

Abstract
Kidney cancer [renal cell carcinoma (RCC)] is the sixth-most-common cancer in the United States, and its incidence is increasing. The current progression-free survival for patients with advanced RCC rarely extends beyond 1–2 yr due to the development of therapeutic resistance. We previously identified peroxisome proliferator-activating receptor-α (PPARα) as a potential therapeutic target for this disease and showed that a specific PPARα antagonist, GW6471, induced apoptosis and cell cycle arrest at G0/G1 in RCC cell lines associated with attenuation of cell cycle regulatory proteins. We now extend that work and show that PPARα inhibition attenuates components of RCC metabolic reprogramming, capitalizing on the Warburg effect. The specific PPARα inhibitor GW6471, as well as a siRNA specific to PPARα, attenuates the enhanced fatty acid oxidation and oxidative phosphorylation associated with glycolysis inhibition, and PPARα antagonism also blocks the enhanced glycolysis that has been observed in RCC cells; this effect did not occur in normal human kidney epithelial cells. Such cell type-specific inhibition of glycolysis corresponds with changes in protein levels of the oncogene c-Myc and has promising clinical implications. Furthermore, we show that treatment with GW6471 results in RCC tumor growth attenuation in a xenograft mouse model, with minimal obvious toxicity, a finding associated with the expected on-target effects on c-Myc. These studies demonstrate that several pivotal cancer-relevant metabolic pathways are inhibited by PPARα antagonism. Our data support the concept that targeting PPARα, with or without concurrent inhibition of glycolysis, is a potential novel and effective therapeutic approach for RCC that targets metabolic reprogramming in this tumor.
Keywords: metabolomics, peroxisome proliferator-activating receptor-α, reprogramming, kidney cancer
we initially utilized urine from patients with kidney cancer [also known as renal cell carcinoma (RCC)] and also performed mouse xenograft metabolomics studies to identify peroxisome proliferator-activating receptor-α (PPARα) as a novel therapeutic target for this disease (1, 8). In addition to altered levels of acylcarnitines, which were identified in urine from RCC patients (7), the mouse xenograft study demonstrated a signature of PPARα alterations, in that the only metabolites that were altered consistently in all three “matrices” [nicotinamide and cinnamoylglycine in tissue, serum, and urine (8)] were previously shown to be altered by a specific PPARα agonist (31). Taken together, these findings suggest that PPARα may be involved in RCC tumorigenesis through its effects on reprogrammed metabolic pathways and prompted us to further explore this nuclear receptor as a possible therapeutic target for advanced RCC, a disease for which there are few satisfactory treatment options.
PPARα is a member of the steroid hormone receptor superfamily. As occurs with other steroid hormone receptors, upon ligand activation, the PPARs heterodimerize with the retinoid X receptor, bind to the specific promoter sequence (the peroxisome proliferator response element), and, as a result, trigger the expression of a variety of target genes, including those involved in glucose, lipid, and amino acid metabolism (18). Fatty acid synthase (FAS), which is highly expressed in many tumors of the urinary tract (13), including RCC (14), and its downstream fatty acid reaction products are the endogenous ligands for PPARα, since selective inactivation of FAS impairs PPARα-dependent gene expression (4). In addition, liver-specific inactivation of FAS results in mice with decreased PPARα-dependent gene expression and a phenotype resembling PPARα deficiency (5). Given that fatty acids are present in cell culture media (with FBS) as well as in the mouse, PPARα is basally activated under living conditions in vitro and in vivo. While tumor suppression by PPARα inhibition has been reported in some cancers, including melanoma (12) and glioblastoma (16), excessive PPARα activation has also been found to lead to progression of tumor growth in other cancers, including hepatocellular carcinoma (22) and breast cancer (24).
To begin to determine the relevance of PPARα to RCC, we previously evaluated this receptor in vitro and showed that PPARα inhibition results in cell cycle arrest and induction of apoptosis in several RCC cell lines, with a pronounced dependence on histological grade of expression and a synergistic effect with glycolysis inhibition (1). The current study extends and supplements this work to focus on specific cancer-relevant metabolic pathways germane to PPARα signaling in RCC. We previously showed that aerobic glycolysis to lactate is overactive in RCC (21), a finding that had been postulated to be due to elevated hypoxia-inducible factor activity (15) and is consistent with the Warburg effect (28). We now show that, in RCC cells, inhibition of glycolysis stimulates fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) and that PPARα inhibition attenuates the enhanced FAO and OXPHOS associated with such glycolysis inhibition. We further show that PPARα antagonism blocks glycolysis in RCC cells, but not in normal renal tubular epithelial cells, and that this difference is associated with corresponding changes in protein levels of the oncogene c-Myc. These data, taken together with the in vivo data also reported here in an RCC xenograft mouse model showing a PPARα inhibitory effect on tumor growth comparable to that of treatment with sunitinib, suggest a new potential target for therapy of advanced RCC that targets metabolic reprogramming.
MATERIALS AND METHODS
Cell Lines
The RCC cell lines, Caki-1 and 786-O, were obtained from the American Type Culture Collection (Rockville, MD); the “normal human kidney” (NHK) proximal tubular epithelial cell line was obtained from Lonza (Basel, Switzerland). NHK, 786-O, and Caki-1 cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml streptomycin, and 100 mg/ml penicillin (complete medium) at 37°C with 5% CO2.
