

Abstract
We have shown a sex-specific effect of fetal programming on Na+ excretion in adult sheep. The site of this effect in the kidney is unknown. Therefore, we tested the hypothesis that renal proximal tubule cells (RPTCs) from adult male sheep exposed to betamethasone (Beta) before birth have greater Na+ uptake than do RPTCs from vehicle-exposed male sheep and that RPTCs from female sheep similarly exposed are not influenced by antenatal Beta. In isolated RPTCs from 1- to 1.5-yr-old male and female sheep, we measured Na+ uptake under basal conditions and after stimulation with ANG II. To gain insight into the mechanisms involved, we also measured nitric oxide (NO) levels, ANG II receptor mRNA levels, and expression of Na+/H+ exchanger 3. Basal Na+ uptake increased more in cells from Beta-exposed male sheep than in cells from vehicle-exposed male sheep (400% vs. 300%, P < 0.00001). ANG II-stimulated Na+ uptake was also greater in cells from Beta-exposed males. Beta exposure did not increase Na+ uptake by RPTCs from female sheep. NO production was suppressed more by ANG II in RPTCs from Beta-exposed males than in RPTCs from either vehicle-exposed male or female sheep. Our data suggest that one site of the sex-specific effect of Beta-induced fetal programming in the kidney is the RPTC and that the enhanced Na+ uptake induced by antenatal Beta in male RPTCs may be related to the suppression of NO in these cells.
Keywords: kidney, angiotensin, programming
developmental programming is a concept that has received increasing acceptance over the last two decades (3). This idea that events occurring at critical times in fetal life have adverse effects on adult health has been established by results from numerous animal and epidemiological studies (1, 2, 4, 18, 46, 47), and data suggest that one common pathway through which programming occurs involves exposure of the fetus to inappropriately high levels of glucocorticoids at some point during development (40, 54, 57, 58). This, in conjunction with the widespread use of synthetic glucocorticoids in women threatening to deliver prematurely to reduce neonatal morbidity and mortality associated with preterm birth (50, 53, 55), has led investigators, including ourselves, to study the effects of antenatal steroid exposure on cardiovascular and renal function (17, 52, 61).
A significant body of literature indicates some programming stimuli (mostly, but not exclusively, associated with dietary manipulations in pregnancy) show sex-specific effects (25, 26, 47, 51). For example, protein restriction during pregnancy in rats produces hypertension in male but not female offspring (68, 69). We have shown that clinically relevant doses of betamethasone (Beta; a synthetic glucocorticoid routinely given to women threatening to deliver prematurely) administered to pregnant sheep at ∼0.6 gestation (term equals 145 days) have different effects on renal function in adult male and female offspring. The ability of male offspring to effectively excrete a Na+ load is markedly compromised by exposure to the steroid, whereas female offspring with identical exposure readily excrete the same Na+ load (61). Considering that the proximal tubule is responsible for reabsorption of ∼65% of filtered Na+ (71), these observations suggest that antenatal Beta exposure may have different effects on Na+ uptake by renal proximal tubule cells (RPTCs) obtained from adult male and female offspring. Thus, the present study was designed to test the basic hypothesis that proximal tubule cells from adult male animals exposed to Beta before birth would demonstrate enhanced Na+ uptake compared with cells from vehicle-treated animals and that the increase in Na+ uptake produced by glucocorticoid exposure in male animals would be absent in female animals. We also wanted to determine whether ANG II-induced Na+ uptake by RPTCs would be differentially affected by antenatal glucocorticoid exposure in male and female animals.
MATERIALS AND METHODS
Animals.
Time-dated pregnant sheep were obtained from local suppliers and maintained in open pasture with free access to food and water during pregnancy and lactation. Animals were randomly assigned to two groups: one group received two 0.17 mg/kg im injections of a 1:1 mixture of betamethasone acetate and betamethasone phosphate (Beta group; Celestone Soluspan, Schering, Kenilworth, NJ) and the other group received two vehicle injections as previously described (60). This regime of Beta administration was chosen because it is equivalent to that currently used in clinical practice (65, 67). A total of 44 animals (1–1.5 yr old) were used: 26 male animals (n = 12 control and 14 Beta-treated animals) and 18 female animals (n = 8 control animals and 10 Beta-treated animals), with roughly equivalent numbers of animals coming from twin pregnancies in the groups (n = 14 male animals and 9 female animals). Animals were euthanized with an overdose of Euthasol (Virbac), and the kidney cortex was either frozen immediately in liquid nitrogen for gene expression assays or dispersed for isolation of RPTCs. All procedures for housing handling, management, and euthanasia of sheep were approved by the Institutional Animal Care and Use Committee of Wake Forest University.
Preparation of primary RTPCs.
The kidney cortex was minced into small pieces and digested by collagenase type II (Worthington Biochemical) and DNAse I (Sigma-Aldrich) solution. After digestion, the solution was passed through a 70-μm mesh sieve (BD Biosciences) and centrifuged at 500 g and 4°C for 10 min. Tissue pellets were then applied to a discontinuous Percoll gradient (Sigma-Aldrich) of 10–35% (vol/vol) and centrifuged at 15,000 g for 60 min at 4°C. The proximal tubule cell pellet was collected. Some cells were put aside for Na+/H+ exchanger 3 (NHE3) protein and mRNA determinations, and the remainder was grown in DMEM-F-12 medium with 15 mM HEPES and 20 mM sodium bicarbonate (pH 7.4). Primary RPTC cultures were maintained at 37°C in a 5% CO2 humidified environment.
Cellular Na+ uptake experiments.
Na+ uptake by RPTCs was determined by measuring the percent change in fluorescence emission of the Na+ dye sodium green (Molecular Probes, Eugene, OR), which reflects changes in intracellular Na+ concentrations (56). Briefly, confluent monolayers were grown for a further 24 h in serum-free medium. Cells were incubated at 37°C for 30 min in loading medium (5 × 10−6 mol/l sodium green in culture medium), and the basal fluorescence signal (excitation: 507 nm and emission: 532 nm) of each well was measured. RPTCs were exposed to different Na+ concentrations in the presence of the Na+-K+-ATPase inhibitor ouabain (5 × 10−5 mol/l), and the fluorescence signal was measured. Solutions with Na+ concentrations of 0, 32, 62, 92, and 142 mmol/l were prepared using NaCl and Na+ substitute (equimolar N-methyl-d-glucamine) mixed in different proportions to maintain osmotic pressure (pH 7.4). For the experiments using ANG II stimulation or blockade of ANG II receptors, immediately before Na+ uptake experiments, RPTCs were incubated with either medium alone (basal) or ANG II (1 × 10−11 mol/l) in the presence or absence of the ANG II type 1 (AT1) receptor blocker candesartan (CS; 1 × 10−6 mol/l) or the ANG II type 2 (AT2) receptor blocker PD-123319 (PD; 1 × 10−6 mol/l) for 1 h. Data from all experiments were normalized to the cellular protein content in each well and expressed as percent changes from basal fluorescence.
