

Abstract
Arterial thrombosis is the primary cause of most cases of myocardial infarction and stroke, the leading causes of death in the developed world. Platelets, highly specialized cells of the circulatory system, are key contributors to thrombotic events. Antiplatelet drugs, which prevent platelets from aggregating, have been very effective in reducing the mortality and morbidity of these conditions. However, approved antiplatelet therapies have adverse side effects, most notably the increased risk of bleeding. Moreover, there remains a considerable incidence of arterial thrombosis in a subset of patients receiving currently available drugs. Thus, there is a pressing medical need for novel antiplatelet agents with a more favorable safety profile and less patient resistance. The discovery of novel antiplatelet targets is the matter of intense ongoing research. Recent findings demonstrate the potential of targeting key signaling molecules, including kinases and phosphatases, to prevent platelet activation and aggregation. Here, we offer perspectives to targeting members of the protein tyrosine phosphatase (PTP) superfamily, a major class of enzymes in signal transduction. We give an overview of previously identified PTPs in platelet signaling, and discuss their potential as antiplatelet drug targets. We also introduce VHR (DUSP3), a PTP that we recently identified as a major player in platelet biology and thrombosis. We review our data on genetic deletion as well as pharmacological inhibition of VHR, providing proof-of-principle for a novel and potentially safer VHR-based antiplatelet therapy.
Keywords: Protein tyrosine phosphatases, PTP, Inhibitor, Thrombosis, Antithrombotic, Platelets, Antiplatelet, Therapy, Drug, GPVI, CLEC-2, DUSP3, VHR, CD148, PTP1B, SHP1, SHP2
1. Introduction
Circulating platelets play a crucial role in hemostasis, acting to maintain the integrity of a closed circulatory system after blood vessel injury.2 They are recruited to the site of vascular damage, where they adhere to the exposed subendothelial matrix and become a major component of the developing thrombus that seals the breach. Under normal conditions, regulatory mechanisms restrain thrombus formation both temporally and spatially. When pathologic processes overwhelm the regulatory mechanisms of hemostasis, excessive quantities of thrombin are formed, initiating thrombosis.1, 2 Thrombosis can occur in the arterial or the venous circulation and has a major medical impact.3 Venous thromboembolism is the third leading cause of cardiovascular-associated death. Acute arterial thrombosis is the primary cause of most cases of myocardial infarction and stroke, collectively the most common causes of death in the developed world.4
Available antiplatelet therapies are used for both the prevention and the acute treatment of arterial thrombosis. These drugs target either one of the three major processes leading to platelet activation and thrombus formation: 1) the initial phase of platelet recruitment and adhesion to the vessel wall, 2) the platelet aggregation phase, and 3) the stabilization of platelet aggregates during the amplification phase (Fig. 1). Aspirin has been used clinically for more than 40 years and is the most commonly used antiplatelet drug.5, 6 It inhibits cyclooxygenase-1, which is required for the synthesis of thromboxane A2 (TXA2), a secondary mediator of platelet aggregation. Thienopyridines, including clopidogrel, ticlopidine, and prasugrel, are irreversible inhibitors of the P2Y12 ADP receptor and also widely used as antiplatelet drugs.7, 8 In fact, clopidogrel (Plavix) is one of the best selling drugs of all time, with peak sales of $9.3 billion in 2011 (source: FirstWord Pharma). Both, aspirin and clopidogrel inhibit the formation of stable platelet aggregates. More recently developed amplification phase inhibitors include ticagrelor,9 a P2Y12 receptor antagonist, vorapaxar10, 11 and atopaxar,12 which are antagonists of the thrombin-activated G-protein coupled receptor (GPCR) PAR-1, and dipyridamole and cilostazol,13 both phosphodiesterase 3 (PDE3) selective inhibitors that increase cAMP levels and block platelet responses to ADP. Investigational drugs include the TXA2 receptor antagonists terutroban and EV-077.14, 15 Platelet aggregation phase inhibitors in clinical use include the monoclonal antibody abciximab, the cyclic heptapeptide eptifibatide, and the small molecule tirofiban.16 These drugs all inhibit the interaction between glycoprotein IIb/IIIa (also known as integrin αIIbβ3) and fibrinogen, thereby potently inhibiting platelet aggregation. At present, there are no drugs in clinical use that block the initial recruitment and binding of platelets to adhesive macromolecules of the subendothelial matrix, such as collagen or von Willebrand factor (vWF). Molecules in clinical studies that inhibit the interaction between vWF and glycoprotein (GP) Ib on the surface of platelets include the aptamer ARC-1779,17 the monoclonal antibody caplacizumab,18 and the snake venom-derived C-type lectin-like protein anfibatide.19 Collagen receptor GPVI inhibitors in development include compounds that induce depletion of GPVI, antibodies that bind to GPVI, and soluble GPVI mimetics that bind to collagen.20
Figure 1.

Platelet activation pathways targeted by current and novel antiplatelet agents.
Antiplatelet therapy has been very effective in reducing the mortality and morbidity of acute myocardial infarction and stroke.4 However, approved antiplatelet agents have adverse side effects, including gastrointestinal toxicity, neutropenia, thrombocytopenia, and bleeding.4 Moreover, there remains a considerable incidence of arterial thrombosis in patients resistant to currently available antiplatelet therapy. For example, the delayed onset of action and the occurrence of poor platelet inhibition responders with clopidogrel is well documented.4 Thus, despite the great success of antiplatelet therapy, there is an unmet medical need for novel antiplatelet agents with a more favorable safety profile, better efficacy in certain patient groups, and rapid onset and offset of action in acute events. Efforts in the pharmaceutical industry have focused on developing new-generation drugs of well-established targets, including the P2Y12 ADP receptor, the PAR-1 thrombin receptor, and the TXA2 receptor.4 In addition, the discovery of novel targets is the matter of intense ongoing research. Recently, various kinases have emerged as promising antiplatelet targets. Proof-of-principle studies have been reported for phosphatidylinositol 3-kinase (PI3K) isoform p110β,21, 22 the non-receptor spleen tyrosine kinase (Syk),23, 24 and Src family kinases (SFKs).25, 26 These findings demonstrate the possibility of targeting key regulators of platelet receptor signaling pathways as novel antiplatelet strategies.
Tyrosine phosphorylation is an important mechanism of signal transduction,27, 28 including in platelets,29, 30 as it crucially activates or inhibits the function of signaling molecules. For instance, tyrosine phosphorylation can result in conformational changes of the tertiary or quaternary structure of proteins, thereby rapidly amplifying or attenuating the catalytic activity of enzymes.31 It also promotes crucial protein-protein interactions, e.g., the recruitment and activation of signaling molecules to the cytoplasmatic tail of cell-surface receptors. Tyrosine phosphorylation is a rapidly reversible post-translational modification, controlled by the opposing activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs).27, 28 Historically, research had long been focused on the role PTKs in signaling and disease, as it was generally believed that PTPs functioned merely as indiscriminate ‘housekeeping’ enzymes with broad specificities. However, we now appreciate that PTPs are not only as numerous and diverse as PTKs, we have also started to understand how highly specific these enzymes indeed act on their substrates in vivo, and how tightly regulated they are to maintain homeostasis.32-36 Indeed, the rapidly increasing number of disorders associated with PTP abnormalities has begun to elicit a growing interest in these enzymes as drug targets, including in major diseases such as cancer,37, 38 diabetes,39 autoimmunity,40, 41 and Alzheimer’s.42 While PTPs have been well studied in numerous cell types, in particular in cells of the immune system,40, 43, 44 the expression, regulation, specificity, and function of PTPs in platelets are largely unknown. Recent proteomic analyses found that 20 out of the 37 classical, phosphotyrosine (pTyr)-specific PTPs are expressed in human platelets.45 Only four of them had been previously identified as critical regulators of platelet function. Platelet expression and function of the 63 dual-specificity phosphatases (DSPs),32, 34 the largest subgroup of PTPs, had been entirely unexplored. A summary of PTPs reported in platelet function is presented in Table 1.
Table 1.