Materials
The PPARα antagonist GW6471 (Tocris, Bristol, UK) was dissolved in DMSO vehicle. It was used at 25 μM, an inhibitory dose established in our previous publications (1, 8), in all RCC cell lines utilized in the current study. 2-Deoxy-d-glucose (2-DG), MTT solution, mouse monoclonal anti-β-actin antibody fatty acid-free BSA, and potassium palmitate-d31 were obtained from Sigma (St. Louis, MO); sunitinib from LC Laboratories (Woburn, MA); rabbit polyclonal anti-c-Myc antibody from Cell Signaling Technology (Beverly, MA); rabbit polyclonal anti-PPARα antibody from Abcam (Cambridge, MA); goat anti-mouse and goat anti-rabbit HRP-conjugated IgG from Bio-Rad (Hercules, CA); ECL Plus solution from Thermo Fisher Scientific (Waltham MA); specific PPARα and scrambled control siRNA from Qiagen (Gaithersburg, MD); and T-PER from Thermo Scientific for tissue protein extraction.
Glucose Measurement
NHK and RCC cells were cultured in six-well plates with complete medium. After 24 h, the cells were washed with PBS and incubated for another 24 h in DMSO (0.5%) or 5 mM 2-DG in complete DMEM containing 5 mM glucose. For glucose measurement, 500 μl of the medium were collected from each group in the previous conditions for all cell lines. The medium was processed on a desktop centrifuge for 5 min to eliminate floating cells and debris. Glucose was measured using the Roche cobas c501 analyzer (6000 series) at the Clinical Diagnostic Laboratories in the University of California, Davis, William R. Pritchard Veterinary Medical Teaching Hospital.
FAO Analysis
Cell culture and media collection.
NHK and RCC cells were cultured in six-well culture plates with complete medium (2.0 ml/well). After 24 h, the cells were washed with PBS and incubated in 2.5% DMSO, 25 μM GW6471, 5 mM 2-DG, or GW6471 + 2-DG in complete medium (2.0 ml/well) containing 200 μM potassium palmitate-d31 conjugated with BSA (3 replicates for each treatment). After 24 h, 1.5 ml of the medium were collected, centrifuged for 5 min to eliminate cells and debris, and kept at −80°C until analysis.
Deuterium enrichment of cell medium.
Water was isolated from the cell medium by microdistillation at 45°C in a refrigerated (4°C) environment. The deuterium content of the distillates was determined using a Thermo Finnigan high-temperature conversion/elemental analyzer coupled with a Thermo Finnigan MAT 253 isotope ratio mass spectrometer via a ConFlo III interface. The deuterium isotope abundance was calculated in δ 2H values relative to the International Vienna Standard Mean Ocean Water standard. Water enrichment results were used to calculate palmitate oxidation rates, which were then normalized to 400,000 cells/well.
Oxidative Metabolism Measurements
Oxygen consumption rates (OCR) were measured using an extracellular flux analyzer (model XF24, Seahorse Bioscience, Chicopee, MA). For experiments utilizing GW6471 or 2-DG, cells were pretreated 30 min before the plate was run on the XF24 analyzer. OCR data represent the calculated average from six wells per group.
Lactate Measurement
An absorbance-based Lactate Assay Kit II (Biovision, Milpitas, CA) was used following the manufacturer's protocol. Briefly, an equal number of cells were seeded in 24-well plates and allowed to settle for 24 h before specific treatments were applied. At the end of the specified treatment times, the cells were standardized using viable cell counts. The medium and cells were mixed with the respective kit reaction mixtures, allowed to sit at room temperature for 30 min, and then read at 450-nm optical density with a microplate reader. The kit allows for quantification of the amount of l-lactate in the samples by generation of a product that interacts with a probe to produce color.
Immunoblotting
Immunoblotting was performed as previously described (1, 8). Briefly, after the indicated treatments, the cells were washed with PBS, lysed, and subjected to immunoblotting. For the xenograft tissue tumors, proteins were extracted with T-PER. The membranes were blocked in 5% nonfat dry milk for 1 h at room temperature, incubated with the designated antibodies, and then probed with HRP-tagged anti-mouse or anti-rabbit IgG antibodies. The signal was detected using ECL Plus solutions. Densitometry was performed using ImageJ software.
siRNA Transfection
Cells were plated in 6-well plates for FAO experiments or 24-well plates for lactate measurement experiments. After 24 h, cell monolayers at ∼75% confluency were subjected to PPARα-specific siRNA or negative siRNA transfection. The transfection mixture was prepared in Opti-MEM GlutaMAX medium (Invitrogen, Carlsbad, CA) with siRNA and Lipofectamine RNAiMAX according to the manufacturer's protocol. The final concentration of siRNA added to the cells was 100 nM. The cells were cultured in the presence of transfection mixture for 24 h; on the following day, the transfection mixture was replaced with fresh DMEM, and cell culture was pursued for an additional 48 h. Extra wells were made for each experiment and collected for PPARα protein quantification by Western blotting to ensure that the transfections were successful.
In Vivo Study
All animal procedures were performed in compliance with the University of California Animal Care and Use Committee. Male athymic Nu/Nu mice (8 wk of age, ∼25 g body wt) were injected with 1 × 105 Caki-1 cells subcutaneously (3:1 DMEM-Matrigel) in the flank region. Tumor progression was monitored weekly by calipers using the following formula: tumor volume (in mm3) = (length × width2)/2. When tumor size reached ∼80–100 mm3, animals were randomly assigned to four groups and treatments were started (day 1). The vehicle group received DMSO (4% in PBS) intraperitoneally and vegetable oil via oral gavage. The PPARα group was injected intraperitoneally with GW6471 in the same vehicle [20 mg/kg body wt; murine dose response is reported elsewhere (6)] every other day. The sunitinib group received sunitinib in vegetable oil via oral gavage (40 mg/kg body wt) 5 days/wk (29). Another group received GW6471 + sunitinib as described above. To determine any potential toxicity of the treatment(s), body weights of the animals were measured and signs of adverse reactions were monitored. On day 28, the mice were euthanized and the tumor mass was determined. Tumor growth rate was calculated as follows: tumor volume on day x/tumor volume on day 1. Serum samples collected from mice at the end of the experiment were analyzed using the Roche cobas c501 analyzer (6000 series) at Clinical Diagnostic Laboratories in the University of California, Davis, using the Small Animal Chem 2 panel. Frozen tumor tissues were also collected at the end of the experiment and homogenized and extracted in T-PER for c-Myc quantification by Western blotting.