Measurement of nitric oxide production by RPTCs.
Cultured RPTCs were preincubated with the fluorescence dye (4-amino-5-methylamino-2′,7′)difluorofluorescein diacetate (2.5 × 10−6 mol/l, Molecular Probes, Invitrogen) in Krebs-Ringer phosphate buffer (KRP buffer) containing (in mM) 140 NaCl, 14 glucose, 4.7 KCl, 2.5 CaCl2, 1.8 MgSO4, and 1.8 KH2PO4 (pH 7.4) for 30 min at 37°C. RPTCs were washed twice in KRP buffer to remove any excess probe and then incubated with KRP buffer for another 20 min. Cells were then treated with ANG II (1 × 10−11 mol/l) alone or with 1 × 10−6 mol/l CS or PD. Background fluorescence was obtained immediately after the addition of peptides or/and inhibitors. The end-point fluorescence was taken after 90 min of incubation. Increases in (4-amino-5-methylamino-2′,7′)difluorofluorescein fluorescence, indicative of nitric oxide (NO) production, were measured using a SpectraMax M2e microplate reader (Molecular Devices, Sunnyvale, CA) at wavelengths of 495 nm (excitation) and 515 nm (emission) as described in manufacturer's instruction. Data were presented as percentages of control. All samples were corrected for background fluorescence and also corrected to the N-nitro-l-arginine methyl ester-suppressible NO signal.
Immunofluorescence microscopy.
RPTCs were washed briefly with PBS, fixed with 2% paraformaldehyde for 15 min, and washed twice with PBS. Cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min and washed three times with PBS. To reduce nonspecific binding of the antibodies, cells were blocked with 3% BSA (Sigma) in PBS for 2 h. Cells were incubated with primary antibody [anti-Na+-dependent glucose cotransporter (SGLT)2 or anti-NHE3, Abcam] for 1 h. Cells were washed and incubated with fluorescein-conjugated secondary antibodies for 30 min. After an additional wash, nuclei were stained with 4′,6-diamidino-2-phenylindole for 1 min. Images were obtained by fluorescence microscopy (Axioplan 2, Carl Zeiss).
ANG II receptors and NHE3 mRNA expression levels in the kidney.
The relative abundance of AT1 and AT2 receptor mRNA and NHE3 mRNA transcripts in the kidney cortex were measured by quantitative real-time RT-PCR using TaqMan PCR as previously described by us (60). Sequences of primers and probes used in real-time PCR assays are shown in Table 1.
Table 1.
Primers and probes used in real-time PCR assays
| Gene | Forward Primer | Reverse Primer | Probe Sequence |
|---|---|---|---|
| ANG II type 1 receptor | 5′-CTGTCATTTACCCCTTTCTGTCTCA-3′ | 5′-AGACAAGCCATACACCAACCAA-3′ | FAM-CCTGGCAAGCATCTTA-MGB |
| ANG II type 2 receptor | 5′-GGCAATCGTGCTTTTCTTTTTCTTC-3′ | 5′-GCCCAACTGAATTAACACATCCAT-3′ | FAM-CCCACCAGATATTCAC-MGB |
| Na+/H+ exchanger 3 | 5′-GCTGCAGCAGTACCTGTACAA-3′ | 5′-CCCATGTCGGCTGTAGAGATG-3′ | FAM-CCCGGCAGGAGTACAAA-MGB |
| GAPDH | 5′-GCATCGTGGAGGGACTTATGAC-3′ | 5′-GGGCCATCCACAGTCTTCTG-3′ | FAM-ACGCCATCACTGCCACCT-MGB |
Western blot analysis for NHE3 protein expression.
RPTC pellets from the above preparation were lysed on ice in RIPA buffer [25 mM Tris·HCl (pH 7.6), 150 mM NaCI, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 2 μg/ml aprotinin, and 0.5 mM PMSF] for 30 min. Samples were then centrifuged at 2,000 g for 15 min, and protein content was assessed using a BCA assay (Pierce, Rockford, IL) as described in the manufacturer's protocol. Western blot analysis using anti-NHE3 antibody (1:100 dilution, Abcam) and signal quantification were performed as previously described (60).
Statistical analysis.
All data analyses were performed using the GraphPad Prism (version 6) statistical analysis package (GraphPad Software, La Jolla, CA). For all tests, significance was set at P ≤ 0.05. Data were analyzed using two-way ANOVA followed by Tukey's post hoc analysis and are expressed as means ± SE.
RESULTS
Identification of RPTCs.
Immunocytochemical staining for SGLT2 revealed that >95% of the cells in culture exhibited positive staining for SGLT2, which is a characteristic marker of RPTCs (Fig. 1A). A high abundance of SGLT2 was apparent in the cell cytoplasm. The controls, which were incubated only with secondary antibodies, showed no detectable immunofluorescence, indicating signal specificity (Fig. 1B).
Fig. 1.
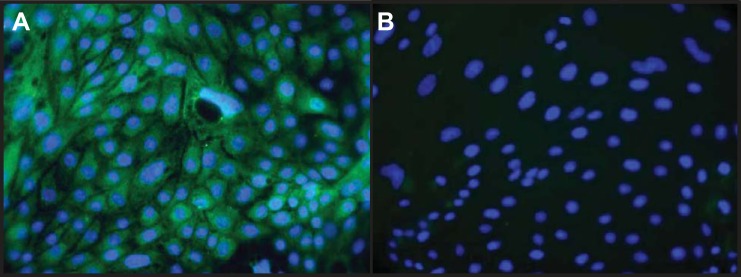
Immunocytochemical identification of cultured renal proximal tubule cells (RPTCs). A: fluorescence image of RPTCs derived from kidneys from control animals stained with an antibody to anti-Na+-glucose cotransporter (SGLT)2, a marker for RPTCs (fluorescein, green), and the nuclear marker 4′,6-diamidino-2-phenylindole (DAPI; blue). B: control incubations where the primary antibody was omitted. Magnification: ×400.
Effects of antenatal Beta on Na+ uptake in RPTCs.
