μl PBS/DTPA at 37°C in the dark. The tissue was then washed with PBS/DTPA, flash frozen in liquid nitrogen, homogenized, and resuspended in 500 μl acetonitrile, lysed by sonication, and centrifuged at 12,000 g for 10 min at 4°C. The supernatants were dried using a speed-vac system. For HLPC analysis, the dried samples were resuspended in 120 μl PBS/DTPA, and 100 μl were injected into a HPLC system (LC-2000 plus series, Jasco Analytical Instruments, Easton, MD) equipped with a C18 column (Nucleosil 100-5, Macherey-Nagel) and with ultraviolet and fluorescence detectors. Elution of analytes was achieved with acetonitrile (solvent A) and water with 0.1% trifluoroacetic acid (vol/vol; solvent B) as a mobile phase, with a flow rate of 0.6 ml/min and with the following solvent A gradient: 10% at time 0 (sample injection) and 60% at minute 10 (linear increase) and through minute 20 (isocratic). Elution of EOH was monitored by fluorescence with emission and excitation wavelengths at 595 and 510 nm, respectively. The retention times for EOH was 12.5–13.5 and 16–16.5 min. The eluate mass was quantified by comparing the peak area of the unknown samples with those of standard EOH (Noxygen, Denzlingen, Germany). EOH and ethidium were monitored by fluorescence detection with excitation at 510 nm and emission at 595 nm.
Western blot analysis.
Western blot analysis was performed as described previously (6, 15–18, 21). The antibodies used were a rabbit polyclonal Nox4 antibody directed against recombinant glutathione S-transferase-mouse Nox4-fragment (299–515, dilution 1:1,000), a rabbit polyclonal anti-Nox1 (dilution 1:500, Santa Cruz Biotechnology), a rabbit polyclonal anti-fibronectin antibody (1:2,500, Sigma-Aldrich), a rabbit polyclonal anti-collagen type IV antibody (1:1,000, Abcam), and rabbit polyclonal anti-GAPDH (1:1,000).
Renal histology.
For morphometric studies, kidneys were fixed in formaldehyde, embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin (H&E) or periodic acid Schiff (PAS) (15, 21). Matrix expansion was evaluated on PAS-stained sections as described previously (15). Determination of glomerular surface area was performed using H&E-stained sections from the different treatment groups as described previously (21).
Immunofluorescence.
Immunofluorescence was performed on 5-μm-thick frozen sections using rabbit polyclonal anti-fibronectin, rabbit polyclonal anti-collagen type IV, or rabbit anti-synaptopodin as described previously (15–17, 21, 31). Dual-label immunohistochemistry was used to identify and count glomerular epithelial cells relative to the glomerular basement membrane (GBM) using a modification of methods described previously (15–17).
Immunohistochemistry.
Localization of fibronectin, collagen IV, and WT-1 was assessed by immunoperoxidase histochemistry in frozen cortical sections (5 μm thick) using polyclonal fibronectin antibodies (1:800, Sigma-Aldrich), collagen type IV antibodies (1:500, Millipore), or WT-1 antibodies (1:400, Santa Cruz Biotechnology) as described previously (15–17, 19, 27, 31). Localization of CD68 (1:100, Serotec) was assessed in paraffin-embedded sections (5 μm thick) by alkaline phosphatase histochemistry.
Statistical analysis.
Data are presented as means ± SE. Statistical comparisons between multiple groups were performed by one-way ANOVA (nonparametric), and posttest analysis was done using Tukey Statistical (GraphPad Prism). A P value of <0.05 was considered statistically significant.
RESULTS
Effect of GKT137831 treatment on blood glucose concentration and body weight.
Table 3 displays blood glucose levels and body and kidney weights in the different groups of mice. Untreated type 1 diabetic OVE26 mice and OVE26 mice treated with either 10 or 40 mg/kg of GKT137831 had equivalently elevated blood glucose concentrations at the end of the study period compared with the FVB control mice. Body weight was similarly reduced in the diabetic mice treated with either 10 or 40 mg/kg GKT13783. Diabetes or treatment of diabetic mice with GKT137831 did not influence total kidney weight. Kidney weight-to-body weight ratio, an indicator of whole kidney hypertrophy, significantly increased in untreated diabetic mice compared with nondiabetic control animals. Although there is a trend toward a decrease, total kidney weight-to-body weight ratio in GKT137831-treated diabetic mice was not significantly reduced compared with that observed for the untreated diabetic group (Table 3). It should be noted that treatment with GKT137831 ameliorated the general appearance of OVE26 mice compared with that of untreated OVE26 mice. GKT137831 also appears to improve the survival of the OVE26 mice. Indeed, 1 of the 12 OVE26 mice treated with GKT137831 died, whereas 3 of the 6 OVE26 mice not on treatment died toward the end of the study.
Table 3.