Statistical Analysis
Mean values were compared using the independent-samples t-test; P < 0.05 was considered significant. Significant differences in OCR in 786-O and Caki-1 cells treated with GW6471 and 2-DG were determined by ANOVA followed by Tukey's test; P < 0.05 was considered significantly different.
RESULTS AND DISCUSSION
Glycolysis Inhibition Results in Enhancement of FAO, Which Is Significantly Decreased by PPARα Inhibition
Inhibition of the FAO metabolic pathway has shown promising results for therapy of prostate cancer (10, 17), and pharmacological inhibition of FAO sensitizes human leukemic cells to apoptosis (23). In addition, proteins involved in FAO, such as carnitine palmitoyltransferase I, have been shown to have an antiapoptotic function that has been attributed to cross talk with proapoptotic proteins (11, 20). However, despite its “clear” cytosol on histology, likely representative of high glycogen, triglyceride, and cholesterol content (hence, the appellation of the most common form of RCC as “clear cell” RCC) (26), the role of FAO in RCC cell survival has not been thoroughly examined. Our previous work showed that blocking glycolysis sensitized RCC cells to loss of viability after PPARα inhibition (1), suggesting that these cells are able to switch between the glycolysis and FAO pathways in response to metabolic stressors (8) and that FAO serves as an alternative energy-generating pathway when the normally overactive (in RCC) glycolysis pathway is inhibited. Accordingly, these two energy pathways have high relevance to RCC metabolism and survival and are worthy of further study in this context.
To begin to evaluate the nature of the FAO pathway and the energy reprogramming that exists in RCC, with an eye toward the discovery of novel therapeutics, we used an in vitro assay of palmitate oxidation to determine how FAO is related to glycolysis in RCC and in “normal” renal epithelial (NHK) cells. We first evaluated the effects of the chemical tools to be used in the subsequent experiments: the glycolysis inhibitor 2-DG and a PPARα-specific siRNA, the latter to check for specificity of GW6471 for PPARα inhibition. When the cells were incubated with 2-DG, there was a marked decrease in glucose uptake, as shown by no change in media glucose under these conditions compared with control cells grown in the absence of 2-DG (Fig. 1A). In Caki-1 and NHK cells transfected with a siRNA specific to PPARα, levels of PPARα protein decreased after 72 h (Fig. 1B). (Transfection of the 786-O RCC cell line was not successful.) Since GW6471 is a competitive inhibitor of PPARα, use of the siRNA is most appropriate for demonstrating specificity of GW6471 to PPARα in vitro.
Fig. 1.
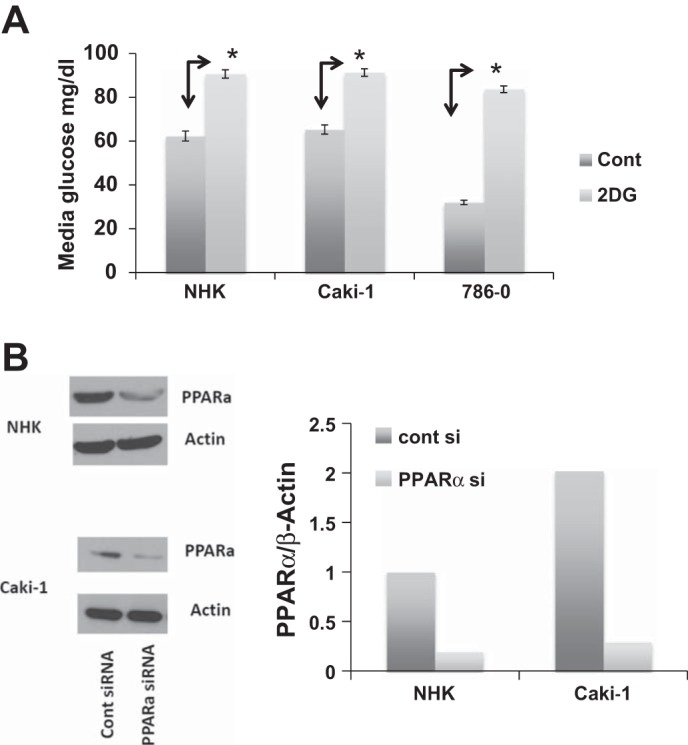
2-Deoxy-d-glucose (2-DG) attenuates glucose uptake, and transfection of siRNA specific to peroxisome proliferator-activating receptor (PPAR)-α attenuates PPARα protein levels. A: normal renal tubular epithelial [normal human kidney (NHK)] cells and 2 renal cell carcinoma (RCC) cell lines (Caki-1 and 786-O) were incubated in 0.5% DMSO [vehicle (Cont)] or 2-DG (5 mM). After 24 h, media glucose was measured. *P < 0.05 vs. Cont. Error bars indicate SD. B: NHK and Caki-1 cells were grown to ∼75% confluence and transfected with a siRNA specific to PPARα or a scrambled-sequence control siRNA (100 nM final concentration). After 72 h, cells were harvested and immunoblotted with PPARα antibody and β-actin (loading control). Data from immunoblots were quantified by densitometry. Results are representative of ≥3 repeats.