There was a highly significant effect of prenatal Beta exposure on Na+ uptake by RPTCs isolated from adult male sheep kidneys. Basal Na+ uptake increased >400% above control (0 Na+) values in Beta-exposed cells from male cells, whereas vehicle-exposed cells increased Na+ uptake by only ∼300% (F = 44.7, P < 0.0001 Na+ effect, F = 10.1, P = 0.0019 Beta effect; Fig. 2A). In contrast, no effect of antenatal steroid treatment was observed in cells from female sheep (Fig. 2B). Cells from both vehicle- and Beta-exposed female sheep increased unstimulated Na+ uptake by 250% between 0 and 140 mmol Na+ (F = 58.6, P < 0.0001 Na+ effect only). ANG II treatment (10−11 mol/1) increased Na+ uptake by RPTCs from both vehicle-treated (F = 11.9, P = 0.0008) and Beta-treated (F = 9.3, P = 0.0027) male sheep compared with cells incubated with Na+ alone (Fig. 2, A and C). The response to ANG II (Fig. 2C) was greatest in cells from Beta-exposed males (F = 9.7, P = 0.0023 Beta effect). These cells increased Na+ uptake by >500%. ANG II also increased Na+ uptake by cells from vehicle-treated female sheep (F = 38.1, P = <0.0001) and Beta-treated female sheep (F = 38.6, P ≤ 0.0001) compared with unstimulated uptake (Fig. 2, C and D) However, as opposed to male cells, Beta exposure did not enhance responses to ANG II by cells from female sheep (Fig. 2D). When equivalent treatment groups (vehicle vs. vehicle, Beta vs. Beta, basal vs. basal, and stimulated vs. stimulated) were compared, Na+ uptake by cells from male sheep was always significantly greater than uptake by cells from female sheep.
Fig. 2.
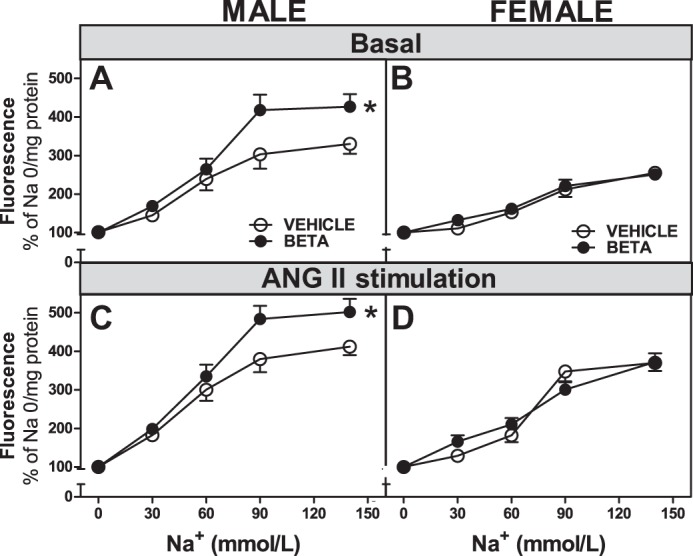
Na+ uptake in RPTCs from male and female vehicle- and betamethasone (Beta)-treated animals. A–D: Na+ uptake in RPTCs from male animals (A and C; n = 12 vehicle-treated animals and 14 Beta-treated animals) and female animals (B and D; n = 8 vehicle-treated animals and 10 Beta-treated animals) expressed as increase in fluorescence relative to a Na+ concentration of 0. A and B: basal conditions; C and D: ANG II-stimulated conditions. Values are means ± SE. *P < 0.05 vs. RPTCs from vehicle-treated animals.
Antenatal Beta alters ANG II-induced Na+ uptake via AT1 receptors in RPTCs.
To explore the contribution of AT1 and AT2 receptor subtypes to the ANG II-mediated increase in Na+ uptake in RPTCs, cells were incubated in media containing the AT1 receptor blocker CS or the AT2 receptor blocker PD and stimulated with 10−11 mol/l ANG II. As shown in Fig. 3A, Na+ uptake by cells from vehicle-treated male sheep increased with increasing concentrations of Na+ (F = 106.1, P < 0.0001), and the stimulatory effect of ANG II was attenuated by CS (F = 3.6, P = 0.041). No significant effect of PD was observed. RPTCs from both vehicle- and Beta-treated female sheep had reduced Na+ uptake when CS was incubated with ANG II compared with uptake with ANG II alone (P < 0.001 for both), whereas PD had no effect.
Fig. 3.
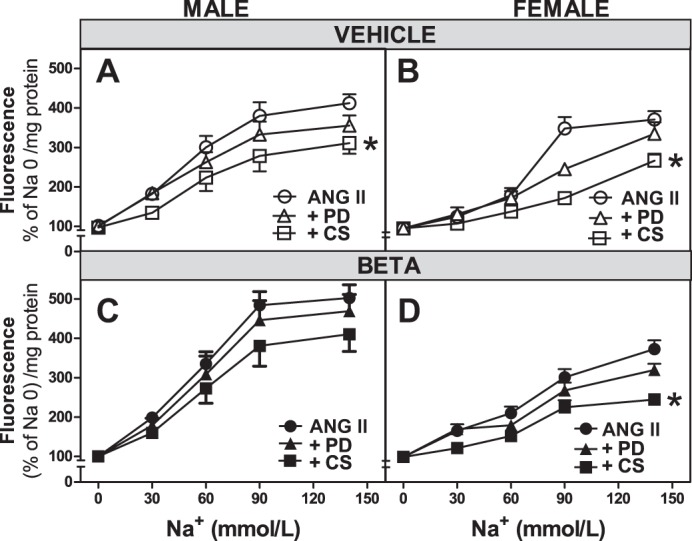
ANG II receptors modulate Na+ uptake in RPTCs from male and female vehicle- and Beta-treated animals. A–D: Na+ uptake in RPTCs from male animals (A and C; n = 12 vehicle-treated animals and 14 Beta-treated animals) and female animals (B and D; n = 8 vehicle-treated animals and 10 Beta-treated animals) expressed as increase in fluorescence relative to a Na+ concentration of 0. In parallel experiments, RPTCs were incubated with ANG II alone (10−11 mol/l) or in the presence of candesartan (+CS; 10−6 mol/1, male/female animals: n = 9/7 in A and B and 9/10 in C and D) or PD-123319 (+PD; 10−6 mol/l, male/female animals: n = 8/5 in A and B and 9/10 in C and D). Values are means ± SE. *P < 0.05 vs. ANG II alone-treated cells.
Effects of antenatal Beta on ANG II-induced NO production by RPTCs.
We observed an ∼27% reduction in NO production upon treatment of RPTCs from vehicle-treated male sheep with ANG II (Fig. 4). This suppression of NO production was enhanced in Beta-treated animals (P < 0.05; Fig. 4). There was a trend (P = 0.07) for modest suppression of NO in ANG II-treated RPTCs from female sheep that was similar in vehicle- or Beta-exposed offspring (Fig. 4). The ANG II-induced suppression of NO was converted to stimulation by coincubation with CS in both vehicle- and Beta-treated male sheep (F = 15.9, P = 0.0002 CS effect; Fig. 5A), whereas coincubation with the AT2 receptor antagonist PD had no significant effect on NO responses to ANG II. Cells from female sheep showed qualitatively similar NO responses as cells from male sheep when ANG II and CS were both present in the incubation media, i.e., suppression was converted to stimulation (F = 5.4, P = 0.016; Fig. 5B).
Fig. 4.