Glucose level, body weight, kidney weight, and kidney weight-to-body weight ratio after 4–5 mo of diabetes
| Group | n | Blood Glucose, mg/dl | Body Weight, g | Kidney Weight, g | Kidney Weight/Body Weight, g/kg |
|---|---|---|---|---|---|
| FVB | 7 | 151 ± 22 | 29 ± 4 | 0.23 ± 0.03 | 7.97 ± 0.81 |
| OVE26 | 5 | 567 ± 14* | 18 ± 2* | 0.25 ± 0.04 | 14.10 ± 0.72* |
| OVE26+GKT 10 mg/kg | 6 | 585 ± 37* | 18 ± 2* | 0.23 ± 0.03 | 12.68 ± 0.68* |
| OVE26+GKT 40 mg/kg | 5 | 583 ± 29* | 17 ± 1* | 0.22 ± 0.01 | 12.51 ± 1.04* |
Values are means ± SE for each group.
P < 0.05 vs. FVB.
Effect of GKT137831 treatment on diabetes-induced ROS generation in the renal cortex.
NADPH-dependent superoxide production was significantly increased in renal cortical homogenates of OVE26 mice compared with controls. Treatment with 10 or 40 mg/kg GKT137831 suppressed diabetes-induced NADPH oxidase activation in cortical homogenates (Fig. 1A). In situ detection of intracellular ROS with DHE staining and confocal microscopy indicated that treatment of OVE26 mice with 10 or 40 mg/kg GKT137831 reduced the diabetes-induced increase in DHE fluorescence in glomerular and tubular compartments (Fig. 1B). HPLC analysis of EOH, the superoxide-specific product of DHE, validated the specificity of superoxide measurements with DHE and confirmed the inhibitory effects of GKT137831 on superoxide production in cortical homogenates from diabetic mice (Fig. 1C). As shown in Fig. 1D, treatment of OVE26 mice with the two doses of the Nox1/Nox4 inhibitor GKT137831 also attenuated the increase in hydrogen peroxide production observed in OVE26 mice in renal cortical homogenates.
Fig. 1.

Treatment of OVE26 type 1 diabetic mice with Nox4/1 inhibitor GKT137831 reduces diabetes-induced reactive oxygen species (ROS) generation in the renal cortex without affecting Nox4 or Nox1 protein expression. A: NADPH oxidase activity in cortical homogenates. Superoxide anion generation was determined by photoemission every 30 s for 5–10 min. The initial rate of enzyme activity was calculated over the first 30–120 s of exposure to NADPH. NADPH-driven superoxide production was expressed as relative light units (RLU)·min−1·mg protein−1. Values are means ± SE. *P < 0.05 vs. FVB. #P < 0.05 vs. OVE26. B: hydrogen peroxide production was assessed using an Amplex Red Assay kit. Values are the mean ± SE. *P < 0.05 vs. FVB. #P < 0.05 vs. OVE26. C: in situ detection of intracellular ROS with dihydroethidium (DHE) staining and confocal microscopy in renal cortex sections from FVB, OVE26, OVE26+GKT137831 10 mg/kg, and OVE26+GKT137831 40 mg/kg groups. Dashed white lines indicate position of glomeruli. D: superoxide generation was evaluated in the renal cortex using DHE and HPLC as described in materials and methods. Superoxide production was expressed as μM 2-hydroxyethidium produced/mg renal cortex. Values are means ± SE from all the animals in each group. *P < 0.01 vs. FVB. #P < 0.05 vs. OVE26. E and F: Nox4 (E) and Nox1 (F) protein expression were determined by direct immunoblotting of cortical lysates. GAPDH was included as a control for loading and the specificity of change in protein expression. Representative results of Western blot analysis were obtained from 3 independent samples from each group. Each histogram at the bottom represents the ratio of the intensity of the Nox4 or Nox1 bands quantified by densitometry factored by the densitometric measurement of the actin band. Values are means ± SE from all the animals in each group. *P < 0.05 vs. FVB.
Effect of GKT137831 treatment on Nox4 and Nox1 protein expression in the diabetic kidney.
The protein levels of Nox4 and Nox1 in renal cortex from the different groups were examined. Western blot analysis showed that Nox4 protein expression was increased in OVE26 mice compared with that in control nondiabetic FVB control mice. GKT137831 administration had no effect on diabetes-induced Nox4 expression (Fig. 1E). The levels of Nox1 were not increased in diabetic animals, and the administration of GKT137831 did not alter Nox1 expression (Fig. 1F). These data indicate that GKT137831 reduces diabetes-induced ROS generation without affecting the expression of Nox4 and Nox1.
Effect of GKT137831 treatment on diabetes-induced extracellular matrix protein accumulation.