Upon incubation with 2-DG to attenuate glycolysis, FAO was significantly increased at 24 h in both RCC cell lines but not in NHK cells (Fig. 2A), supporting the concept that RCC cells are reprogrammed to be highly dependent on aerobic glycolysis (the Warburg effect) at baseline (15, 21) but that normal kidney epithelial cells, from which RCC are derived, are not. Inhibition of PPARα using GW6471 + 2-DG showed nearly complete abolition of FAO in Caki-1 cells, a significant decrease in FAO in 786-O RCC cells, and a trend toward a decrease in FAO in NHK cells; since these RCC cells were not able to “escape” glycolysis inhibition in the presence of PPARα inhibition by utilizing FAO, this finding suggests obvious clinical utility with combined therapy of PPARα and glycolysis inhibition. Furthermore, the finding that GW6471 showed a trend toward an increase in FAO in Caki-1 cells suggests that this inhibitor might also affect glycolysis itself (see below). Caki-1 and NHK cells showed similar changes, with cells transfected with PPARα-specific siRNA (Fig. 2B), confirming specificity of GW6471.
Fig. 2.
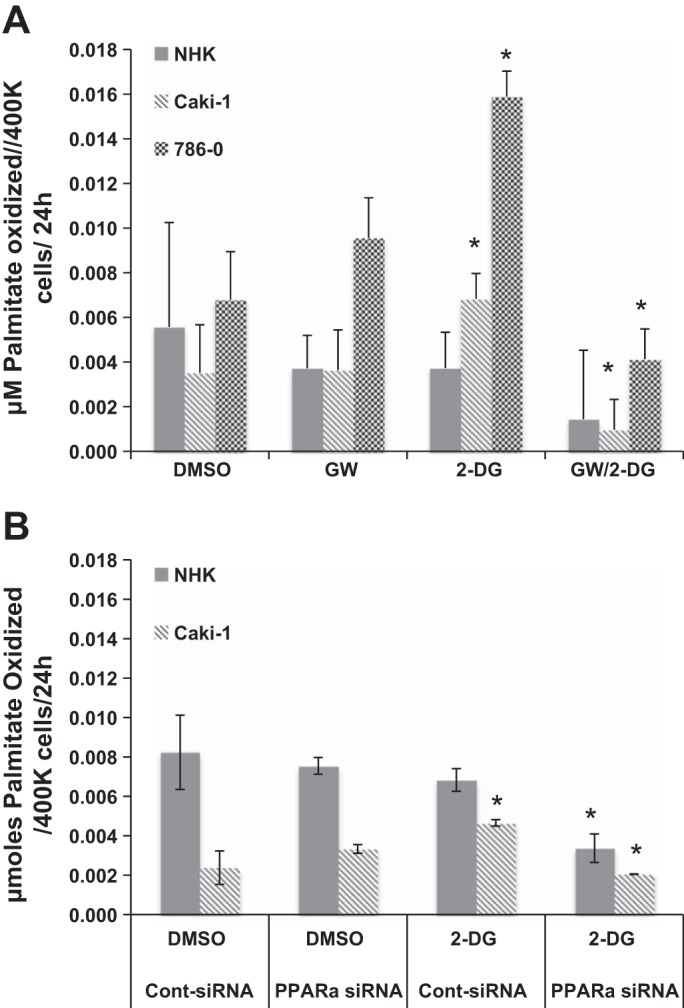
Fatty acid oxidation was increased when glycolysis was inhibited and reduced by PPARα inhibition in RCC, but not NHK, cells. A: NHK cells and 2 RCC cell lines (Caki-1 and 786-O) were incubated in 2.5% DMSO (vehicle), 25 μM GW6471 (GW), 5 mM 2-DG, or 2-DG + GW6471 in DMEM containing 200 μM potassium palmitate-d31. After 24 h of incubation, conditioned medium was withdrawn and analyzed for deuterated water as a measure of palmitate oxidation. B: NHK and Caki-1 cells were grown to 75% confluence and transfected with a siRNA specific to PPARα or a scrambled-sequence control siRNA (100 nM final concentration). After 72 h in transfection medium containing 200 μM potassium palmitate-d31, cells were treated with 0.5 DMSO or 5 mM 2-DG. After 24 h of incubation, conditioned medium was withdrawn and analyzed for deuterated water as a measure of palmitate oxidation. *P < 0.05 vs. DMSO alone (A) or DMSO and control siRNA (B) for each group. Error bars indicate SD.
The finding that FAO in NHK cells was not significantly different from the control group in most conditions evaluated suggests that the normal renal epithelial cells may be less dependent on a single energy pathway (i.e., aerobic glycolysis) and further suggests that PPARα inhibition would be less toxic in normal kidney epithelial cells than RCC cells in vivo. Furthermore, the data confirm that RCC cells are highly dependent on glycolysis and are unable to adapt by maintaining FAO upon PPARα inhibition; hence, a clinically useful PPARα inhibitor would be expected to be associated with fewer adverse effects once translated to the clinic.