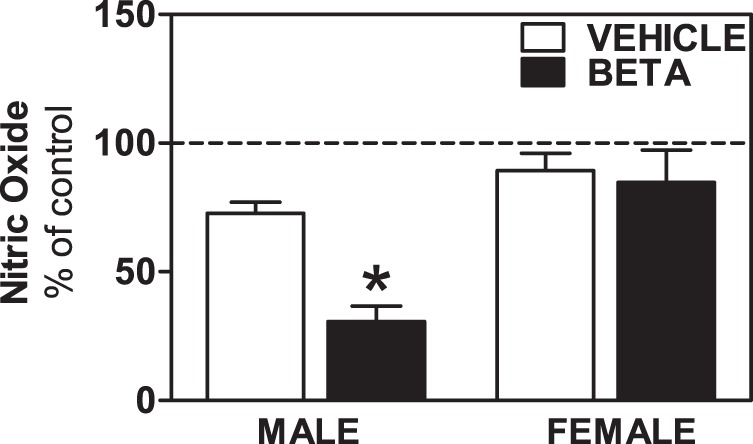
Nitric oxide (NO) production by RPTCs from male and female vehicle- and Beta-treated animals. NO production was expressed as a percentage of basal levels (% of control) in RPTCs from male animals (n = 5 vehicle-treated animals and 5 Beta-treated animals) and female animals (n = 5 vehicle-treated animals and 5 Beta-treated animals) treated with ANG II (10−11 mol/l). *P < 0.05 vs. vehicle treatement.
Fig. 5.
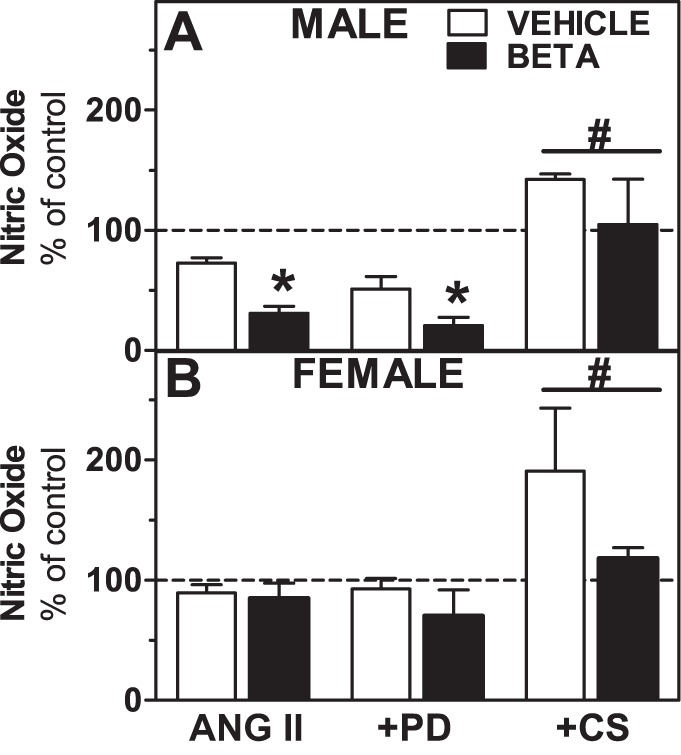
Role of ANG II receptors in modulating NO production by RPTCs from male (top) and female (bottom) vehicle- and Beta-treated animals. NO production was expressed as a percentage of basal levels (% of control) in RPTCs from male animals (n = 5 vehicle-treated animals and 5 Beta-treated animals) and female animals (n = 5 vehicle-treated animals and 5 Beta-treated animals) treated with ANG II (10−11 mol/l). In parallel experiments, RPTCs were incubated with ANG II alone or +PD (10−6 mol/l) or +CS (10−6 mol/l) as indicated. Values are means ± SE. *P < 0.05 vs. ANG II alone-treated cells.
Effects of antenatal Beta on ANG II receptor mRNA levels in the kidney cortex.
As shown in Fig. 6A, prenatal Beta exposure significantly increased AT1 receptor mRNA expression in male and female kidneys. There were sex (male > female, F = 20.4, P = 0.0002) and treatment (Beta > vehicle, F = 5.2, P = 0.033) effects. AT2 receptor mRNA levels were also higher in male cells compared with female cells (F = 5.7, P = 0.0265; Fig. 6B). There was no overall effect of steroid exposure on AT2 receptor mRNA expression.
Fig. 6.
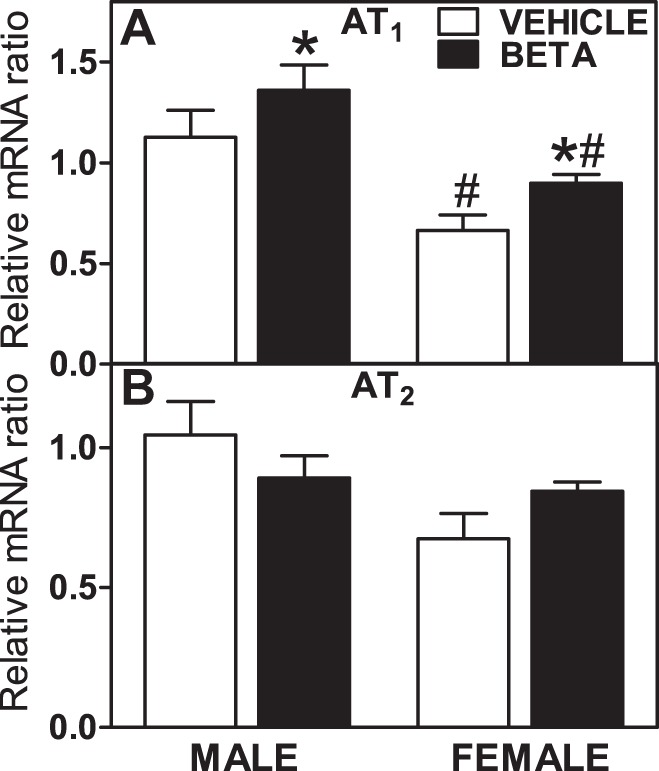
Expression levels of ANG II type 1 (AT1) and type 2 (AT2) receptors in kidney tissue. A and B: relative gene expression for AT1 (A) and AT2 (B) II receptors in the kidney in male animals (n = 6 vehicle-treated animals and 6 Beta-treated animals) and female animals (n = 6 vehicle-treated animals and 6 Beta-treated animals). Values are means ± SE. *P < 0.05 vs. vehicle treatment, #P < 0.05 vs. male animals.
Effects of antenatal Beta on NHE3 mRNA and protein expression in RPTCs.