As depicted in the immunoblot in Fig. 2A, expression of fibronectin protein was significantly increased in the renal cortex of OVE26 mice compared with FVB controls. The increased expression of fibronectin was markedly reduced in the renal cortex of diabetic mice treated with 10 mg/kg (Fig. 2A, left) or 40 mg/kg (Fig. 2A, right) of GKT137831. GKT137831 also significantly inhibited diabetes-induced cortical collagen IV protein expression (Fig. 2B).
Fig. 2.

Treatment of OVE26 mice with GKT137831 reduces diabetes-induced extracellular matrix protein expression in the renal cortex including glomeruli. A: representative Western blot for fibronectin protein expression in the renal cortex. Representative results of Western blot analysis were obtained from 3 independent samples from each group. Each histogram at the bottom represents the ratio of the intensity of the fibronectin bands quantified by densitometry factored by the densitometric measurement of the actin band. Values are means ± SE from all the animals in each group. *P < 0.05 vs. FVB. #P < 0.05 vs. OVE26. B: representative Western blot for collagen IV protein expression in the renal cortex. Representative results of Western blot analysis were obtained from 2 independent samples from each group. The histogram on the right represents the ratio of the intensity of collagen IV bands quantified by densitometry factored by the densitometric measurement of GAPDH band. The data are expressed as percentage of control (FVB), where the ratio in the control was defined as 100%. Values are means ± SE from all the animals in each group. *P < 0.05 vs. FVB. #P < 0.05 vs. OVE26. C: fibronectin expression was detected by immunoperoxidase (top) and immunofluorescence (bottom) staining of kidney sections from FVB, OVE26, OVE26+GKT137831 10 mg/kg, and OVE26+GKT137831 40 mg/kg groups. D: quantitation of glomerular collagen fibronectin expression. The histograms represent means ± SE of glomeruli in sections from individual mice in each group. *P < 0.001 vs. FVB. #P < 0.001 vs. OVE26. E: collagen type IV was detected by immunoperoxidase (top) and immunofluorescence (bottom) staining of kidney sections from FVB, OVE26, OVE26+GKT137831 10 mg/kg, and OVE26+GKT137831 40 mg/kg groups. F: quantitation of glomerular collagen type IV expression. The histograms represent means ± SE of glomeruli in sections from individual mice in each group. *P < 0.001 vs. FVB. #P < 0.001 vs. OVE26.
The protective effects of GKT137831 on extracellular matrix accumulation were confirmed by immunohistochemical and immunofluorescence analyses of fibronectin and collagen IV expression. As shown in Fig. 2, C and E, the amount of fibronectin and collagen IV was increased in the diabetic group, and treatment with both doses of GKT137831 decreased the expression of fibronectin and collagen IV induced by diabetes in the glomerular and tubular compartments. Figure 2, D and F, represents the quantitation of the immunohistochemical staining of glomerular fibronectin and collagen IV expression, respectively.
Effect of GKT137831 treatment on diabetes-induced glomerular hypertrophy.
Glomerular hypertrophy was assessed by quantifying glomerular surface area in histological sections of kidneys removed from FVB control mice, diabetic OVE26 mice, and GKT137831-treated OVE26 mice. Figure 3A shows that glomeruli of OVE26 mice are significantly larger compared with those of FVB control mice. Treatment with 10 mg/kg of GKT137831 resulted in a decrease in glomerular size. There is a trend toward a decrease in glomerular hypertrophy with the 40 mg/kg dose.
Fig. 3.

Treatment of OVE26 mice with GKT137831 reduces diabetes-induced glomerular hypertrophy and mesangial expansion. A: representative photomicrographs of kidney sections stained with hematoxylin and eosin from FVB, OVE26, OVE26+GKT137831 10 mg/kg, and OVE26+GKT137831 40 mg/kg groups. Right: quantitation of glomerular size. Glomerular cross-sectional areas were measured from kidney sections stained with hematoxylin and eosin by using ImagePro Plus 4.5 software. The histograms represent means ± SE from 25 individual glomeruli in sections from 6 individual rats in each group. B: representative photomicrographs of kidney sections stained with periodic acid- Schiff from FVB, OVE26, OVE26+GKT137831 10 mg/kg, and OVE26+GKT137831 40 mg/kg groups. Right: evaluation of mesangial matrix expansion index. The histograms represent means ± SE from 25 individual glomeruli in the individual mice for each group. *P < 0.01 vs. FVB. #P < 0.05 vs. OVE26.
Effect of GKT137831 treatment on mesangial matrix expansion in diabetic mice.
Histopathological features of glomerular injury were also defined by assessing mesangial matrix expansion. Mesangial matrix expansion was examined by subjecting sections of paraffin-embedded kidneys to PAS staining. Untreated OVE26 mice exhibited enlarged glomeruli with expanded mesangial matrix compared with FVB control mice (Fig. 3B). In OVE26 mice treated with 10 mg/kg or 40 mg/kg of GKT137831, mesangial matrix accumulation was significantly reduced (Fig. 3B).