Protein tyrosine phosphatases (PTPs) implicated in platelet function
| PTP | Functions | Evidence | References |
|---|---|---|---|
| CD148 (PTPRJ) | -positive regulator of GPVI- and CLEC-2-mediated platelet activation, aggregation and αIIbβ3-mediated spreading -positive and negative regulator of SFKs |
-CD148-deficient mouse model -transiently transfected cell line -in vitro biochemical |
Senis Y et al. Blood. 2009, 113:4942-54; Ellison S et al. J Thromb Haemost. 2010, 8:1575-83; Mori J et al. Arterioscl Thromb Vasc Biol. 2012, 32:2956-65 |
| PTP1B (PTPN1) | -positive regulator of late stage platelet activation and aggregation -positive regulator of SFKs -implicated in dephosphorylating LAT |
-PTP1B-deficient mouse models -transiently transfected cell line -in vitro biochemical |
Frangioni JV et al. EMBO J. 1993, 12:4843-56; Ragab A et al. J Biol Chem. 2003, 278:40923-32; Arias-Salgado EG et al. J Cell Biol. 2005, 170:837-45; Kuchay SM et al. Mol Cell Biol. 2007, 27:6038-52 |
| SHP1 (PTPN6) | -positive regulator of GPVI-mediated platelet aggregation and αIIbβ3-mediated spreading -positive regulator of SFKs -negative regulator of GPCR signaling -mediates release of RGS10 and RGS18 from sphinophilin -dephosphorylates actinin downstream of αIIbβ3 |
-SHP1-deficient mouse models -transiently transfected cell line -pharmacological (inhibitors: NSC87877, PTPI-1) -in vitro biochemical |
Pasquet JM et al. J Biol Chem. 2000, 275:28526-31; Lin SY et al. J Biol Chem. 2004, 279:25755-64; Tadokoro S et al. Blood. 2011, 117:250-8; Ma P et al. Blood. 2012, 119:1935-45; Mazharian A et al. Blood. 2013, 121:4205-20 |
| SHP2 (PTPN11) | - negative regulator of platelet GPVI- and CLEC-2-mediated activation, aggregation and αIIbβ3-mediated spreading | -SHP2-deficient mouse model -transiently transfected cell line -in vitro biochemical |
Jackson DE et al. J Biol Chem. 1997, 272:6986-93; Newman DK et al. Blood. 2001, 97:2351-7; Mazharian A et al. Blood. 2013, 121:4205-20 |
| PTP-MEG2 (PTPN9) | - biogenesis and fusion of vesicle membranes with the plasma membrane | - PTP-MEG2-deficient mouse model | Wang Y et al. J Exp Med. 2005, 202:1587-97 |
| VHR (DUSP3) | - positive regulator of GPVI- and CLEC-2-mediated platelet activation and aggregation | -VHR-deficient mouse model -pharmacological (inhibitor: MLS-0437605) |
Musumeci L et al. Circulation. 2015, 131:656-68 |
| PTEN | - negative regulator of GPVI-mediated platelet activation and aggregation | - PTEN-deficient mouse model | Weng Z et al. Blood. 2010, 116:2579-81 |
| LMPTP (ACP1) | - implicated in down-regulating FcγRIIA-mediated platelet activation | -transiently transfected cell line -in vitro biochemical |
Mancini F et al. Blood. 2007, 110:1871-8 |
Our laboratories recently published work investigating the role of DSPs in human platelets, implicating the Vaccinia H1-related (VHR) phosphatase (also known as DUSP3) as a key positive regulator of platelet signaling through the GPVI collagen receptor and the C-type lectin-like 2 (CLEC-2) podoplanin receptor.46 More importantly, we found that VHR-deficient mice were more resistant to collagen- and epinephrine-induced thromboembolism, compared to wild-type (WT) mice, and showed severely impaired thrombus formation upon FeCl3-induced carotid artery injury.46 Intriguingly, bleeding times were not altered in VHR-deficient mice. To investigate VHR function in human platelets, we developed a specific small-molecule inhibitor of VHR. This compound specifically inhibited GPVI- and CLEC-2-induced human platelet aggregation, thereby phenocopying the effect of VHR deficiency in murine cells. This was the first time a specific platelet PTP had been targeted with a small-molecule drug. Our findings, which we discuss in more detail in Section 3 of this perspective article, may lead to a novel, effective, and safer antiplatelet therapy.
2. Classical PTPs in platelet signaling
Four classical PTPs have been identified as critical regulators of platelet function, namely: the transmembrane receptor-like PTP CD148 and the intracellular non-receptor like PTPs PTP1B, SHP1, and SHP2. Below we discuss the main functions of these PTPs in platelet reactivity, as well as their potential as antithrombotic drug targets.
2.1. CD148 (PTPRJ, DEP-1): Master regulator of platelet reactivity
CD148 is a fundamental regulator of platelet reactivity (Fig. 2).47 Platelets rely heavily on CD148 function to regulate SFK activity and signaling from immunoreceptor tyrosine-based activation motif (ITAM)-containing receptors and integrins.48, 49 CD148 consists of a large, highly glycosylated ectodomain (containing eight fibronectin type III domains), a single transmembrane domain, and a single PTP domain in its cytoplasmic tail. Physiologically relevant ligands of CD148 remain ambiguous, although syndecan-2 and thrombospondin-1 were recently reported to bind CD148.50, 51 The C-terminal inhibitory tyrosine residue of SFKs is the most well established substrate of CD148. However, CD148 also attenuates SFK activity by dephosphorylating the activation loop tyrosine residue, thus acting as a molecular rheostat, optimizing SFK activity under resting and activated conditions.49
Figure 2. CD148 is a critical regulator of platelet Src family kinases and platelet reactivity.

Src family kinases (SFKs) are essential for initiating and propagating signals from a variety of platelet receptors, including: the low affinity immunoglobulin receptor FcγRIIA; the von Willebrand factor (vWF) receptor complex GPIb-IX-V; the collagen integrin α2β1; the collagen activation receptor complex GPVI-FcRγ; the integrin αIIbβ3, which binds a variety of extracellular matrix and plasma proteins including fibrinogen and vWF; and the podoplanin receptor CLEC-2. Dark green boxes in the cytoplasmic regions of FcγRIIA and the FcRγ denote immunoreceptor tyrosine-based activation motifs (ITAMs); the light green box in the cytoplasmic tail of CLEC-2 denotes a hemi-ITAM.
Targeted deletion of CD148 in mice has profound consequences on platelet reactivity and thrombosis due to a dramatic reduction in SFK activity both in resting and activated platelets.47 The capacity of CD148 to attenuate SFK activity is masked in CD148-deficient mice, where the net effect is a reduction in platelet reactivity. CD148-deficient platelets exhibit impaired responses to collagen and fibrinogen.47 However, responses to thrombin and TXA2 are only marginally impaired, while responses to ADP are normal. Thrombus formation is markedly reduced in CD148 constitutive knockout mice, following laser- and FeCl3-induced injury of arterioles, with a minor prolongation of bleeding. CD148-deficient platelets also exhibit a concomitant 50% reduction in GPVI expression, which contributes to the reduced reactivity to collagen.47 These findings clearly establish CD148 as a critical positive regulator of platelet activation and aggregation.
CD148 expression is tightly regulated in human platelets, in which little inter-individual variability is observed.47 Specific polymorphisms identified in the extracellular region of CD148 (Q276R and R326Q) have a protective effect in the development of heparin-induced thrombocytopenia by reducing platelet reactivity to antibody-platelet factor 4/heparin complexes, which activate platelets via clustering of the ITAM-containing Fc-γ receptor IIA (FcγRIIA).52 It is proposed that these mutations introduce torsional stress and a loss of positive charge in the second fibronectin type III domain of CD148 that may affect ligand binding or membrane compartmentalization of CD148.52 Collectively, these findings suggest CD148 as a potential antiplatelet drug target.