We examined the effect of antenatal Beta on NHE3 mRNA expression in RPTCs before and after stimulation with ANG II. There was a tendency for prenatal Beta exposure to increase NHE3 mRNA expression in RPTCs from male sheep compared with RPTCs from vehicle-exposed male sheep, but the differences were not statistically significant (F = 4.17, P = 0.0513; Fig. 7A). No differences were observed in NHE3 mRNA expression in RPTCs from female sheep (Fig. 7B). No effects of ANG II stimulation on NHE3 mRNA expression were observed. As shown in Fig. 8A, NHE3 protein expression was detected by immunostaining in the cytoplasm and associated with the plasma membrane in RPTCs. Protein expression was increased significantly by Beta exposure in cells from both sexes (F = 12.9, P = 0.0024; Fig. 8B).
Fig. 7.
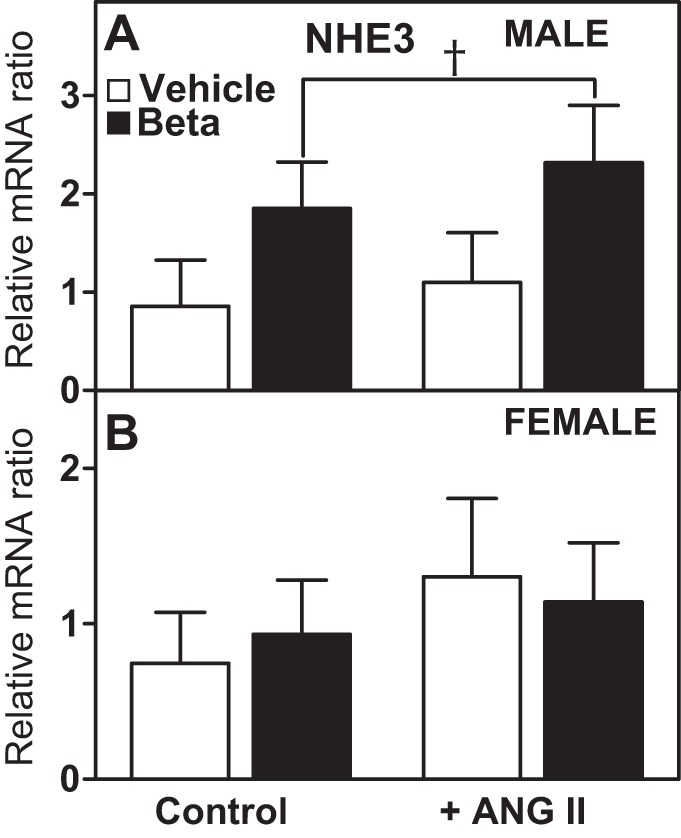
Gene expression of Na+/H+ exchanger 3 (NHE3) in sheep RPTCs: effects of ANG II treatment. A and B: relative gene expression of NHE3 in RPTCs from male animals (A; n = 5 vehicle-treated animals and 6 Beta-treated animals) and female animals (B; n = 5 vehicle-treated animals and 4 Beta-treated animals). RPTCs were also treated with ANG II (10−11 mol/l) as indicated. Values are means ± SE. † P = 0.05 vs. vehicle treatment.
Fig. 8.
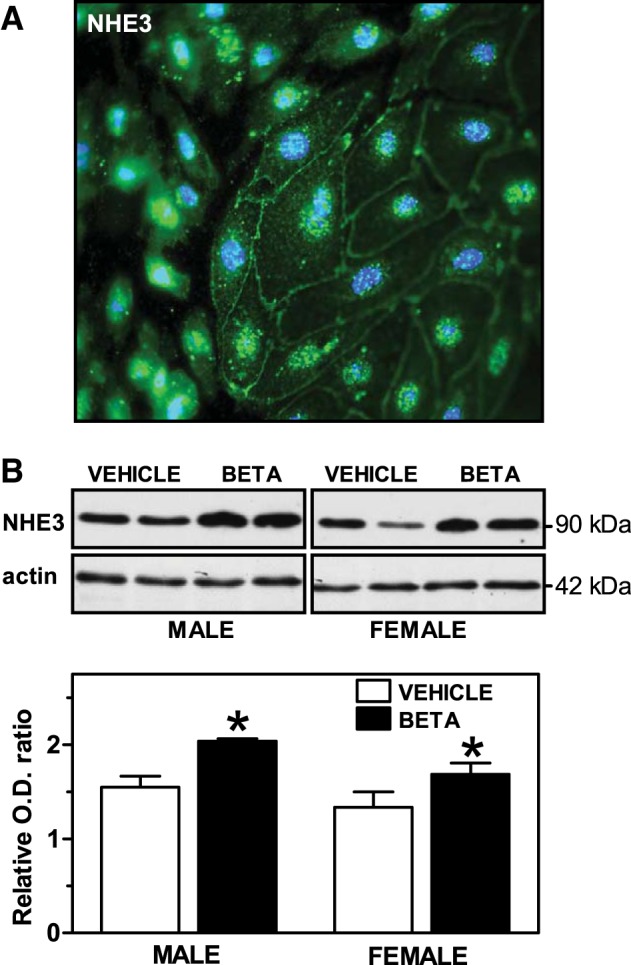
Protein expression of NHE3 in sheep RPTCs. A: immunocytochemical identification of NHE3 in RPTCs from a control animal. Shown is a fluorescence image of RPTCs stained with an antibody to anti-NHE3 (fluorescein, green) and the nuclear marker DAPI (blue). Note the cytoplasmic and plasma membrane localization of NHE3. Magnification: ×800. B: representative Western blots of protein extracts obtained from RPTCs from male animals (n = 5 vehicle-treated animals and 6 Beta-treated animals) and female animals (n = 5 vehicle-treated animals and 4 Beta-treated animals). A densitometric analysis of the Western blot signals is also included. Values are mean ± SE. *P < 0.05. Beta effect: F = 12.9, P = 0.0024; sex effect: F = 5.9, P = 0.027.
DISCUSSION
The primary purpose of this study was to determine if antenatal Beta exposure would have a sex-specific effect on Na+ handling by RPTCs obtained from adult offspring. We found that exposure to a clinically relevant dose of Beta at 0.6 gestation enhanced the uptake of Na+ by RPTCs from adult male offspring under basal conditions and after stimulation with ANG II. This effect was absent in cells from female animals similarly exposed, thereby demonstrating sex specificity in the effects of antenatal Beta exposure on Na+ handling by RPTCs from adult offspring. The greater ANG II-induced uptake of Na+ by cells from Beta-treated male animals was associated with more pronounced suppression of NO production by the peptide, and this effect of Beta was absent in cells from female animals. The sex-related differences in Na+ uptake may also be related to greater expression of the AT1 receptor and NHE3 in cells from Beta-exposed male animals compared with cells from female animals. To our knowledge, these are the first experiments that have demonstrated a sex-specific effect of a programming stimulus on RPTCs. Since RPTCs from male animals have increased Na+ uptake compared with cells from female animals, the results also suggest that the mechanisms involved in hypertension, in particular the hypertension produced by antenatal steroid exposure, may be more related to alterations in Na+ handling by the kidney in male than female animals. To date, the mechanisms responsible for these sex differences are unknown.