Effect of GKT137831 treatment on albuminuria in diabetic mice.
Urine was collected, and albumin levels were measured. OVE26 mice showed significant albuminuria (Fig. 4A). Treatment of the mice with either 10 or 40 mg/kg of GKT137831 for 4 wk resulted in a significant decrease in albumin excretion compared with control diabetic mice receiving vehicle (Fig. 4A).
Fig. 4.

Treatment of OVE26 mice with GKT137831 ameliorates urine albumin excretion and reduces podocyte loss. A: urine albumin excretion was evaluated using a mouse albumin ELISA quantification kit and expressed as μg albumin/24 h. B: representative immunoperoxidase images of glomeruli stained with podocyte marker WT-1 (brown). Right: quantitation of WT-1-positive cells (WT-1+) in glomeruli using ImagePro Plus 4.5 software. The histograms represent means ± SE of 25 individual glomeruli in each section. Values are means ± SE. *P < 0.001 vs. FVB. #P < 0.05 vs. OVE26. &P < 0.01 vs. OVE26. C: representative immunofluorescence images of glomeruli stained with synaptopodin (red), collagen IV (green), and 4′,6-diamidino-2-phenylindole (DAPI) (blue), and quantification of podocytes per glomerulus. Values are means ± SE. *P < 0.001 vs. FVB.
Effect of GKT137831 treatment on podocyte loss in diabetic mice.
Podocyte loss was assessed by enumeration of WT-1- or synaptopodin-positive cells in the glomeruli. As shown in Fig. 4B, WT-1 staining is markedly reduced in glomeruli from OVE26 mice. The two doses of GKT137831 significantly prevented the decrease in WT-1-positive cells induced by diabetes (Fig. 4B). Dual-label immunohistochemistry was used to identify and count podocytes relative to the glomerular basement membrane (GBM). Representative immunofluorescent images of glomeruli stained with collagen IV (marker of GBM, green), synaptopodin (podocyte marker, red), and DAPI (blue) showed that administration of GKT137831 (10 or 40 mg/kg) tends to reduce the loss of podocytes in diabetic OVE26 mice, as evidenced by the increased number of synaptopodin-positive cells in the glomeruli from treated animals (Fig. 4C).
Effect of GKT137831 treatment on inflammation in diabetic mice.
One of the features of diabetic kidney disease is a high degree of inflammation, characterized by macrophage infiltration that contributes to renal injury. Macrophage infiltration was examined in glomeruli and tubulointerstitium using CD68 staining. The number of CD68-positive cells was increased in both glomeruli and tubules from OVE26 mice, indicating that diabetes elicited macrophage infiltration (Fig. 5, A and B).
Fig. 5.

Treatment of OVE26 mice with GKT137831 reduces diabetes-induced monocyte infiltration in the interstitial area and glomeruli. A: representative immunohistochemistry images of interstitial area stained with CD68 (pink). Right: quantitation of CD68-positive cells (CD68+) in interstitial area using ImagePro Plus 4.5 software. Values are means ± SE. *P < 0.01 vs. FVB. #P < 0.01 vs. OVE26. B: representative immunohistochemistry images of glomeruli stained with CD68. Right: quantitation of CD68-positive cells (CD68+) in glomeruli using ImagePro Plus 4.5 software. The histograms represent means ± SE of 25 individual glomeruli in each section. Values are means ± SE. *P < 0.01 vs. FVB. #P < 0.01 vs. OVE26.
Treatment with both doses of GKT137831 markedly attenuated diabetes-induced macrophage infiltration in the glomeruli (Fig. 5A) and the tubulointerstitial compartment (Fig. 5B), as evidenced by the reduction in CD68-positive cells.
DISCUSSION
Here, we provide evidence that a relatively short-term therapeutic administration of GKT137831 markedly attenuates the severity of diabetic kidney disease in the OVE26 model. Oral administration of GKT137831 reduces Nox-dependent ROS generation and attenuates diabetes-induced albuminuria, glomerular and tubular injury, as well as macrophage infiltration. Importantly, treatment was initiated in 4- to 5-mo-old mice, at a time when overt diabetes and kidney disease are already present (49). In OVE26 mice, significant hyperglycemia and albuminuria are already present at 2 mo of age, and urinary albumin excretion reaches ∼1,000 μg/24 h at 5 mo of age (49). In line with these published data, in our study albuminuria was approximatively 1,300 μg/24 h at the time of euthanasia in untreated mice. This indicates that Nox1/4 inhibition is effective in mice with established kidney disease.
Many of the rodent models of type 1 or type 2 diabetes have their limitations in that proteinuria and the renal injury are modest and certain histological features do not mimic DN in humans (7–9). OVE26 mice exhibit many hallmarks of human diabetic kidney disease, including severe proteinuria, and glomerular and tubulointerstitial injury (15–17, 49). We have previously reported that NADPH oxidase activity as well as Nox4 and Nox1 protein expression are increased in glomeruli isolated from OVE26 mice (15–17). We now show that Nox4 but not Nox1 expression is increased in the renal cortex, likely reflecting tubular epithelial cells. Therefore, the OVE26 mice model represents a suitable model for investigating the effect of Nox1/4 inhibitors.