OXPHOS in RCC Cells Is Attenuated by PPARα Inhibition but Lacks Synergy With Glycolysis Inhibition
Nonmalignant cells under nonhypoxic conditions, in which glycolysis is a mitochondrial process utilizing the tricarboxylic acid (TCA) cycle and the electron transport chain, rely primarily on mitochondrial OXPHOS to generate ATP for cellular energetic processes; on the other hand, many cancer cells display enhanced aerobic glycolysis (pyruvate-to-lactate conversion, the Warburg effect) at the expense of OXPHOS capacity (30). In fact, glycolysis and OXPHOS are tightly coupled in normal cellular metabolism and serve as a molecular interconversion system (27). In “normal” aerobic metabolism in nonmalignant cells, glycolysis occurs in the cytoplasm, leading to generation of pyruvate, which in turn is a fuel for OXPHOS in the mitochondria via the TCA cycle; FAO is also a major provider of OXPHOS intermediates. β-Oxidation of fatty acids consists of a series of cyclical reactions resulting in the shortening of fatty acids by two carbons per cycle. These reactions occur in mitochondria (short- and long-chain fatty acids) and peroxisomes (very-long-chain fatty acids) and generate acetyl-CoA, which can also feed the TCA cycle and, thereby, contribute to OXPHOS. We next probed the nature of the OXPHOS response under conditions of glycolysis inhibition to determine whether RCC cells are able to upregulate OXPHOS under these conditions, a reprogramming condition that might make them responsive to PPARα inhibition.
RCC cells were incubated with 2-DG, GW6471, or 2-DG + GW6471, and the OCR, a proxy for OXPHOS (30), was measured. Similar to the FAO experiments, RCC cells incubated with 2-DG to inhibit glycolysis showed increased OXPHOS levels (Fig. 3), likely because they were highly dependent on glycolysis at baseline (as shown by FAO measurements; Fig. 2) and were thus required to switch their metabolism to FAO and/or amino acid oxidation upon glycolysis inhibition. However, in contrast to the effect on FAO, in RCC cells, incubation with 2-DG + GW6471 did not significantly reduce levels of OXPHOS compared with control cells, likely due to the contribution of amino acid or other nonglycolytic metabolic pathways (e.g., glutamine metabolism) to OXPHOS (but not to FAO). Thus, while PPARα inhibition combined with glycolysis inhibition markedly attenuated FAO, the same cannot be said for OXPHOS, because the latter process is downstream of several interrelated metabolic pathways, in contrast to glycolysis. Furthermore, PPARα inhibition alone significantly attenuated OXPHOS, but not FAO, in both cell lines, when measured under the same conditions. This was likely due to additional OCR reduction at the peroxisomal level by GW6471 (due to inhibition of very-long-chain fatty acid β-oxidation) and, consequently, the decrease in acetyl-CoA input to the TCA cycle.
Fig. 3.

Oxidative phosphorylation (OXPHOS) is attenuated by PPARα inhibition but increased by glycolysis inhibition in RCC (Caki-1 and 786-O) cells. Caki-1 and 786-O cells were incubated in 2.5% DMSO, 25 μM GW6471, 5 mM 2-DG, or 2-DG + GW6471. Data for each cell line represent average of 6 replicates. Oxygen consumption rate (OCR) was measured using the XF24 (Seahorse) extracellular flux analyzer. Data are presented as percent increase in OCR in response to indicated conditions and times (top). Data were further analyzed, and area under the curve (AUC) for each condition was determined (bottom). OCR was increased by 2-DG in both cell lines but decreased by GW6471. *P < 0.05 vs. DMSO (control). Error bars indicate SE.
PPARα Inhibition Blocks Glycolysis in RCC, but not NHK, Cells and Is Correlated With c-Myc Expression
Because of the Warburg effect (9, 25), which has been shown to be prominent in RCC cells (Fig. 2) (21), most of the pyruvate produced by RCC cells is reduced to lactate by lactate dehydrogenase in the cytoplasm, regardless of oxygen availability, and then lactate is excreted into the extracellular space through monocarboxylate transporters (2). By contrast, in normal renal epithelial (NHK) cells, which are not prone to the Warburg effect, pyruvate reduction would be expected to occur only under anaerobic conditions.
To evaluate the role of PPARα inhibition in glycolysis under “normal” nonhypoxic conditions, RCC and NHK cells were incubated with GW6471, 2-DG, or 2-DG + GW6471, and, after 24 h, the lactate produced concurrently in cells and medium was determined as an assay of aerobic glycolysis (pyruvate-to-lactate conversion). Parallel experiments were conducted with a siRNA specific to PPARα. As expected, lactate levels in cells and medium were reduced in all cell lines upon inhibition of glycolysis by 2-DG (Fig. 4, A and B), consistent with the lower uptake of glucose from the medium (Fig. 1A), which confirms that glucose metabolism results in a considerable amount of lactate in RCC cells and a lesser amount in normal epithelial cells. However, PPARα inhibition alone (using GW6471 or PPARα-specific siRNA) decreased lactate levels in both RCC cell lines, but not in normal (NHK) cells (Fig. 4, A and B), confirming our finding of high basal dependence of RCC, but not NHK, cells on aerobic glycolysis or on diversion of pyruvate into nonglycolysis mitochondrial oxidation pathways consequent to inhibition of FAO. These findings additionally suggest a potential beneficial effect of PPARα inhibition on glycolysis, specifically in RCC, in addition to its effect on FAO (Fig. 2), as evidenced by its concurrent effect on lactate production in cells highly dependent on the Warburg effect.
Fig. 4.