While there is little doubt of the multifactorial nature of hypertension, there is general consensus that the kidney plays a significant role in the development of this disease (13). In particular, it is the action of the kidney in balancing salt and water excretion with intake that is important for the regulation of blood pressure (28, 29). There is also a large body of evidence indicating that the kidney is one prime target for the effects of fetal programming due to a variety of causes (12, 39, 41, 64). These effects frequently are related to disturbances in renal development, in particular a reduction in the number of nephrons during early development (62, 63). More often than not, the reductions in nephron number are associated with elevations in blood pressure in the offspring, and alterations in renal function have also been noted (4, 62, 63). For example, we have found that antenatal Beta exposure in sheep reduces nephron number and increases blood pressure in both male and female adult offspring. The elevations in blood pressure are accompanied by an inability to excrete a Na+ load in male offspring, whereas female offspring maintain their capability to excrete Na+ (61). These data suggest that a potential explanation for the elevated blood pressure observed in adult male offspring after antenatal Beta exposure is related to altered Na+ handling by the kidney. The results of our experiments on Na+ uptake by RPTCs support this possibility. Thus, under basal conditions and after ANG II stimulation, Na+ uptake was significantly enhanced in RPTCs from male animals exposed to Beta before birth compared with cells from vehicle-exposed male animals. In contrast, Beta exposure did not increase basal Na+ uptake in cells from female animals or change responses to ANG II. Considering the above, it is reasonable to propose that the elevations in blood pressure observed in adult male offspring exposed to Beta before birth may be related to enhanced Na+ reabsorption by proximal tubule cells, whereas the increased blood pressure in female offspring exposed to the steroid are not the result of disturbances in proximal tubular Na+ reabsorption. While it is conceivable that Na+ uptake by RPTCs from Beta-exposed female animals could increase with age and exacerbate their existing hypertension, we know of no evidence in the literature to indicate that this occurs. Other evidence favoring the idea that different mechanisms may be responsible for the development of hypertension in males and females has been nicely summarized in a recent review (72).
Our prior work suggests that there is an imbalance in the intrarenal renin-angiotensin system resulting from the antenatal glucocorticoid treatment that favors AT1 receptor activation relative to ANG(1–7)-Mas receptor stimulation or AT2 receptor-mediated actions (10, 15, 30, 59). It has been known for some time that ANG II promotes Na+ reabsorption in the proximal tubule (14, 24, 31) and that the role of the intrarenal renin-angiotensin system has become increasingly prominent, particularly relative to hypertension (8, 11, 38, 49). Indeed, ANG II activation of AT1 receptors on proximal tubule cells appears essential for the development of ANG II-induced hypertension (27, 42, 43). The enhanced Na+ uptake in response to ANG II by RPTCs from Beta-exposed male offspring fits nicely with a crucial role for AT1 receptors and emphasizes the potential importance of the sex-related difference in responsiveness in terms of the difference in Na+ excretion between male and female Beta-exposed offspring (61).
The role of NO in influencing Na+ handling by the kidney has been studied for over a decade, and there is general consensus that NO inhibits Na+ absorption in the proximal tubule, although there is some evidence there may be a biphasic effect (23, 34, 44). The increased ANG II-induced Na+ uptake we observed in male RPTCs was associated with a decrease in NO production after ANG II treatment. This inhibition was greater in RPTCs from Beta-treated male animals compared with cells from vehicle-treated animals and was blocked by CS, suggesting that 1) ANG II suppresses NO and the suppression promotes increased Na+ uptake in RPTCs and 2) the effect of ANG II on NO is mediated by activation of the AT1 receptor. There was a dichotomy in the suppression of NO by ANG II in cells from Beta-exposed male animals compared with Beta-exposed female animals, which was consistent with the differences in Na+ uptake in response to ANG II in these groups. Thus, there was enhanced NO suppression and Na+ uptake in cells from Beta-treated male animals compared with cells from vehicle-treated animals, whereas cells from vehicle-treated and Beta-treated female animals showed similar NO suppression and Na+ uptake. Taken together, the data suggest that lack of an effect of Beta exposure on Na+ uptake by cells from female animals is related to a lack of enhanced suppression of NO by ANG II in these cells.
The mechanisms by which prenatal Beta exposure enhances ANG II-induced NO suppression in RPTCs from males and how females are protected from this are not known but may be related to differences in the generation of ROS. ANG II stimulates ROS production in the kidney via activation of NAD(P)H oxidases, which increase levels of superoxide and superoxide can effectively reduce levels of NO while inducing oxidative stress (21, 22, 66). We have shown that ANG II stimulates 8-isoprostane (a marker of oxidative stress) production by RPTCs from Beta-exposed male animals and that this effect, associated with increased levels of p47phox, a subunit of NAD(P)H oxidase, was absent in female animals, in which increased SOD activity was observed (5). Thus, it is possible that antenatal treatment with glucocorticoids increased the generation of ROS by ANG II in RPTCs from male animals, which limited the bioavailability of NO, whereas female animals were protected by higher levels of SOD, which scavenges ROS. Additional work is needed to establish if this is the explanation for the different NO responses in cells from Beta-exposed male and female animals.
A somewhat unexpected finding was that ANG II in the presence of the AT1 receptor antagonist CS stimulated NO production compared with inhibition produced by ANG II alone in both male and female cells. This stimulation was associated with a reduction in Na+ uptake by RPTCs. Recently, there has been significant interest in the role of the AT2 receptor in promoting natriuresis (9, 32, 33, 37), and it has been suggested that selective stimulation of the AT2 receptor might be a therapeutic approach for the treatment of some forms of hypertension (33, 35, 37). Activation of the AT2 receptor with a nonpeptide-specific agonist increases NO levels, which then inhibit Na+ uptake by proximal tubule cells (37), which results in natriuresis. One simple explanation for the increased NO levels we observed in cells treated with the combination of ANG II and CS is that blockade of the AT1 receptor allows ANG II to stimulate only the AT2 receptor and this increases NO in RPTCs. Alternatively, ANG II might have been converted to other peptides, such as ANG III, which stimulates the AT2 receptor (7, 36) or ANG(1–7), which stimulates the Mas receptor (10), to produce the effect on NO that was unmasked by blockade of the AT1 receptor. Both angiotensin-converting enzyme 2 and aminopeptidase A, enzymes responsible for the generation of ANG(1–7) and ANG III, respectively, are primarily expressed on the apical membrane of the proximal tubule (70).