We find that GKT137831 protects against the pathological features of diabetes in the kidney of OVE26 mice without altering blood glucose levels, suggesting that the inhibitor targets the kidney directly. It is likely that GKT137831 reduces the level of ROS in the kidney and subsequently the damage mediated by oxidative stress through inhibition of Nox oxidase activity. This is supported by the observations that treatment of OVE26 mice with the two doses of the Nox1/Nox4 inhibitor GKT137831 abolished NADPH-dependent superoxide generation and hydrogen peroxide production in renal cortical homogenates. GKT137831 had no effect on Nox4 or Nox1 protein expression in OVE26 mice cortex. These observations suggest that the enzyme activity of Nox4 or Nox1 is targeted by the inhibitor and not the expression level, consistent with the mechanism of action of GKT137831, an allosteric inhibitor. Competition binding experiments carried out by Genkyotex on membranes prepared from Chinese hamster ovary cells overexpressing Nox1 show that GKT137831 is a noncompetitive inhibitor for NADPH, i.e., it binds to the same subunit as NADPH but on another binding site distant from that of the NADPH. Furthermore, results obtained in whole cells with nonpermeable analogs indicate that GKT137831 binds to the extracellular part of the catalytic subunit of the Nox enzymes. Since Nox4 is constitutively active (4, 5, 22, 23, 33), the overall ROS output of Nox4 is at least partially dependent on its expression level. In contrast, Nox1 requires association with regulatory subunits to be activated (4, 5, 22, 23, 33).
The present study clearly demonstrates that diabetes-induced glomerular injury, including mesangial matrix accumulation, podocyte injury, as well as tubular injury are significantly attenuated by the inhibitor. Although GKT137831 is a dual Nox1 and Nox4 inhibitor, there is some indication that its actions are due to the targeting of Nox4 in the kidney. The homolog Nox4 has been shown to be critical for glomerular and tubular pathology associated with diabetes, including glomerular hypertrophy and matrix accumulation in the mesangium and tubulointerstitial compartment (21–23, 27). In cultured renal cells, Nox4 is required for high glucose-induced ROS production and subsequent cell injury (6, 16, 18, 19, 21–23, 27, 36, 39). In mesangial or tubular epithelial cells, ROS generated by Nox4 mediate high glucose-induced extracellular matrix protein expression (6, 18, 21–23, 36, 39), and, in podocytes, Nox4 contributes to apoptotic cell death (15–17). Studies by our group using a Nox4 antisense oligonucleotide strategy established a causal relationship between Nox4-dependent ROS generation and renal hypertrophy, mesangial matrix accumulation, and interstitial matrix protein expression in a rat model of early type 1 diabetes (18, 21). Two recent studies that addressed the role of Nox4 in mediating proteinuria emphasized that functional and structural manifestations in the kidney of diabetic mice using whole body Nox4 knockout mice yielded conflicting results (3, 27). In C57BL/6J mice rendered diabetic with streptozotocin (STZ), deletion of Nox4 resulted in no change in proteinuria or in matrix protein expression (3). However, this study was a short-term study (60 days) conducted on a genetic background (3) that is resistant to the development of DN. Thus diabetes induction resulted in hyperfiltration but failed to induced significant kidney injury, as shown by the lack of kidney hypertrophy and the modest extracellular matrix protein accumulation (3). Also, renal ROS production in diabetic mice was not reported. Consequently, this model may not be ideal for studying the role of Nox4 in DN. Importantly, in a more recent study in STZ-induced diabetic mice on a C57BL/6J background (44) genetic deletion of Nox4 showed that Nox4 deficiency is renoprotective. The duration of diabetes in this study was 20 wk, and the animals had marked injury compared with that of the 60-day study.
Another study compared the effects of Nox1 or Nox4 gene deletion, as well as pharmacological Nox1/4 inhibition with GKT137831, on the development of DN (27). Apolipoprotein E-deficient mice were made diabetic with STZ and assessed for up to 20 wk. In this model of accelerated diabetic complications, diabetes induction lead to kidney injury, as shown by sustained albuminuria, glomerular hypertrophy and mesangial expansion, macrophage infiltration, profibrogenic gene expression, glomerulosclerosis, and tubulointerstitial injury (27). Deletion of Nox4, but not Nox1, resulted in amelioration of proteinuria, preserved renal structure, reduced glomerular accumulation of extracellular matrix proteins, and reduced renal ROS production and inflammation (27). Similar improvement was achieved by the administration of GKT137831, suggesting that Nox4, but not Nox1, is the source of ROS contributing to albuminuria, glomerular and tubular accumulation of extracellular matrix proteins in this model (27). It should be emphasized that unlike the previous studies using GKT inhibitors (27, 40, 44), our present findings indicate that Nox1/4 inhibition achieves protective effects when administered to type 1 diabetic mice with well-established nephropathy. Importantly, these results were observed in 5-mo-old OVE26 mice, at a time where severe diabetes and albuminuria are present. These results suggest that Nox1/4 inhibition with GKT137831 could be effective in reversing pathological changes in patients with established diabetic kidney disease.