Because CD148 not only regulates the basal level of SFK activity, but also the threshold of signaling from a number of platelet receptors (Fig. 2), we anticipate that inhibiting CD148 is likely to have dramatic effects on thrombus formation. Thus, we believe it would be an appropriate target in the most severe thrombotic cases, such as disseminated intravascular coagulation or post-operatively, when thrombus formation is a high risk. As with all antiplatelet therapies, bleeding side effects are a distinct possibility, which may be minimized by titrating the dose of a reversible inhibitor. The ectodomain of CD148 is an obvious target, owing to the ease of access. Blocking ligand binding would presumably alter the threshold of platelet activation. Indeed, a monoclonal antibody targeting the extracellular domain of CD148 has been shown to inhibit CD148-dependent endothelial cell growth and angiogenesis in mouse cornea, providing proof-of-principle of this strategy.53 The cytoplasmatic juxtamembrane region, PTP domain, and C-terminal tail region are also potential targets, with the added challenge of the drug needing to cross the plasma membrane. Currently, no CD148-specific inhibitors are available.
2.2. PTP1B (PTPN1): Positive regulator of outside-in integrin signaling
PTP1B is highly expressed in platelets and is a positive regulator of outside-in signaling from the integrin αIIbβ3.54, 55 Specifically, PTP1B dephosphorylates the C-terminal inhibitory tyrosine of β3-associated SFKs, initiating downstream signaling (Fig. 3).55, 56 Outside-in signaling regulates cytoskeletal remodeling and is essential for platelet spreading on fibrinogen, as well as clot retraction, two essential processes for stable thrombus formation and cessation of bleeding. Interestingly, mice lacking PTP1B have normal platelet counts and show no apparent bleeding tendency, suggesting that PTP1B is dispensable for primary hemostasis.55 However, they exhibit a dramatic reduction in thrombus formation following laser-induced injury of arterioles. Whether or not the reduced thrombus formation is due exclusively to the absence of PTP1B in platelets has yet to be elucidated. PTP1B-deficient platelets exhibit reduced spreading on fibrinogen,55, 57 yet they bind soluble fibrinogen and aggregate normally in response to the GPVI agonists collagen-related peptide (CRP) and convulxin, the P2Y12 and P2Y1 agonist ADP, and the PAR-4 agonist thrombin.55, 57 In line with these observations, PTP1B-deficient platelets aggregate normally on collagen under intermediate shear conditions, but aggregate volume is dramatically reduced at high shear conditions (3,000 s−1), typically found in stenotic arteries prone to occlusion.57 This provides further compelling evidence for PTP1B as a potential anti-thrombotic target. The highly specific role of PTP1B in platelets may in fact make it a better target than the more globally acting CD148, predicting fewer bleeding side effects compared to inhibiting CD148.
Figure 3. PTP1B is a positive regulator of outside-in signaling from the integrin αIIbβ3.

Inside-out signals from various activation receptors induce the integrin αIIbβ3 to undergo a conformational change from a low-affinity to a high-affinity state. C-terminal Src kinase (Csk), associated with the β3 subunit, maintains SFKs in an inactive state. Fibrinogen-mediated clustering of the high-affinity form of the intregrin triggers dissociation of Csk and association with PTP1B, which dephosphorylates the C-terminal inhibitory tyrosine residue of SFKs and initiates outside-in signaling.
PTP1B has been considered a potential drug target in the treatment of type 2 diabetes since its role as a negative regulator of insulin receptor signaling was published some 15 years ago.58 More recently, PTP1B has also been implicated in the development of breast59, 60 and prostate61 cancer. Despite a tremendous effort put into the development of specific PTP1B inhibitors, both in academia and industry, only a few compounds progressed to clinical trials, and none have advanced beyond phase II (Table 2).62-65 This is largely due to the conventional approach taken to target the highly conserved PTP1B active site.32 However, novel strategies of inhibiting PTP1B have emerged, including the use of cell-permeable antisense oligonucleotides (ISIS-PTP1BRX, ISIS Pharmaceuticals), as well as the development of inhibitors that target less conserved, allosteric sites in PTP1B (e.g., trodusquemine, Table 2).66, 67 These novel and promising approaches may result in the first PTP1B inhibitors with clinical potential. Importantly, targeting PTP1B may have secondary health benefits in conditions such as diabetes and cancer where thrombosis is a known consequence.
Table 2.
Selected pharmacological inhibitors of PTP1B and SHP2 with demonstrated activity in cells and/or in vivo.
| Structure/Name | Potency | Activity in cells | Activity in vivo | Notes and references |
|---|---|---|---|---|
| PTP1B Inhibitors | ||||
|
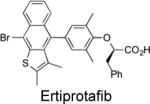
IC50 = 384 nM | Increased phosphorylation of insulin receptor (IR) and IR substrate (IRS) 1 in 3T3-L1 adipocytes. | Lowered glucose, insulin, and triglycerides in rodent diabetes models. | Progressed to phase II clinical trials for treatment of type 2 diabetes (Wyeth Research). Erbe D et al. Mol Pharmacol. 2005, 67:69-77 |
|
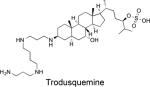
IC50 = 1,000 nM | Increased insulin-stimulated phosphorylation of IR in HepG2 cells | Caused obesity-dependent body weight loss. | Lantz KA et al. Obesity. 2010, 18:1516-23. Trodusquemine is an allosteric PTP1B inhibitor (Krishnan N et al. Nat Chem Biol. 2014, 10:558-66). It was tested in phase I clinical trials for treatment of type 2 diabetes (Genaera Corp.). A Phase I clinical trial in HER2-positive breast cancer patients is planned (DepYmed, Inc.). |
|
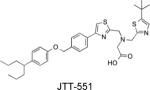
Ki = 220 nM | Increased insulin-stimulated glucose uptake in L6 cells. | Improved glucose metabolism in rodents by enhancing insulin signaling. | Fukuda S et al. Diabetes Obes Metab. 2010, 12:299-306 |
|
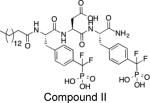
Ki = 26 nM | Increased phosphorylation of IR and IRS-1, enhanced glucose uptake, Elk1 phosphorylation, and cell proliferation, and activated Akt and ERK1/2 in CHO/HIRc cells. | Restored leptin sensitivity in mature, leptin-resistant rats. | Xie L et al. Biochemistry. 2003, 42:12792-804 Morrison C et al. Endocrinology. 2007, 148:433-40 |
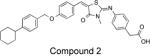
|
IC50 = 240 nM | Improved oral glucose tolerance and decreased plasma glucose and triglyceride levels in diet-induced obese mice. | Arora SK et al. PCT Int Appl, WO 2009/109998 | |

|
IC50 = 82 nM | Caused reduction in body weight, fed-state whole blood glucose (WBG), fasting WBG, plasma glucose, and plasma cholesterol levels in ob/ob mice. | Lakshminarayana N et al. Eur J Med Chem. 2010, 45:3709-18 | |
|
IC50 = 120 nM | Inhibited glucose excursion in DIO mice. Caused a delay in the onset of tumor development in NDL2 Ptpn1 transgenic mice. | Han Y et al. Bioorg Med Chem Lett. 2008, 18:3200-5 | |
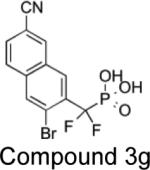
|
IC50 = 35 nM | Increased phosphorylation of IR in HEK293 cells transfected with plasmids encoding human IR. | Combs AP et al. J Med Chem. 2006, 49:3774-89 | |
| SHP2 Inhibitors | ||||
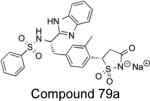
|
IC50 = 2,100 nM | Inhibited hepatocyte growth factor/scatter factor (HGF/SF)-induced epithelial cell scattering and branching morphogenesis. Blocked HGF/SF-induced sustained phosphorylation of ERK1/2 and dephosphorylation of paxillin. Blocked anchorage-independent growth of tumor cell lines. | Hellmuth K et al. Proc Natl Acad Sci U S A. 2008, 105:7275-80. | |
|
|
IC50 = 2,900 nM | Blocked T cell receptor–mediated ERK1/2 activation and SHP2 gain-of-function mutant-induced hematopoietic progenitor hyperproliferation and monocytic differentiation. | Liu S et al. Chem Biol. 2011, 18:101-10. | |