We have previously reported on the basis of binding assays that antenatal Beta increased receptors thought to be AT1 and decreased receptors thought to be AT2 in the renal cortex of adult offspring (30). However, the data were not stratified by sex. The increase in mRNA levels for the AT1 receptor on RPTCs from both male and female Beta-exposed animals is consistent with that previous study, but the increase in mRNA levels for the AT2 receptor is not reflected by the binding experiments. The cause for this discrepancy is unclear, but it may represent an example of lack of parallelism between gene and protein expression, as has been previously observed in the kidney (16). However, if there is some quantitative relationship between expression of AT1 receptor mRNA and receptor expression, then the highest levels of the AT1 receptor would be expected on RPTCs from Beta-exposed male animals. This may contribute to the increased Na+ uptake by them.
Another factor that likely contributes to the increase Na+ uptake by male cells induced by Beta exposure is NHE3, which is responsible for the reabsorption of 50–60% of filtered Na+ in the proximal tubule (71). Prenatal dexamethasone exposure increases NHE3 activity and protein expression in proximal tubules from young male rat offspring (16). Our data indicate that prenatal Beta exposure increases NHE3 mRNA in cells from male but not female animals and that protein expression tends to be higher in male cells as well. This is consistent with observations in male rats. The other important determinant of NHE3 activity is trafficking from the apical membrane of the proximal tubule cell to subapical locations (38, 70). It will be of interest in future studies to ascertain if antenatal exposure to glucocorticoids results in programmed trafficking of NHE3 in cells from adults and if any effect is sex specific.
Perspectives
In the United States alone, at least 100,000 people are born each year after exposure to antenatal glucocorticoids (6, 20, 45, 50, 53). While the beneficial effects of antenatal steroid treatment are clear and robust, there have been some unexpected side effects reported, such as elevations in blood pressure (19). Studies in animals have clearly demonstrated effects on the kidney and on blood pressure (4, 47, 48, 62). To our knowledge, this report is the first to show sex-specific effects of Beta exposure on Na+ uptake by proximal tubule cells in adult offspring. The data emphasize that even in the absence of the neural and humoral input that affect proximal tubule function in vivo, antenatal Beta exposure has a marked sex-specific effect on the handling of Na+ by RPTCs. The data suggest that the mechanisms responsible for the hypertensive effects of antenatal steroid exposure may differ in males and females. If this is so, more attention must be given to deciphering the explanations for these sex-related differences to develop rational approaches to interventions that will effectively reduce any negative consequences of antenatal steroid exposure in both men and women.
GRANTS
This work was supported by National Institute of Child Health and Human Development Grants P01-HD-047584 and R01-HD-017644.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.S. and J.B. performed experiments; Y.S., J.B., and J.C.R. analyzed data; Y.S., J.B., and V.M.P. prepared figures; Y.S. and V.M.P. drafted manuscript; Y.S., V.M.P., M.C.C., and J.C.R. edited and revised manuscript; Y.S., V.M.P., J.P.F., M.C.C., and J.C.R. approved final version of manuscript; J.B., V.M.P., and J.C.R. interpreted results of experiments; J.C.R. conception and design of research.
REFERENCES
- 1.Barker DJ, Bagby SP. Developmental antecedents of cardiovascular disease: a historical perspective. J Am Soc Nephrol 16: 2537–2544, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol 2: 700–707, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Barker DJP. The origins of the developmental origins theory. J Intern Med 261: 412–417, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Baum M. Role of the kidney in the prenatal and early postnatal programming of hypertension. Am J Physiol Renal Physiol 298: F235–F247, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bi J, Contag SA, Chen K, Su Y, Figueroa JP, Chappell MC, Rose JC. Sex specific effect of antenatal betamethasone exposure on renal oxidative stress induced by angiotensins in adult sheep. Am J Physiol Renal Physiol 307: F1013–F1022, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonanno C, Wapner RJ. Antenatal corticosteroid treatment: what's happened since Drs Liggins and Howie? Am J Obstet Gynecol 200: 448–457, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Carey RM. Newly discovered components and actions of the renin-angiotensin system. Hypertension 62: 818–822, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Carey RM. The intrarenal renin-angiotensin and dopaminergic systems: control of renal sodium excretion and blood pressure. Hypertension 61: 673–680, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carey RM, Padia SH. Angiotensin AT2 receptors: control of renal sodium excretion and blood pressure. Trends Endocrinol Metab 19: 84–87, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the angiotensin converting enzyme 2-angiotensin (1–7)-Mas receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne); doi: 10.3389/fendo.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1–7)-Mas receptor axis: more than regulation of blood pressure? Hypertension 50: 596–599, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Chong E, Yosypiv IV. Developmental programming of hypertension and kidney disease. Int J Nephrol 2012: 760580, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest 124: 2341–2347, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cogan MG. Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension 15: 451–458, 1990. [DOI] [PubMed] [Google Scholar]
- 15.Contag SA, Bi J, Chappell MC, Rose JC. Developmental effect of antenatal exposure to betamethasone on renal angiotensin II activity in the young adult sheep. Am J Physiol Renal Physiol 298: F847–F856, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dagan A, Gattineni J, Cook V, Baum M. Prenatal programming of rat proximal tubule Na+/H+ exchanger by dexamethasone. Am J Physiol Regul Integr Comp Physiol 292: R1230–R1235, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodic M, May CN, Wintour EM, Coghlan JP. An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin Sci 94: 149–155, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Dotsch J, Plank C, Amann K, Ingelfinger J. The implications of fetal programming of glomerular number and renal function. J Mol Med 87: 841–848, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Doyle LW, Ford GW, Davis NM, Callanan C. Antenatal corticosteroid therapy and blood pressure at 14 years of age in preterm children. Clin Sci (Lond) 98: 137–142, 2000. [PubMed] [Google Scholar]
- 20.Fanaroff AA, Stoll BJ, Wright LL, Carlo WA, Ehrenkranz RA, Stark AR, Bauer CR, Donovan EF, Korones SB, Laptook AR, Lemons JA, Oh W, Papile LA, Shankaran S, Stevenson DK, Tyson JE, Poole WK. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol 196: 147–148, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Fridovich I. Superoxide anion radical (O2−), superoxide dismutases, and related matters. J Biol Chem 272: 18515–18517, 1997. [DOI] [PubMed] [Google Scholar]
- 22.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302: 148–158, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu Rev Physiol 73: 359–376, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Geibel J, Giebisch G, Boron WF. Angiotensin II stimulates both Na+-H+ exchange and Na+/HCO3 cotransport in the rabbit proximal tubule. Proc Natl Acad Sci USA 87: 7917–7920, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert JS, Nijland MJ. Sex differences in the developmental origins of hypertension and cardiorenal disease. Am J Physiol Regul Integr Comp Physiol 295: R1941–R1952, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gender Med 5: S121–S132, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guyton AC. Blood pressure control–Special role of the kidneys and body fluids. Science 252: 1813–1816, 1991. [DOI] [PubMed] [Google Scholar]