The idea that Nox4 is the primary target of GKT137831 in DN is reinforced by the fact that the other Nox homologs that could potentially be affected by the inhibitor were either not found to be implicated in a mice model of DN (Nox2) (48) or are not expressed in mice (Nox5) (5, 22, 23).
In our study, GKT137831 markedly reduced macrophage infiltration in glomeruli and interstitium. These results are consistent with the reduction of monocyte chemotactic protein-1 (MCP-1) and macrophage infiltration observed in the aorta and kidneys of diabetic mice treated with GKT137831 (24, 37). Collectively, these results indicate that Nox-derived ROS play an important role in chemokine expression and macrophage infiltration in several micro- and macrovascular diabetic complications. The magnitude of macrophage infiltration correlates with disease progression in human DN. Furthermore, pharmacological blockade of MCP-1 or its receptor chemokine receptor 2 have been shown to be effective in animal models of DN and are being evaluated in patients with DN (20, 29, 35, 37, 41, 43). Therefore, the marked effect of GKT137831 on MCP-1 expression and macrophage infiltration likely contributes to its therapeutic efficacy. Furthermore, Nox1/4 inhibition attenuated the severity of structural abnormalities, as shown by the reduction in glomerular hypertrophy, fibronectin and collagen type IV expression, and mesangial expansion.
As anticipated, GKT137831 treatment did not affect the expression of Nox4 or Nox1, indicating that pharmacological inhibition of these Nox isoforms does not cause a compensatory increase in their expression level. Of note, a compensatory increase in renal Nox4 expression was reported in mice lacking Nox2 (48).
An intriguing and novel finding was the restoration of cells positive for podocyte markers in glomeruli of OVE26 mice treated with GKT137831. Podocytes are known to be exquisitely sensitive to oxidative stress (8, 16, 17, 22, 23, 30, 42). It is remarkable that a relatively short-term inhibition of Nox4 and Nox1 achieves a protective effect in mice with established kidney disease. GKT137831 attenuates ROS production in human podocytes exposed to high glucose (27). Our results indicate that this in vitro finding translates into podocyte protection in vivo. Further work is required to determine whether these results are related to increased cell survival, reduced loss of specific podocyte markers, or regeneration of new cell populations of podocytes. Incidental findings included an improved general appearance and a trend for improved survival in OVE26 mice treated with GKT137831. The cause of death in the untreated OVE26 mice was not identified. Circulatory collapse secondary to osmotic diuresis and proteinuria, or severe metabolic disturbances related to sustained hyperglycemia, might all underlie the increased mortality. While Nox1 and Nox4 inhibition could modulate some of these metabolic abnormalities, further work is required to confirm these findings and delineate the underlying mechanism(s). Figure 6 summarizes the renoprotective actions of GKT137831 described in the present study.
Fig. 6.
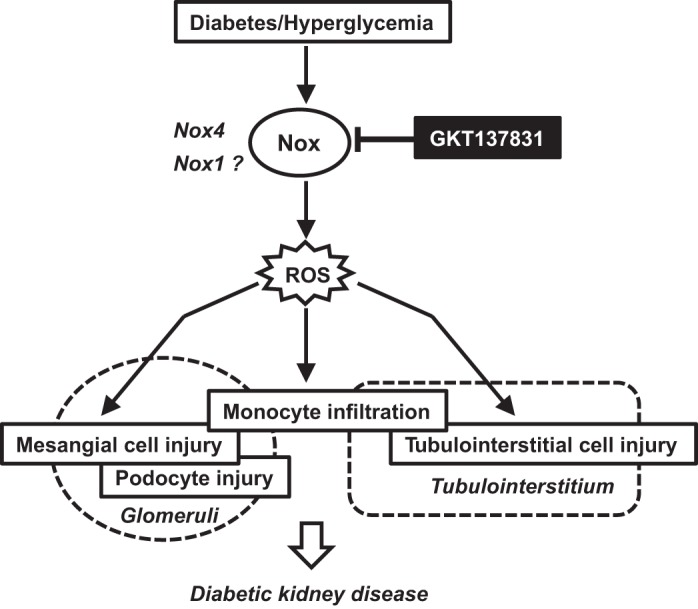
Speculative schematic for the renoprotective effects of small-molecule Nox1/4 inhibitor GKT137831 in diabetic kidney disease.