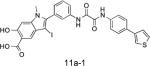
|
IC50 = 200 nM | Reduced lung cancer cell (H1975) proliferation and blocked SHP2-dependent signaling. Inhibited ERK1/2 and Akt activity and ErbB2+ breast cancer cell growth (SKBR3). | Zheng LF et al. J Med Chem. 2014, 57:6594-609. | |
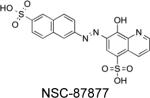
|
IC50 = 318 nM | Inhibited EGF-induced activation of SHP2, Ras, and ERK1/2 in HEK293 cells. | Chen L et al. Mol Pharmacol. 2006, 70:562-70 NSC-87877 does inhibit SHP1 with a similar IC50 value. |
|
2.3. SHP1 (PTPN6) and SHP2 (PTPN11): Critical regulators of platelet homeostasis
The closely related Src homology 2 (SH2) domain-containing phosphatases SHP1 and SHP2 use their tandem N-terminal SH2 domains to bind to phosphorylated immunoreceptor tyrosine-based inhibition motifs (ITIMs) associate with the platelet endothelial cell adhesion molecule 1 (PECAM-1), which functions as a negative regulator of platelet reactivity and thrombosis,68 but has also been implicated as a positive regulator of the outside-in signaling properties of αIIbβ3.69 PECAM-1-mediated membrane localization of SHP1 and SHP2 is thought to allow them to access and dephosphorylate key signaling and cytoskeletal molecules. Work done using SHP1 and SHP2 conditional knockout mice revealed complex phenotypes that are not compatible with a simple model of SHP1 and SHP2 solely acting as negative regulators of platelet signaling.70 Table 1 and Figure 4 highlight putative functions attributed to SHP1 and SHP2 in platelets and megakaryocytes. Mice lacking either SHP1 or SHP2 in the megakaryocyte lineage (i.e., in platelets and their progenitors megakaryocytes) exhibited distinct phenotypes that were exacerbated in double knockout mice, demonstrating non-redundant functions that collectively regulate platelet production and homeostasis. Deletion of both SHP1 and SHP2 resulted in a critical block in megakaryocyte development and survival and almost a complete absence of platelets in the circulation. As a result, SHP1/2 double knockout mice had a severe bleeding diathesis. Interestingly, a similar phenotype was seen in mice lacking G6b-B,70 an ITIM-containing receptor that was previously shown to be associated with SHP1 and SHP2.71
Figure 4. Putative functions of SHP1 and SHP2 in platelets and megakaryocytes.




(A/B) SHP1 and SHP2 have been implicated in regulating proximal signaling events downstream of the immunoreceptor tyrosine-based activation motif (ITAM)-containing collagen receptor complex GPVI-FcRγ, and the hemi-ITAM-containing podoplanin receptor CLEC-2. (C) SHP2 has also been implicated as a negative regulator of the integrin αIIbβ3 in mice. It should be noted that the ITAM-containing low affinity immunoglobulin receptor FcγRIIA acts as a docking site for Syk downsream of αIIbβ3 in human platelets, but not in mouse platelets, which do not express FcγRIIA. (D) The best-characterized function of SHP2 in the megakaryocyte lineage is its positive regulatory role of the Ras-ERK1/2 pathway, downstream of the thrombopoietin (Tpo) receptor MPL. However, the exact mechanism remains ambiguous. Functions illustrated in this figure are mainly based on findings from SHP1 and SHP2 conditional knockout mice and need to be validated in mouse and human platelets through the use of SHP1- and SHP2-specific inhibitors.
Based on these findings, targeting SHP2 may be beneficial in treating conditions characterized by overproduction of platelets such as thrombocythemia or polycythemia vera, both myeloproliferative disorders that are frequently caused by elevated signaling via the thrombopoietin receptor MPL (also known as myeloproliferative leukemia protein). Indeed, SHP2 inhibitors (Table 2) are currently being developed for the treatment of various leukemias and other forms of cancer,72, 73 where elevated SHP2 activity contributes to oncogenesis.74, 75 In contrast, inhibition of SHP1, for which no specific inhibitors have been reported, may result in undesirable side effects, given its important negative regulatory role in many survival and growth signaling pathways in hematopoietic cells,43 and the fact that mice deficient in SHP1 display a severe autoimmune and immunodeficiency syndrome (motheaten mouse).76, 77 Thus, one of the main challenges in targeting SHP2 is to generate inhibitors with sufficient selectivity for SHP2 over its closely related cousin SHP1.
3. VHR (DUSP3): Major positive regulator of platelet signaling and thrombosis
VHR, first described by Ishibashi et al.,78 and named for its sequence similarity with the VH1 gene in Vaccinia virus, is a member of the dual-specificity phosphatases. The DSP subfamily of PTPs is the most diverse group in terms of substrate specificity, ranging from pTyr-specific to phosphothreonine (pThr)/pTyr-specific, to phosphoserine (pSer)-specific, to pSer/pThr-specific DSPs. Additionally, some DSPs can also dephosphorylate mRNA or phosphatidylinositol phosphates.32, 34 VHR is a small DSP (185 amino acids; 20.5 kDa), containing only a catalytic PTP domain, with a preference for dephosphorylating pTyr over pSer/pThr.79 Reported substrates of VHR in vitro are the receptor tyrosine kinases EGFR, KGFR, PDGFR, and the insulin receptor,78 as well as the MAP kinases ERK1/2 and JNK1/2.80, 81 However, dephosphorylation of MAP kinases by VHR remains controversial.46, 82, 83 Several reports implicated VHR in cell cycle regulation and cancer.84-87 For instance, RNA interference-mediated knockdown of VHR in HeLa cells caused cell cycle arrest at the G1-S and G2-M transitions and also induced cell senescence.84 In addition, VHR was found upregulated in prostate and cervix carcinomas,86, 87 and pharmacological inhibition of VHR decreased the proliferation of HeLa and other cervix cancer cell lines.85 Finally, in the LNCaP prostate cancer cell line, androgen-induced VHR expression reduced 12-O-tetradecanoylphorbol-13-acetate- and thapsigargin-induced apoptosis.87 Based on these reports, VHR behaves like a prooncogene.88 However, VHR also acts as a tumor-suppressor in breast cancer89 and non-small cell lung cancers.82, 90 Regardless, the actual biological functions of VHR are still poorly understood. (A more detailed survey of the literature can be found in a recent review by Pavic et al.91)
3.1. VHR is a key positive regulator of the GPVI and CLEC-2 pathways in platelets
We recently found that VHR is highly expressed in human and mouse platelets.46 We previously generated VHR-knockout (VHR-KO) mice92 and utilized these animals to study the role of VHR in hemostasis and thrombosis.46 We found that VHR-KO mice were more resistant to pulmonary thromboembolism than their WT littermates. Moreover, thrombus formation was strongly impaired after FeCl3-induced injury of carotid arteries in a platelet-specific manner. Intriguingly, bleeding was not affected by VHR deficiency, suggesting that VHR plays a key role in arterial thrombosis, but is dispensable for primary hemostasis.46 Ex vivo, VHR deficiency resulted in defective platelet aggregation, granule secretion, and integrin αIIbβ3 inside-out activation, specifically in response to GPVI and CLEC-2 stimulation (Fig. 5). Platelet activation mediated by GPCRs (i.e., the P2Y12, TXA2, and thrombin receptors) was not affected. Impaired thrombus formation in VHR-KO mice was comparable to the previously reported GPVI-KO/FcRβ–KO,93-95 CLEC-2-KO,96 CLEC-2-depleted,97 GPVI-depleted,98 and CLEC-2/GPVI-depleted mice.99 The finding that VHR-deficient mice show no apparent bleeding tendency was in line with what was observed previously in GPVI-KO and CLEC-2-KO mice.93, 96, 100 VHR-KO mice were also protected against pulmonary thromboembolism, similar to GPVI-KO mice.100
Figure 5. VHR is a positive regulator of platelet activation in response to GPVI and CLEC-2 stimulation.