- 29.Guyton AC, Coleman TG, Cowley AV Jr, Scheel KW, Manning RD Jr, Norman RA Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972. [DOI] [PubMed] [Google Scholar]
- 30.Gwathmey TM, Shaltout HA, Rose JC, Diz DI, Chappell MC. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 57: 620–626, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris PJ, Young JA. Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Arch 367: 295–297, 1977. [DOI] [PubMed] [Google Scholar]
- 32.Hilliard LM, Nematbakhsh M, Kett MM, Teichman E, Sampson AK, Widdop RE, Evans RG, Denton KM. Gender differences in pressure-natriuresis and renal autoregulation: role of the angiotensin type 2 receptor. Hypertension 57: 275–282, 2011. [DOI] [PubMed] [Google Scholar]
- 33.Hilliard LM, Chow CLE, Mirabito KM, Steckelings UM, Unger T, Widdop RE, Denton KM. Angiotensin type 2 receptor stimulation increases renal function in female, but not male, spontaneously hypertensive rats. Hypertension 64: 378–383, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Horita S, Nakamura M, Shirai A, Yamazaki O, Satoh N, Suzuki M, Seki G. Regulatory roles of nitric oxide and angiotensin II on renal tubular transport. World J Nephrol 3: 295–301, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.James GD, Sealey JE, Muller F, Alderman M, Madhavan S, Laragh JH. Renin relationship to sex, race and age in a normotensive population. J Hypertens Suppl 4: S387–S389, 1986. [PubMed] [Google Scholar]
- 36.Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, Padia SH, Carey RM. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension 60: 387–395, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Res 115: 388–399, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Koleganova N, Benz K, Piecha G, Ritz E, Amann K. Renal, cardiovascular and metabolic effects of fetal programming. Nephrol Dial Transplant 27: 3003–3007, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Langley-Evans SC. Intrauterine programming of hypertension by glucocorticoids. Life Sci 60: 1213–1221, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Lankadeva YR, Singh RR, Tare M, Moritz KM, Denton KM. Loss of a kidney during fetal life: long-term consequences and lessons learned. Am J Physiol Renal Physiol 306: F791–F800, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, Cassis LA, Allen AM, Sigmund CD. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol 301: R1067–R1077, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol 304: R588–R598, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang M, Knox FG. Production and functional roles of nitric oxide in the proximal tubule. Am J Physiol Regul Integr Comp Physiol 278: R1117–R1124, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Kirmeyer S, Matthews T. Births: final data for 2006 (online). http://www.cdc.gov/nchs/data/nvsr/nvsr57/nvsr57_07.pdf [9 April 2015]. [Google Scholar]
- 46.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85: 571–633, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Moritz KM, Cuffe JS, Wilson LB, Dickinson H, Wlodek ME, Simmons DG, Denton KM. Review: Sex specific programming: a critical role for the renal renin-angiotensin system. Placenta Suppl 31: S40–S46, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Moritz KM, Cullen-McEwen LA. Kidney development and fetal programming. In: Early Life Origins of Health and Disease, edited by Wintour EM, Owens JA. New York: Springer, 2006. [Google Scholar]
- 49.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. Effects of corticosteroids for fetal maturation on perinatal outcomes. JAMA 273: 413–418, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Ojeda NB, Intapad S, Alexander BT. Sex differences in the developmental programming of hypertension. Acta Physiol (Oxf) 210: 307–316, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortiz LA, Quan A, Weinberg A, Baum M. Effect of prenatal dexamethasone on rat renal development. Kidney Int 59: 1663–1669, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polyakov A, Cohen S, Baum M, Trickey D, Jolley D, Wallace EM. Patterns of antenatal corticosteroid prescribing 1998–2004. Aust N Z J Obstet Gynaecol 47: 42–45, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Reynolds RM. Programming effects of glucocorticoids. Clin Obstet Gynecol 56: 602–609, 2013. [DOI] [PubMed] [Google Scholar]
- 55.Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev: CD004454, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Sasaki S, Siragy HM, Gildea JJ, Felder RA, Carey RM. Production and role of extracellular guanosine cyclic 3′,5′ monophosphate in sodium uptake in human proximal tubule cells. Hypertension 43: 286–291, 2004. [DOI] [PubMed] [Google Scholar]
- 57.Seckl JR. Physiologic programming of the fetus. Clin Perinatol 25: 939–962, 1998. [PubMed] [Google Scholar]
- 58.Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab 3: 479–488, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Shaltout HA, Figueroa JP, Rose JC, Diz DI, Chappell MC. Alterations in circulatory and renal angiotensin-converting enzyme and angiotensin-converting enzyme 2 in fetal programmed hypertension. Hypertension 53: 404–408, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su Y, Carey LC, Rose JC, Pulgar VM. Antenatal glucocorticoid exposure enhances the inhibition of adrenal steroidogenesis by leptin in a sex-specific fashion. Am J Physiol Endocrinol Metab 304: E1404–E1411, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang L, Carey LC, Bi J, Valego N, Sun X, Deibel P, Perrott J, Figueroa JP, Chappell MC, Rose JC. Gender differences in the effects of antenatal betamethasone exposure on renal function in adult sheep. Am J Physiol Regul Integr Comp Physiol 296: R309–R317, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vehaskari VM. Developmental origins of adult hypertension: new insights into the role of the kidney. Pediatr Nephrol 22: 490–495, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Vehaskari VM. Prenatal programming of kidney disease. Curr Opin Pediatr 22: 176–182, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Vehaskari VM, Woods LL. Prenatal programming of hypertension: lessons from experimental models. J Am Soc Nephrol 16: 2545–2556, 2005. [DOI] [PubMed] [Google Scholar]
- 65.Wapner RJ, Sorokin Y, Mele L, Johnson F, Dudley DJ, Spong CY, Peaceman AM, Leveno KJ, Malone F, Caritis SN, Mercer B, Harper M, Rouse DJ, Thorp JM, Ramin S, Carpenter MW, Gabbe SG. Long-term outcomes after repeat doses of antenatal corticosteroids. N Engl J Med 357: 1190–1198, 2007. [DOI] [PubMed] [Google Scholar]
- 66.Welch WJ. Angiotensin II-dependent superoxide: effects on hypertension and vascular dysfunction. Hypertension 52: 51–56, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wirtschafter DD, Danielsen BH, Main EK, Korst LM, Gregory KD, Wertz A, Stevenson DK, Gould JB. Promoting antenatal steroid use for fetal maturation: results from the California Perinatal Quality Care Collaborative. J Pediatr 148: 606–612, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 49: 460–467, 2001. [DOI] [PubMed] [Google Scholar]
- 69.Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol 289: R1131–R1136, 2005. [DOI] [PubMed] [Google Scholar]
- 70.Zhuo JL, Li XC. New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides 32: 1551–1565, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhuo JL, Li XC. Proximal nephron. Compr Physiol 3: 1079–1123, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zimmerman MA, Sullivan JC. Hypertension: what's sex got to do with it? Physiology (Bethesda) 28: 234–244, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]