GW6471 decreases lactate levels in RCC, but not NHK, cells and is associated with c-Myc expression. A: NHK cells and 2 RCC cell lines were incubated in 2.5% DMSO [vehicle (Cont)], 25 μM GW6471 (GW), 5 mM 2-DG, or 2-DG + GW6471 in DMEM for 24 h, and lactate was measured in cells and conditioned medium. B: NHK and Caki-1 cells were grown to 75% confluence and transfected with a PPARα-specific siRNA or a scrambled-sequence control siRNA (100 nM final concentration). After 72 h, medium was changed to conditioned medium with 2.5% DMSO or 5 mM 2-DG. After 24 h, medium and cells were harvested, and lactate was measured as described in A. *P < 0.05 vs. DMSO alone (A) or DMSO and control siRNA (B) for each group. Error bars indicate SD. C: immunoblots for conditions shown for c-Myc and the loading control β-actin and densitometry data normalized to intensity of actin bands. *P < 0.05 DMSO vs. GW6471. Data are representative of ≥3 repeats.
In most cancer cells, increased glucose uptake and enhanced glycolytic flux result from hyperactivity of the protein product of the oncogene c-Myc (19). Our previous work showed that the PPARα antagonist GW6471 resulted in downregulation of c-Myc (1), which would be expected to contribute to the beneficial effect of PPARα inhibition in cancer. We next asked whether c-Myc is linked to the glycolysis data in the normal and RCC cell lines as a potential mechanism for the metabolic differences between malignant and normal renal epithelial cells. After 24 h of incubation with GW6471, c-Myc showed a trend toward an increase in protein levels in NHK cells and a significant decrease in both RCC cell lines (Fig. 4C), suggesting that PPARα inhibition is mediating its downstream effects via c-Myc and intimating an explanation of the difference in GW6471-mediated lactate levels between RCC and normal epithelial cells.
In Vivo Treatment of a Xenograft Mouse Model With the PPARα Antagonist GW6471 Attenuates RCC Growth
To test the antitumor activity of PPARα antagonism in vivo, we used a subcutaneous xenograft mouse model. Caki-1 cells were implanted subcutaneously in nude (Nu/Nu) mice. After tumor masses reached ∼5 mm in diameter, GW6471 was administrated intraperitoneally every other day for 4 wk at a dose (20 mg/kg mouse body wt) that was described to be effective in an in vivo dose-response study (6) and confirmed here to be efficacious. Sunitinib, an drug approved by the US Food and Drug Administration for metastatic RCC, was used alone at a dose that was previously reported effective in mice (29) was confirmed in the present study as a positive control or in combination with GW6471 to evaluate synergistic effects on tumor growth. There were significant differences in tumor growth between vehicle- and GW6471-treated animals (Fig. 5), with results comparable to those from the sunitinib-treated group; no synergy was detected when sunitinib and GW6471 were administered together. Similar results were obtained with an orally available PPARα inhibitor (data not shown) structurally related to published compounds (3). No toxicity was observed at the doses of GW6471 based on weights of the animals (Fig. 6), and laboratory values, including kidney and liver function tests, were not adversely affected (Table 1). To demonstrate on-target effects of GW6471, we evaluated c-Myc levels in the tumors, which showed significant decreases in the GW6471-treated animals (Fig. 7). Given that no adverse effects were observed in these animals and that sunitinib is associated with very high levels of drug resistance, we propose PPARα as a potential novel drug for RCC that, since it targets reprogrammed tumor metabolism, is likely to be associated with minimal resistance.
Fig. 5.
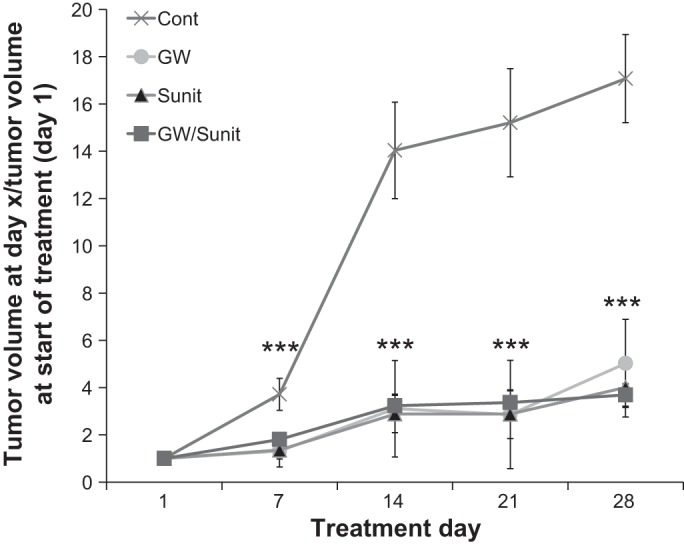
Treatment with the PPARα antagonist GW6471 attenuates RCC growth in a xenograft mouse model. Male athymic Nu/Nu mice (8 wk old) were injected subcutaneously with Caki-1 cells and treated as follows: control mice (Cont) received the vehicles vegetable oil (200 μl/mouse po) and 4% DMSO in PBS (200 μl/mouse ip) 5 days/wk; GW mice received GW6471 (20 mg/kg body wt) in 4% DMSO in PBS every other day (200 μl/mouse ip); Sunit mice received sunitinib (40 mg/kg body wt po) in vegetable oil (200 μl/mouse) 5 days/wk; the GW/Sunit group received GW6471 and sunitinib simultaneously in doses, via routes, and at frequencies described above. All treatment groups were significantly different from vehicle-treated (control) group from posttreatment day 7 to the end of the experiment. ***P < 0.05 vs. control. Error bars indicate SE. Data represent fold change in tumor volume between treatment days 7, 14, 21, and 28 and tumor volume at initiation of treatment (day 1).
Fig. 6.