Upon GPVI (A) and CLEC-2 (B) receptor stimulation, VHR positively regulates the phosphorylation of GPVI-FcRγ-associated ITAMs (A, dark green) and CLEC-2-associated hemi-ITAMs (B, light green), resulting in the recruitment of Syk to these receptors, and subsequent platelets activation.
At the molecular level, phosphorylation of the previously reported VHR substrates ERK1/2 and JNK1/2 was not affected by VHR deficiency in platelets. However, we found significantly reduced pTyr levels in the tyrosine kinase Syk, a key signaling molecule in GPVI- and CLEC-2-mediated platelet activation (Fig. 5). Upon stimulation, Syk phosphorylation was reduced on the activatory residues Tyr-525/526, while phosphorylation on Tyr-323, which is implicated in Syk ubiquitination/degradation,101 was not affected. Previous data suggested that Syk is recruited to the GPVI/Fc receptor γ-chain (FcRγ) complex via phosphorylation of FcRγ-associated ITAMs by SFKs.102, 103 We found that phosphorylation of FcRγ-associated ITAMs was reduced, and recruitment of Syk to FcRγ was impaired in VHR-KO compared to WT platelets. Additionally, inducible tyrosine phosphorylation in phospholipase-C (PLC) γ2, a key signaling molecule downstream of Syk, was also reduced in VHR-deficient compared to WT platelets. In contrast, phosphorylation of inhibitory or activatory tyrosine residues in SFKs, including Lyn, Fyn, and Src, was not altered, indicating that the reduced activation and recruitment of Syk in VHR-deficient platelets was not due to aberrant activation of SFKs. At this point we do not know the direct target(s) of VHR in platelets. Given that VHR deficiency led to a decrease of pTyr in Syk, and no proteins with an increase in pTyr could be identified, our data raise the question whether pSer/pThr in Syk, SFKs, or other protein(s) may be dephosphorylated by VHR. However, due to the limited usability of available pSer/pThr antibodies, future studies using quantitative phospho-proteomics will be necessary to address this question.
3.2. Pharmacological inhibition of VHR in human platelets phenocopies the effect of VHR deficiency
Platelets derive from cytoplasmatic fragments of megakaryocytes and hence are anucleate cells that are not amenable to RNA interference or recombinant DNA technologies. Tool compounds to pharmacologically inhibit (or activate) the function of a specific signaling molecule in human platelets are invaluable to corroborate findings that originate from animal models. In our study, we utilized a chemical genomics approach to develop a specific small-molecule inhibitor of VHR (Fig. 6).46 High-throughput screening (HTS) of ~300,000 drug-like molecules (from the NIH Molecular Libraries Program) culminated in 67 hits with confirmed activities in two orthogonal PTP assays (colorimetric and fluorimetric),104, 105 and dose-dependent responses with IC50 values <20 μM. Based on compound potency and selectivity (derived from subsequent counter-screens against four related PTPs), two scaffolds were selected for structure-activity relationship (SAR) studies: MLS-0103602 (IC50 = 0.37 μM) and MLS-0049585 (IC50 = 2.68 μM). Analogs of MLS-0103602 included 37 benzothioamide derivatives that were tested against VHR. All analogs were at least an order of magnitude less potent than the original hit with no improvement of selectivity, leading to the termination of this series. In contrast, several analogs of MLS-0049585 with similar or even better potency and selectivity could be identified. Testing of these inhibitors on human platelets collected from healthy donors revealed an efficient inhibition of platelet aggregation by compound MLS-0437605. This effect of MLS-0437605 was specific to platelets activated through the GPVI and CLEC-2 receptors, equivalent to the effect of VHR deficiency in murine cells. Tests on platelets from WT mice yielded similar results, while MLS-0437605 only minimally affected aggregation of VHR-deficient platelets. In selectivity profiling studies against a larger panel of PTPs, MLS-0437605 showed excellent selectivity for VHR over the vast majority of PTPs tested, including good selectivity over VHX (DUSP22), another DSP that we found highly expressed in platelets.46 Similar to VHR deficiency, inhibition of VHR by MLS-0437605 in human platelets reduced tyrosine phosphorylation of immunoprecipitated Syk and PLCγ2 in response to GPVI and CLEC-2 stimulation; global tyrosine phosphorylation was not affected. Collectively, our data demonstrate that pharmacological inhibition of VHR activity in human platelets phenocopies the effect of VHR deficiency in murine platelets. Given that VHR-KO mice remain healthy and do not suffer from increased bleeding events, MLS-0437605 may serve as the basis for the development of a novel antiplatelet strategy in the treatment of arterial thrombosis.
Figure 6. Development of a VHR-specific inhibitor.


High-throughput screening (HTS) of the Molecular Libraries Probe Production Centers Network (MLPCN) compound collection, using a colorimetric phosphatase assay with p-nitrophenyl phosphate (pNPP) as substrate, resulted in 1524 primary hits. Of these hits, 1048 compounds were available for dose-response (DR) hit confirmation assays using two orthogonal assay formats, the colorimetric pNPP assay, as well as a fluorimetric assay using O-methylfluorescein phosphate (OMFP) as substrate. 67 cross-active hits with IC50 values <20 μM in both assays were taken into selectivity profiling experiments (SEL) against four additional PTPs (HePTP, STEP, LYP, and MKP-3). Based on potency and selectivity, two scaffolds (MLS-0103602 and MLS-0049585) were selected for structure-activity relationship (SAR) studies. Several analogs with similar or improved potency/selectivity could be identified for MLS-0049585. The four most active analogs were chosen for cell-based assays (CELL) and tested on human platelets freshly isolated from healthy donors. Compound MLS-0437605, which inhibited recombinant VHR with an IC50 of 3.7 μM, effectively inhibited platelet aggregation at IC50 concentration, specifically in response to platelet stimulation with collagen-related peptide (CRP; GPVI stimulation) and rhodocytin (CLEC-2 stimulation), but not after stimulation with the TXA2 analog U46619, thereby phenocopying the effect of VHR deficiency on mouse platelets. Selectivity of MLS-0437605 for VHR was evaluated against a total of 10 recombinant PTPs, as well as under physiological conditions on VHR-deficient platelets, whose aggregation was only minimally affected by MLS-0437605. Finally, MLS-0437605 (3.7 μM) decreased tyrosine phosphorylation of Syk and PLCγ2 in human platelets in response to CRP and rhodocytin, similar to the effect of VHR deficiency on mouse platelets. (Please see Musumeci et al.46 to review the actual data.)
4. Challenges in targeting PTPs
PTPs are the largest class of protein phosphatases with over 100 members in humans.32-36 They are defined by their active-site signature motif C(X)5R, which is part of the phosphate-binding loop (P-loop) that, together with additional conserved loops, forms the catalytic pocket.32 PTPs share a common catalytic mechanism that is based on key amino acids within these loops, most importantly a nucleophilic cysteine and an invariant arginine that are part of the signature motif. The vast majority of reported small-molecule PTP inhibitors target the catalytic site. These compounds are usually potent, but poorly selective, due to the fact that they often carry a charged pTyr-mimicking group that provides most of the binding energy through interactions with the highly conserved P-loop.32, 64, 105-110 High-affinity peptides carrying nonhydrolyzable (fluoro)phosphonate group(s) exhibit incredible potency and selectivity, but little to no cell-based activity, owing to their multi-charged nature and poor drug-likeness, resulting in the lack of cell membrane permeability.111 Peptidic tool compounds that cross cell membranes were generated by the addition of long-chain fatty acids112 or cell-penetrating peptides such as polyarginines.113 Additionally, prodrug strategies to deliver charged compounds more easily into cells have been utilized.114-116 However, generating small-molecule drug-like PTP inhibitors with significant selectivity for a particular PTP over related enzymes remains challenging. Selectivity of active site inhibitors can be increased by tailoring compounds to interact with unique amino acids and surface features peripheral of the catalytic pocket, an approach that is supported by large-scale structural analyses of PTPs.117 Because such compounds are designed to interact with both the catalytic site and additional peripheral sites, the trade-off is usually expanded size and molecular weight of inhibitors, resulting in lower ligand efficiency,118 and compounds that are less likely to enter cells or to be absorbed in the gut. Several new approaches to overcome the selectivity hurdle have been proposed. These include inhibitors that target a less conserved inactive-state PTP conformation, inhibitors that target allosteric sites in PTPs, as well as inhibitors that specifically bind a PTP in its oxidized state. (For more information on these new approaches please see our recent review article.32) Interestingly, Michaelis-Menten kinetic studies suggest a noncompetitive inhibition mode for our VHR inhibitor MLS-0437605, a compound that lacks any pTyr-mimicking group (unpublished data). Noncompetitive inhibition is typical for allosteric inhibitors; however, further studies are needed to conclusively determine the mode of action of MLS-0437605. Certainly, allosteric inhibition of PTPs has been gaining significant traction recently,66, 67, 119, 120 and further success could result in a major paradigm shift in how we target PTPs in the future. Finally, targeting the extracellular domains of receptor-like PTPs such as CD148 has been under-exploited to date, but may provide unique opportunities in terms of drug specificity and target accessibility. In a recent study by Takahashi and co-workers, antibody-induced cross-linking of CD148 resulted in inhibition of endothelial cell growth and angiogenesis.53 It is conceivable that a similar approach may inhibit activation of platelets.