Overall, our results provide compelling evidence for a beneficial effect of Nox1/4 inhibition in the OVE26 model of DN, which reproduces the key features of human DN. Importantly, our study suggests that a relatively short-term treatment can be effective even when initiated in mice with established diabetes and diabetic kidney disease. However, further investigations in animal models of diabetes are needed, including the role of other Nox enzymes such as Nox5. Nox5 has recently been suggested to play a role in human DN (23, 26), but the lack of expression of Nox5 in rodents has made it difficult to assess the role of this Nox isoform in DN. Transgenic expression of human Nox5 in mouse podocytes resulted in mild proteinuria (26). It is worth noting that GKT137831 possesses substantial inhibitory activity on Nox5 (Ki ∼0.5 μM) and was shown to attenuate glucose-induced ROS production in human podocytes that express Nox5 (26).
In conclusion, there is accumulating evidence supporting a role for Nox4 as the major Nox responsible for the increased generation of ROS in DN. Since Nox1 mediates accelerated atherosclerosis in diabetes (24), pharmacological inhibition of Nox4 and Nox1 represents an attractive therapeutic strategy for patients with longstanding diabetes, to attenuate the development of nephropathy as well as macrovascular complications. Our data support the clinical evaluation of Nox1/4 inhibitors in diabetic patients. GKT137831 is currently being evaluated in a phase 2 clinical trial in patients with type 2 diabetes and albuminuria (ClinicalTrials.gov reference number NCT02010242). Our work suggests that such studies should be extended to patients with DN secondary to type 1 diabetes.
GRANTS
This work was supported by Juvenile Diabetes Research Foundation Multiproject Grants (Y. Gorin, K. Block, J. L. Barnes, and H. E. Abboud), the National Center for Advancing Translational Sciences, the National Institutes of Health (NIH) through Grants UL1 TR001120 (Y. Gorin), RO1 DK 079996 (Y. Gorin), RO1 CA 131272 (K. Block), RO1 DK 78971 (H. E. Abboud), and the Department of Veterans Affairs (K. Block, J. L. Barnes, and H. E. Abboud). GKT137831 was provided by Genkyotex through funds from Juvenile Diabetes Research Foundation Multiproject Grants.
DISCLOSURES
C. Szyndralewiez is a paid employee and owns shares of Genkyotex SA.
AUTHOR CONTRIBUTIONS
Author contributions: Y.G., C.S., and H.E.A. provided conception and design of research; Y.G., R.C.C., K.K., C.S., J.L.B., and H.E.A. analyzed data; Y.G., C.S., J.L.B., K.B., and H.E.A. interpreted results of experiments; Y.G., R.C.C., and C.S. prepared figures; Y.G. drafted manuscript; Y.G., C.S., and H.E.A. edited and revised manuscript; Y.G., C.S., and H.E.A. approved final version of manuscript; R.C.C., K.K., D.Y.L., F.B., S.T., P.F., C.S., and J.L.B. performed experiments.
ACKNOWLEDGMENTS
We thank Philippe Wiesel, MD, PhD for a critical reading of the manuscript.
The contributions of our late friend and colleague, Dr. Hannah E. Abboud, are gratefully acknowledged.
REFERENCES
- 1.Abboud HE. Mesangial cell biology. Exp Cell Res 318: 979–985, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56: 2316–2327, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schröder K, Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53: 842–853, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int 79: 944–956, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA 106: 14385–14390, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breyer MD, Böttinger E, Brosius FC 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K; Animal Models of Diabetic Complications Consortium. Mouse models of diabetic nephropathy. J Am Soc Nephrol 16: 27–45, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Breyer MD, Böttinger E, Brosius FC, Coffman TM, Fogo A, Harris RC, Heilig CW, Sharma K. Diabetic nephropathy: of mice and men. Adv Chronic Kidney Dis 12: 128–145, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Brosius FC 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications Consortium. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi Pache JC B, Barazzone-Argiroffo C, Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15: 607–619, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen AF, Chen DD, Daiber A, Faraci FM, Li H, Rembold CM, Laher I. Free radical biology of the cardiovascular system. Clin Sci (Lond) 123: 73–91, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Cooper ME, Jandeleit-Dahm K, Thomas MC. Targets to retard the progression of diabetic nephropathy. Kidney Int 68: 1439–1445, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Di Marco E, Gray SP, Chew P, Koulis C, Ziegler A, Szyndralewiez C, Touyz RM, Schmidt HH, Cooper ME, Slattery R, Jandeleit-Dahm KA. Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune-inflammatory responses in diabetic Apoe(−/−) mice. Diabetologia 57: 633–642, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10: 453–471, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eid AA, Ford BM, Bhandary B, Cavagliery R, Block K, Barnes JL, Gorin Y, Choudhury GG, Abboud HE. mTOR regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes 62: 2935–2947, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, Barnes JL, Abboud HE. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem 285: 37503–37512, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 58: 1201–1211, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol 33: 3439–3460, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ford BM, Eid AA, Göőz M, Barnes JL, Gorin YC, Abboud HE. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am J Physiol Renal Physiol 305: F323–F332, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giunti S, Barutta F, Perin PC, Gruden G. Targeting the MCP-1/CCR2 system in diabetic kidney disease. Curr Vasc Pharmacol 8: 849–860, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Gorin Y, Block K. Nox as a target for diabetic complications. Clin Sci (Lond) 125: 361–382, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Gorin Y, Block K. Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 61C: 130–142, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127: 1888–1902, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, Thannickal VJ. Reversal of persistent fibrosis in aging by targeting nox4-nrf2 redox imbalance. Sci Transl Med 6: 231ra47, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holterman CE, Thibodeau JF, Towaij C, Gutsol A, Montezano AC, Parks RJ, Cooper ME, Touyz RM, Kennedy CR. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol 25: 784–797, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang JX, Chen X, Serizawa N, Szyndralewicz C, Page P, Schroder K, Brandes RP, Devaraj S, Torok NJ. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radical Biol Med 53: 289–296, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK, Kim SH, Han SY, Han KH, Lee JE, Han JY, Kim HK, Cha DR. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int 78: 883–894, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol 6: 395–423, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khazim K, Gorin Y, Cavaglieri RC, Abboud HE, Fanti P. The antioxidant silybin prevents high glucose-induced oxidative stress and podocyte injury in vitro and in vivo. Am J Physiol Renal Physiol 305: F691–F700, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem 53: 7715–7730, 2010. [DOI] [PubMed] [Google Scholar]
- 33.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol 302: C597–C604, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, Eulberg D, Buchner K, Selve N, Klussmann S, Anders HJ. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3'PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol 172: 628–637, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papadimitriou A, Peixoto EB, Silva KC, Lopes de Faria JM, Lopes de Faria JB. Increase in AMPK brought about by cocoa is renoprotective in experimental diabetes mellitus by reducing NOX4/TGFβ-1 signaling. J Nutr Biochem 25: 773–784, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Grüner S, Green L, Mattei P, Hartmann G, Anders HJ. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 80: 68–78, 2011. [DOI] [PubMed] [Google Scholar]
- 38.Schramm A, Matusik P, Osmenda G, Guzik TJ. Targeting NADPH oxidases in vascular pharmacology. Vasc Pharmacol 56: 216–231, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, Hebert RL. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol 299: F1348–F1358, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, Burns KD, Cooper ME, Jandeleit-Dahm K, Page P, Szyndralewiez C, Heitz F, Hebert RL, Touyz RM. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) 124: 191–202, 2013. [DOI] [PubMed] [Google Scholar]
- 41.Seok SJ, Lee ES, Kim GT, Hyun M, Lee JH, Chen S, Choi R, Kim HM, Lee EY, Chung CH. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol Dial Transplant 28: 1700–1710, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol 7: 176–184, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan T, Miao Z, Dairaghi DJ, Krasinski A, Wang Y, Zhao BN, Baumgart T, Ertl LS, Pennell A, Seitz L, Powers J, Zhao R, Ungashe S, Wei Z, Boring L, Tsou CL, Charo I, Berahovich RD, Schall TJ, Jaen JC. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am J Physiol Renal Physiol 305: F1288–F1297, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HH, Coughlan MT, Cooper ME, Forbes JM, Jandeleit-Dahm KA. Nox-4 deletion reduces oxidative stress and injury by PKC-α-associated mechanisms in diabetic nephropathy. Physiol Rep 2: e12192, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vendrov AE, Madamanchi NR, Niu XL, Molnar KC, Runge M, Szyndralewiez C, Page P, Runge MS. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem 285: 26545–26557, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiesel P, Hovsepian L, Mutch PJ, Herve J, Heitz F, Page P. Safety and pharmacokinetics of single and multiple doses of a first in class dual NADPH oxidase 1 and 4 inhibitor administered orally in healthy subjects. J Am Soc Nephrol 23: 559A, 2012. [Google Scholar]
- 47.Wilkinson-Berka JL, Deliyanti D, Rana I, Miller AG, Agrotis A, Armani R, Szyndralewiez C, Wingler K, Touyz RM, Cooper ME, Jandeleit-Dahm KA, Schmidt HH. NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid Redox Signal 20: 2726–2740, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You YH; Okada S; Ly S; Jandeleit-Dahm KA; Barit D; Namikoshi T; Sharma K. Role of Nox2 in diabetic kidney disease. Am J Physiol Renal Physiol 304: F840–F848, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng S, Noonan WT, Metreveli NS, Coventry S, Kralik PM, Carlson EC, Epstein PN. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes 53: 3248–3257, 2004. [DOI] [PubMed] [Google Scholar]