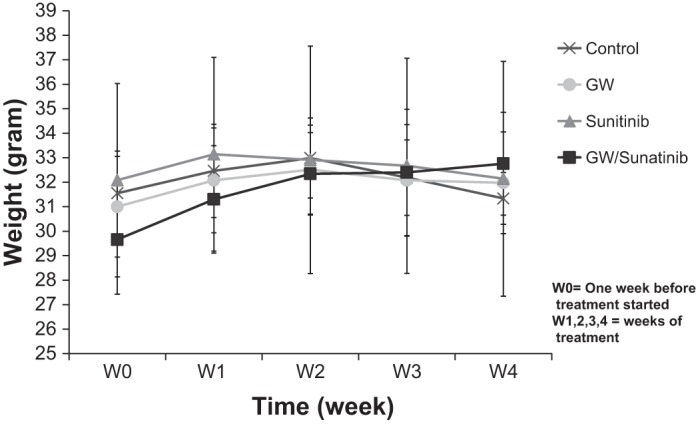
Weights of animals did not differ among treatment groups. All mice from the experiment described in Fig. 5 were weighed each week and at euthanasia. Values are means ± SE.
Table 1.
Blood chemistry analysis
| Control |
GW |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Test | Cont1 | Cont2 | Cont3 | Cont4 | Cont Avg | GW1 | GW2 | GW3 | GW4 | GW Avg |
| Anion gap, mmol/l | 90.2 | 96 | 121 | 118 | 106.30 | 98 | 98 | 92 | 131 | 104.75 |
| Sodium, mmol/l | 130 | 128 | 126 | 124 | 127.00 | 130 | 130 | 135 | 121 | 129 |
| Potassium, mmol/l | 57.2 | 64 | 84 | 86 | 72.80 | 65 | 65 | 59 | 95 | 71 |
| Chloride, mmol/l | 88 | 85 | 81 | 83 | 84.25 | 85 | 85 | 90 | 79 | 84.75 |
| Bicarbonate, mmol/l | 11 | 11 | 8 | 9 | 9.75 | 12 | 12 | 12 | 6 | 10.5 |
| Phosphorus, mg/dl | 4.8 | 5.1 | 7.1 | 5.2 | 5.55 | 5.5 | 5.5 | 6.9 | 7.6 | 6.375 |
| BUN, mg/dl | 20 | 21 | N/A | 21 | 20.67 | 16 | 16 | 17 | 21 | 17.5 |
| Glucose, mg/dl | 194 | 213 | 99 | 207 | 178.25 | 181 | 181 | 166 | 138 | 166.5 |
| Total protein, g/dl | 3.9 | 3.8 | 4.1 | 3.7 | 3.88 | 4.1 | 4.1 | 4.1 | 3.8 | 4.025 |
| Albumin, g/dl | 2.5 | 2.5 | 2.6 | 2.7 | 2.58 | 2.8 | 2.8 | 2.8 | 2.6 | 2.75 |
| Globulin, g/dl | 1.4 | 1.3 | 1.5 | 1 | 1.30 | 1.3 | 1.3 | 1.3 | 1.2 | 1.275 |
| ALT, U/l | 77 | 120 | 65 | 149 | 102.75 | 121 | 121 | 74 | 72 | 97 |
| AST, U/l | 522 | 355 | 629 | 473 | 494.75 | 327 | 327 | 364 | 858 | 469 |
| CK, U/l | 6,347 | 8,360 | 10,373 | 10,780 | 8,965.00 | 7,117 | 8,580 | 6,875 | 1,2364 | 8,734 |
| Alkaline phosphatase, U/l | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | ||
| GGT, U/l | <3 | <3 | <3 | <3 | <3 | <3 | <3 | <3 | ||
| Cholesterol, mg/dl | 119 | 110 | 84 | 113 | 106.50 | 101 | 101 | 112 | 93 | 101.75 |
| Bilirubin total, mg/dl | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | ||
| Magnesium, mg/dl | <0.2 | <0.2 | N/A | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | ||
Results from analysis of serum from 4 randomly selected vehicle (Cont1, Cont2, Cont3, Cont4) and 4 randomly selected GW6471-treated (GW1, GW2, GW3, GW4) animals with the Small Animal Chemistry Panel 2. Values show no differences between control and GW6471-treated animals. BUN, blood urea nitrogen; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; GGT, γ-glutamyltransferase.
Fig. 7.

GW6471 exhibits on-target effects on PPARα in vivo. Renal tumors from all vehicle- and GW6471-treated animals (n = 8) were frozen immediately at euthanasia. A: proteins were extracted using T-PER, and 40 μg of total protein from each sample/lane were used for immunoblots for c-Myc, a known target of PPARα, and β-actin, a loading control. Two separate gels were run. B: results from immunoblots were quantified by densitometry. C: means ± SE (n = 8 experimental and 8 controls, both gels) of results in B. *P = 0.0019.
Conclusion
Our group initially identified PPARα from nontargeted metabolomics as a potential new target for RCC (1). In the current study we have extended our previous work to show that GW6471, a specific antagonist of PPARα, decreases FAO in the presence of glycolysis inhibition in RCC cells and has a predilection for RCC cells over normal renal tubular epithelial cells. The combined effect of simultaneous administration of PPARα and glycolysis inhibition is likely due to inhibition of β-oxidation in a FAO-preferential state, and the glycolysis inhibition by PPARα inhibition is likely mediated by c-Myc. Furthermore, in vivo activity of GW6471 with respect to tumor attenuation is equivalent to that of sunitinib, with no adverse effects and appropriate on-target findings. Thus, PPARα inhibition is a promising new therapy for RCC, acting upon two arms of energy metabolism.