5. Conclusion
Antiplatelet therapy has been very successful in the prevention and treatment of arterial thrombosis, the primary cause of most cases of myocardial infarction and stroke. However, approved antiplatelet drugs have adverse side effects and are not effective in a considerable subset of patients. In the search for novel antiplatelet agents with a more favorable safety profile and better efficacy, the discovery of new targets is the matter of intense research. Protein tyrosine phosphatases are important molecules for receptor signal transduction and platelet activation. Previous research implicated CD148, PTP1B, SHP1, and SHP2 as critical regulators in platelet signaling. Recent findings from our laboratories show that VHR, a dual-specificity phosphatase, plays a selective and essential role in GPVI- and CLEC-2-mediated platelet activation and thrombus formation in vivo. In contrast to current antiplatelet therapies, which completely inhibit a specific receptor or signaling pathway, findings from the CD148, PTP1B, and VHR knockout mouse studies suggest that inhibiting a specific PTP can reduce platelet function, but not completely abrogate it. Thus, platelets can still respond to all agonists, albeit less strongly, which, in turn, may result in fewer bleeding side effects. However, because CD148 is a global regulator of platelet reactivity, inhibition of CD148 is more likely to have bleeding consequences than, for instance, targeting PTP1B or VHR. Effective targeting of PTPs with drug-like molecules has been very difficult to achieve. Specifically, the generation of selective PTP inhibitors remains challenging. However, recent developments, in particular in regards to allosteric mechanisms, demonstrate the possibility to overcome the selectivity hurdle, which so far has impeded the success of PTP inhibitors in the clinic. Our novel VHR inhibitor efficiently inhibited human platelet activation in vitro. These data provide, for the first time, proof-of-principle for a PTP-based antiplatelet strategy using a specific small-molecule inhibitor.
Acknowledgments
This work was supported by the American Heart Association (Innovative Research Grant 14IRG18980075 to LT), the National Institutes of Health (Grants U54 HG005033 to JC Reed/CPCCG and R21 CA132121 and R03 MH084230 to LT), the Belgian National Fund for Scientific Research (F.R.S.-FNRS: PDR N° T.0105.13), the University of Liège (Fonds Spéciaux pour la Recherche to CO and SR), and the British Heart Foundation (Senior Fellowship FS/13/1/29894 to YAS). S.R. and C.O. are Research Associates at the Belgian Fund for Scientific Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Furie B, Furie BC. N Engl J Med. 2008;359:938. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 2.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. Physiol Rev. 2013;93:327. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 3.Mackman N. Nature. 2008;451:914. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michelson AD. Nat Rev Drug Discov. 2010;9:154. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- 5.Franchi F, Angiolillo DJ. Nature reviews. Cardiology. 2015;12:30. doi: 10.1038/nrcardio.2014.156. [DOI] [PubMed] [Google Scholar]
- 6.Becattini C, Agnelli G. Blood Rev. 2014;28:103. doi: 10.1016/j.blre.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Oury C, Toth-Zsamboki E, Vermylen J, Hoylaerts MF. Curr Pharm Des. 2006;12:859. doi: 10.2174/138161206776056029. [DOI] [PubMed] [Google Scholar]
- 8.Raju NC, Eikelboom JW, Hirsh J. Nat Clin Pract Cardiovasc Med. 2008;5:766. doi: 10.1038/ncpcardio1372. [DOI] [PubMed] [Google Scholar]
- 9.Mahaffey KW, Held C, Wojdyla DM, James SK, Katus HA, Husted S, Steg PG, Cannon CP, Becker RC, Storey RF, Khurmi NS, Nicolau JC, Yu CM, Ardissino D, Budaj A, Morais J, Montgomery D, Himmelmann A, Harrington RA, Wallentin L. J Am Coll Cardiol. 2014;63:1493. doi: 10.1016/j.jacc.2014.01.038. [DOI] [PubMed] [Google Scholar]
- 10.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. J Med Chem. 2008;51:3061. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 11.Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, Ziada KM, Berman G, Strony J, Joseph D, Mahaffey KW, Van de Werf F, Veltri E, Harrington RA. Lancet. 2009;373:919. [Google Scholar]
- 12.Ungerer M, Munch G. Thromb Haemost. 2013;110:868. doi: 10.1160/TH13-02-0084. [DOI] [PubMed] [Google Scholar]
- 13.Bender AT, Beavo JA. Pharmacol Rev. 2006;58:488. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 14.Maalej N, Osman HE, Shanmuganayagam D, Shebuski RJ, Folts JD. J Cardiovasc Pharmacol. 2005;45:389. doi: 10.1097/01.fjc.0000157439.49612.83. [DOI] [PubMed] [Google Scholar]
- 15.Richardson A, Sakariassen KS, Meyer JP, Alberts P, Sorensen AS. European journal of clinical pharmacology. 2013;69:459. doi: 10.1007/s00228-012-1348-9. [DOI] [PubMed] [Google Scholar]
- 16.Estevez B, Shen B, Du X. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:24. doi: 10.1161/ATVBAHA.114.303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, Boufakhreddine S, Holohan TV, Schaub RG. Circulation. 2007;116:2678. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 18.Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, Priem S, Lauwereys M, Casteels P, Van Bockstaele F, Verschueren K, Stanssens P, Baumeister J, Holz JB. Blood. 2011;118:757. doi: 10.1182/blood-2010-11-317859. [DOI] [PubMed] [Google Scholar]
- 19.Lei X, Reheman A, Hou Y, Zhou H, Wang Y, Marshall AH, Liang C, Dai X, Li BX, Vanhoorelbeke K, Ni H. Thromb Haemost. 2014;111:279. doi: 10.1160/TH13-06-0490. [DOI] [PubMed] [Google Scholar]
- 20.Dutting S, Bender M, Nieswandt B. Trends Pharmacol Sci. 2012;33:583. doi: 10.1016/j.tips.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Sturgeon SA, Jones C, Angus JA, Wright CE. European journal of pharmacology. 2008;587:209. doi: 10.1016/j.ejphar.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 22.Bird JE, Smith PL, Bostwick JS, Shipkova P, Schumacher WA. Thrombosis research. 2011;127:560. doi: 10.1016/j.thromres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Reilly MP, Sinha U, Andre P, Taylor SM, Pak Y, Deguzman FR, Nanda N, Pandey A, Stolla M, Bergmeier W, McKenzie SE. Blood. 2011;117:2241. doi: 10.1182/blood-2010-03-274969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andre P, Morooka T, Sim D, Abe K, Lowell C, Nanda N, Delaney S, Siu G, Yan Y, Hollenbach S, Pandey A, Gao H, Wang Y, Nakajima K, Parikh SA, Shi C, Phillips D, Owen W, Sinha U, Simon DI. Blood. 2011;118:5000. doi: 10.1182/blood-2011-06-360743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gratacap MP, Martin V, Valera MC, Allart S, Garcia C, Sie P, Recher C, Payrastre B. Blood. 2009;114:1884. doi: 10.1182/blood-2009-02-205328. [DOI] [PubMed] [Google Scholar]