GRANTS
This work was supported by National Institutes of Health Grants 1R01 CA-135401-01A1 and 1R01 DK-082690-01A1 and the Medical Service of the US Department of Veterans Affairs (all to R. H. Weiss) and the Lawrence Livermore National Laboratory-University of California Davis Cancer Center Fitzpatrick Award (to O. Abu Aboud).
DISCLOSURES
M. Hellerstein and T. Riif are partly employed by KineMed; however, KineMed had absolutely no influence on the design, results, or interpretation of the data.
AUTHOR CONTRIBUTIONS
O.A.A. and R.H.W. developed the concept and designed the research; O.A.A., D.D., M.F., and T.R. performed the experiments; O.A.A., D.D., S.B., M.F., T.R., M.H., and R.H.W. analyzed the data; O.A.A., D.D., S.B., M.F., T.R., M.H., and R.H.W. interpreted the results of the experiments; O.A.A., M.F., and R.H.W. prepared the figures; O.A.A., D.D., S.B., M.F., M.H., and R.H.W. edited and revised the manuscript; O.A.A., D.D., S.B., M.F., T.R., M.H., and R.H.W. approved the final version of the manuscript; R.H.W. drafted the manuscript.
REFERENCES
- 1.Abu Aboud O, Wettersten HI, Weiss RH. Inhibition of PPAR-α induces cell cycle arrest and apoptosis and synergizes with glycolysis inhibition in kidney cancer cells. PLoS One 8: e71115, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergersen LH. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 145: 11–19, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bravo Y, Baccei CS, Broadhead A, Bundey R, Chen A, Clark R, Correa L, Jacintho JD, Lorrain DS, Messmer D, Stebbins K, Prasit P, Stock N. Identification of the first potent, selective and bioavailable PPARα antagonist. Bioorg Med Chem Lett 24: 2267–2272, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, Semenkovich CF. “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab 1: 309–322, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF. Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J Clin Invest 117: 2539–2552, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cluny NL, Keenan CM, Lutz B, Piomelli D, Sharkey KA. The identification of peroxisome proliferator-activated receptor-α-independent effects of oleoylethanolamide on intestinal transit in mice. Neurogastroenterol Motil 21: 420–429, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Ganti S, Taylor S, Kim K, Hoppel CL, Guo L, Yang J, Evans C, Weiss RH. Urinary acylcarnitines are altered in kidney cancer. Int J Cancer 130: 2791–2800, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganti S, Taylor SL, Abu AO, Yang J, Evans C, Osier MV, Alexander DC, Kim K, Weiss RH. Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res 72: 3471–3479, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891–899, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Mendrinos S, Sivridis E. The metabolic interactions between tumor cells and tumor-associated stroma (TAS) in prostatic cancer. Cancer Biol Ther 13: 1284–1289, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MA, Bonelli P, Calarco A, Peluso G. tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1. Cell Death Differ 12: 603–613, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor-α activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res 12: 3028–3036, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Hamada S, Horiguchi A, Asano T, Kuroda K, Asakuma J, Ito K, Asano T, Miyai K, Iwaya K. Prognostic impact of fatty acid synthase expression in upper urinary tract urothelial carcinoma. Jpn J Clin Oncol 44: 486–492, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J Urol 180: 1137–1140, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12: 9–22, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu DC, Zang CB, Liu HY, Possinger K, Fan SG, Elstner E. A novel PPARα/γ dual agonist inhibits cell growth and induces apoptosis in human glioblastoma T98G cells. Acta Pharmacol Sin 25: 1312–1319, 2004. [PubMed] [Google Scholar]
- 17.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 9: 230–234, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor-α target genes. Cell Mol Life Sci 61: 393–416, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortega AD, Sanchez-Arago M, Giner-Sanchez D, Sanchez-Cenizo L, Willers I, Cuezva JM. Glucose avidity of carcinomas. Cancer Lett 276: 125–135, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Paumen MB, Ishida Y, Han H, Muramatsu M, Eguchi Y, Tsujimoto Y, Honjo T. Direct interaction of the mitochondrial membrane protein carnitine palmitoyltransferase I with Bcl-2. Biochem Biophys Res Commun 231: 523–525, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Perroud B, Ishimaru T, Borowsky AD, Weiss RH. Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics 8: 971–985, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-α and liver cancer: where do we stand? J Mol Med 83: 774–785, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120: 142–156, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-activated receptor-α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog 34: 165–171, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Unwin RD, Craven RA, Harnden P, Hanrahan S, Totty N, Knowles M, Eardley I, Selby PJ, Banks RE. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics 3: 1620–1632, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Van HM, Garmo H, Hammar N, Jungner I, Walldius G, Lambe M, Holmberg L. The interplay between lipid profiles, glucose, BMI and risk of kidney cancer in the Swedish AMORIS study. Int J Cancer 130: 2118–2128, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Warburg O. On respiratory impairment in cancer cells. Science 124: 269–270, 1956. [PubMed] [Google Scholar]
- 28.Warburg O. On the origin of cancer cells. Science 123: 309–314, 1956. [DOI] [PubMed] [Google Scholar]
- 29.Wettersten HI, Landesman Y, Friedlander S, Shacham S, Kauffman M, Weiss RH. Specific inhibition of the nuclear exporter exportin-1 attenuates kidney cancer growth. PLoS One 9: e113867, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 292: C125–C136, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Zhen Y, Krausz KW, Chen C, Idle JR, Gonzalez FJ. Metabolomic and genetic analysis of biomarkers for peroxisome proliferator-activated receptor-α expression and activation. Mol Endocrinol 21: 2136–2151, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]