- 26.Mazharian A, Ghevaert C, Zhang L, Massberg S, Watson SP. Blood. 2011;117:5198. doi: 10.1182/blood-2010-12-326850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter T. Cell. 2000;100:113. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 28.Pawson T, Scott J. Trends Biochem Sci. 2005;30:286. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 29.Watson SP, Auger JM, McCarty OJ, Pearce AC. Journal of thrombosis and haemostasis : JTH. 2005;3:1752. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 30.Stalker TJ, Newman DK, Ma P, Wannemacher KM, Brass LF. Handbook of experimental pharmacology. 2012:59. doi: 10.1007/978-3-642-29423-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter T. Curr Opin Cell Biol. 2009;21:140. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tautz L, Critton D, Grotegut S. Methods Mol Biol. 2013;1053:179. doi: 10.1007/978-1-62703-562-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tonks N. Nat Rev Mol Cell Biol. 2006;7:833. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 34.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Andersen J, Mortensen O, Peters G, Drake P, Iversen L, Olsen O, Jansen P, Andersen H, Tonks N, Moller N. Mol Cell Biol. 2001;21:7117. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tonks N. FEBS J. 2012 [Google Scholar]
- 37.Julien S, Dube N, Hardy S, Tremblay M. Nat Rev Cancer. 2011;11:35. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 38.Ostman A, Hellberg C, Bohmer F. Nat Rev Cancer. 2006;6:307. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 39.Tiganis T. FEBS J. 2013;280:445. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 40.Vang T, Miletic A, Arimura Y, Tautz L, Rickert R, Mustelin T. Annu Rev Immunol. 2008;26:29. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 41.Rhee I, Veillette A. Nat Immunol. 2012;13:439. doi: 10.1038/ni.2246. [DOI] [PubMed] [Google Scholar]
- 42.Goebel-Goody S, Baum M, Paspalas C, Fernandez S, Carty N, Kurup P, Lombroso P. Pharmacol Rev. 2012;64:65. doi: 10.1124/pr.110.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mustelin T, Vang T, Bottini N. Nat Rev Immunol. 2005;5:43. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- 44.Mustelin T, Alonso A, Bottini N, Huynh H, Rahmouni S, Nika K, Louis- dit-Sully C, Tautz L, Togo S, Bruckner S, Mena-Duran A, al-Khouri A. Mol Immunol. 2004;41:687. doi: 10.1016/j.molimm.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 45.Senis YA. Journal of thrombosis and haemostasis : JTH. 2013;11:1800. doi: 10.1111/jth.12359. [DOI] [PubMed] [Google Scholar]
- 46.Musumeci L, Kuijpers MJ, Gilio K, Hego A, Theatre E, Maurissen L, Vandereyken M, Diogo CV, Lecut C, Guilmain W, Bobkova EV, Eble JA, Dahl R, Drion P, Rascon J, Mostofi Y, Yuan H, Sergienko E, Chung TD, Thiry M, Senis Y, Moutschen M, Mustelin T, Lancellotti P, Heemskerk JW, Tautz L, Oury C, Rahmouni S. Circulation. 2015;131:656. doi: 10.1161/CIRCULATIONAHA.114.010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senis YA, Tomlinson MG, Ellison S, Mazharian A, Lim J, Zhao Y, Kornerup KN, Auger JM, Thomas SG, Dhanjal T, Kalia N, Zhu JW, Weiss A, Watson SP. Blood. 2009;113:4942. doi: 10.1182/blood-2008-08-174318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellison S, Mori J, Barr AJ, Senis YA. Journal of thrombosis and haemostasis : JTH. 2010;8:1575. doi: 10.1111/j.1538-7836.2010.03865.x. [DOI] [PubMed] [Google Scholar]
- 49.Senis YA, Mazharian A, Mori J. Blood. 2014;124:2013. doi: 10.1182/blood-2014-01-453134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi K, Mernaugh RL, Friedman DB, Weller R, Tsuboi N, Yamashita H, Quaranta V, Takahashi T. Proc Natl Acad Sci U S A. 2012;109:1985. doi: 10.1073/pnas.1106171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whiteford JR, Xian X, Chaussade C, Vanhaesebroeck B, Nourshargh S, Couchman JR. Mol Biol Cell. 2011;22:3609. doi: 10.1091/mbc.E11-02-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rollin J, Pouplard C, Gratacap MP, Leroux D, May MA, Aupart M, Gouilleux-Gruart V, Payrastre B, Gruel Y. Blood. 2012;120:1309. doi: 10.1182/blood-2012-04-424044. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi T, Takahashi K, Mernaugh RL, Tsuboi N, Liu H, Daniel TO. Blood. 2006;108:1234. doi: 10.1182/blood-2005-10-4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frangioni JV, Oda A, Smith M, Salzman EW, Neel BG. EMBO J. 1993;12:4843. doi: 10.1002/j.1460-2075.1993.tb06174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arias-Salgado EG, Haj F, Dubois C, Moran B, Kasirer-Friede A, Furie BC, Furie B, Neel BG, Shattil SJ. J Cell Biol. 2005;170:837. doi: 10.1083/jcb.200503125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Proc Natl Acad Sci U S A. 2003;100:13298. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mori J, Wang YJ, Ellison S, Heising S, Neel BG, Tremblay ML, Watson SP, Senis YA. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2956. doi: 10.1161/ATVBAHA.112.300447. [DOI] [PubMed] [Google Scholar]
- 58.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy A, Normandin D, Cheng A, Himms-Hagen J, Chan C, Ramachandran C, Gresser M, Tremblay M, Kennedy B. Science. 1999;283:1544. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 59.Julien S, Dube N, Read M, Penney J, Paquet M, Han Y, Kennedy B, Muller W, Tremblay M. Nat Genet. 2007;39:338. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 60.Bentires-Alj M, Neel B. Cancer Res. 2007;67:2420. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- 61.Lessard L, Labbe DP, Deblois G, Begin LR, Hardy S, Mes-Masson AM, Saad F, Trotman LC, Giguere V, Tremblay ML. Cancer Res. 2012;72:1529. doi: 10.1158/0008-5472.CAN-11-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang S, Zhang Z. Drug Discov Today. 2007;12:373. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 63.Lee S, Wang Q. Med Res Rev. 2007;27:553. doi: 10.1002/med.20079. [DOI] [PubMed] [Google Scholar]
- 64.Barr A. Future Med Chem. 2010;2:1563. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 65.Popov D. Biochem Biophys Res Commun. 2011;410:377. doi: 10.1016/j.bbrc.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 66.Wiesmann C, Barr K, Kung J, Zhu J, Erlanson D, Shen W, Fahr B, Zhong M, Taylor L, Randal M, McDowell R, Hansen S. Nat Struct Mol Biol. 2004;11:730. doi: 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- 67.Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, Jensen MR, Gauss CM, Page R, Blackledge M, Muthuswamy SK, Peti W, Tonks NK. Nat Chem Biol. 2014;10:558. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Falati S, Patil S, Gross PL, Stapleton M, Merrill-Skoloff G, Barrett NE, Pixton KL, Weiler H, Cooley B, Newman DK, Newman PJ, Furie BC, Furie B, Gibbins JM. Blood. 2006;107:535. doi: 10.1182/blood-2005-04-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wee JL, Jackson DE. Blood. 2005;106:3816. doi: 10.1182/blood-2005-03-0911. [DOI] [PubMed] [Google Scholar]
- 70.Mazharian A, Mori J, Wang YJ, Heising S, Neel BG, Watson SP, Senis YA. Blood. 2013;121:4205. doi: 10.1182/blood-2012-08-449272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazharian A, Wang YJ, Mori J, Bem D, Finney B, Heising S, Gissen P, White JG, Berndt MC, Gardiner EE, Nieswandt B, Douglas MR, Campbell RD, Watson SP, Senis YA. Sci Signal. 2012;5:ra78. doi: 10.1126/scisignal.2002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butterworth S, Overduin M, Barr AJ. Future Med Chem. 2014;6:1423. doi: 10.4155/fmc.14.88. [DOI] [PubMed] [Google Scholar]
- 73.Zeng LF, Zhang RY, Yu ZH, Li S, Wu L, Gunawan AM, Lane BS, Mali RS, Li X, Chan RJ, Kapur R, Wells CD, Zhang ZY. J Med Chem. 2014;57:6594. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan R, Feng G. Blood. 2007;109:862. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan G, Kalaitzidis D, Neel B. Cancer Metastasis Rev. 2008;27:179. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 76.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR. Cell. 1993;73:1445. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 77.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Nat Genet. 1993;4:124. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 78.Ishibashi T, Bottaro DP, Chan A, Miki T, Aaronson SA. Proc Natl Acad Sci U S A. 1992;89:12170. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schumacher MA, Todd JL, Rice AE, Tanner KG, Denu JM. Biochemistry. 2002;41:3009. doi: 10.1021/bi015799l. [DOI] [PubMed] [Google Scholar]
- 80.Todd JL, Tanner KG, Denu JM. J Biol Chem. 1999;274:13271. doi: 10.1074/jbc.274.19.13271. [DOI] [PubMed] [Google Scholar]
- 81.Todd JL, Rigas JD, Rafty LA, Denu JM. Oncogene. 2002;21:2573. doi: 10.1038/sj.onc.1205344. [DOI] [PubMed] [Google Scholar]
- 82.Wang JY, Yeh CL, Chou HC, Yang CH, Fu YN, Chen YT, Cheng HW, Huang CY, Liu HP, Huang SF, Chen YR. J Biol Chem. 2011;286:10177. doi: 10.1074/jbc.M110.163295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amand M, Erpicum C, Bajou K, Cerignoli F, Blacher S, Martin M, Dequiedt F, Drion P, Singh P, Zurashvili T, Vandereyken M, Musumeci L, Mustelin T, Moutschen M, Gilles C, Noel A, Rahmouni S. Mol Cancer. 2014;13:108. doi: 10.1186/1476-4598-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rahmouni S, Cerignoli F, Alonso A, Tsutji T, Henkens R, Zhu C, Louis- dit-Sully C, Moutschen M, Jiang W, Mustelin T. Nat Cell Biol. 2006;8:524. doi: 10.1038/ncb1398. [DOI] [PubMed] [Google Scholar]
- 85.Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, Vazquez-Rodriguez J, Cerignoli F, Arimura Y, Williams S, Hayes T, Moutschen M, Vasile S, Pellecchia M, Mustelin T, Tautz L. J Med Chem. 2009;52:6716. doi: 10.1021/jm901016k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Henkens R, Delvenne P, Arafa M, Moutschen M, Zeddou M, Tautz L, Boniver J, Mustelin T, Rahmouni S. BMC Cancer. 2008;8:147. doi: 10.1186/1471-2407-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arnoldussen YJ, Lorenzo PI, Pretorius ME, Waehre H, Risberg B, Maelandsmo GM, Danielsen HE, Saatcioglu F. Cancer Res. 2008;68:9255. doi: 10.1158/0008-5472.CAN-08-1224. [DOI] [PubMed] [Google Scholar]
- 88.Julien SG, Dube N, Hardy S, Tremblay ML. Nat Rev Cancer. 2011;11:35. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 89.Hao L, ElShamy WM. Int J Cancer. 2007;121:39. doi: 10.1002/ijc.22597. [DOI] [PubMed] [Google Scholar]
- 90.Wagner KW, Alam H, Dhar SS, Giri U, Li N, Wei Y, Giri D, Cascone T, Kim JH, Ye Y, Multani AS, Chan CH, Erez B, Saigal B, Chung J, Lin HK, Wu X, Hung MC, Heymach JV, Lee MG. J Clin Invest. 2013;123:5231. doi: 10.1172/JCI68642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pavic K, Duan G, Kohn M. FEBS J. 2015 doi: 10.1111/febs.13263. [DOI] [PubMed] [Google Scholar]
- 92.Amand M, Erpicum C, Bajou K, Cerignoli F, Blacher S, Martin M, Dequiedt F, Drion P, Singh P, Zurashvili T, Vandereyken M, Musumeci L, Mustelin T, Moutschen M, Gilles C, Noel A, Rahmouni S. Molecular cancer. 2014;13:108. doi: 10.1186/1476-4598-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kato K, Kanaji T, Russell S, Kunicki TJ, Furihata K, Kanaji S, Marchese P, Reininger A, Ruggeri ZM, Ware J. Blood. 2003;102:1701. doi: 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- 94.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Blood. 2006;107:3902. doi: 10.1182/blood-2005-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Konishi H, Katoh Y, Takaya N, Kashiwakura Y, Itoh S, Ra C, Daida H. Circulation. 2002;105:912. doi: 10.1161/hc0802.105256. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, Shin Y, Hirashima M, Ozaki Y. J Biol Chem. 2010;285:24494. doi: 10.1074/jbc.M110.130575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.May F, Hagedorn I, Pleines I, Bender M, Vogtle T, Eble J, Elvers M, Nieswandt B. Blood. 2009;114:3464. doi: 10.1182/blood-2009-05-222273. [DOI] [PubMed] [Google Scholar]
- 98.Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, Heinzmann U, Nieswandt B. J Exp Med. 2003;197:41. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, Vogtle T, Remer K, Braun A, Bosl M, Watson SP, Nieswandt B. Arterioscler Thromb Vasc Biol. 2013;33:926. doi: 10.1161/ATVBAHA.112.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lockyer S, Okuyama K, Begum S, Le S, Sun B, Watanabe T, Matsumoto Y, Yoshitake M, Kambayashi J, Tandon NN. Thromb Res. 2006;118:371. doi: 10.1016/j.thromres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Mocsai A, Ruland J, Tybulewicz VL. Nat Rev Immunol. 2010;10:387. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ezumi Y, Shindoh K, Tsuji M, Takayama H. J Exp Med. 1998;188:267. doi: 10.1084/jem.188.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watson SP, Asazuma N, Atkinson B, Berlanga O, Best D, Bobe R, Jarvis G, Marshall S, Snell D, Stafford M, Tulasne D, Wilde J, Wonerow P, Frampton J. Thrombosis and haemostasis. 2001;86:276. [PubMed] [Google Scholar]
- 104.Tautz L, Sergienko E. Methods Mol Biol. 2013;1053:223. doi: 10.1007/978-1-62703-562-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tautz L, Mustelin T. Methods. 2007;42:250. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 106.Rios P, Nunes-Xavier CE, Tabernero L, Kohn M, Pulido R. Antioxidants & redox signaling. 2014;20:2251. doi: 10.1089/ars.2013.5709. [DOI] [PubMed] [Google Scholar]
- 107.He R, Zeng L, He Y, Zhang S, Zhang Z. FEBS J. 2012 [Google Scholar]
- 108.Vintonyak VV, Waldmann H, Rauh D. Bioorg Med Chem. 2011;19:2145. doi: 10.1016/j.bmc.2011.02.047. [DOI] [PubMed] [Google Scholar]
- 109.Tautz L, Pellecchia M, Mustelin T. Expert Opin Ther Targets. 2006;10:157. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 110.Bialy L, Waldmann H. Angew Chem Int Ed Engl. 2005;44:3814. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 111.Burke TJ, Kole H, Roller P. Biochem Biophys Res Commun. 1994;204:129. doi: 10.1006/bbrc.1994.2435. [DOI] [PubMed] [Google Scholar]
- 112.Xie L, Lee S, Andersen J, Waters S, Shen K, Guo X, Moller N, Olefsky J, Lawrence D, Zhang Z. Biochemistry. 2003;42:12792. doi: 10.1021/bi035238p. [DOI] [PubMed] [Google Scholar]
- 113.Meyer C, Hoeger B, Temmerman K, Tatarek-Nossol M, Pogenberg V, Bernhagen J, Wilmanns M, Kapurniotu A, Kohn M. ACS Chem Biol. 2014;9:769. doi: 10.1021/cb400903u. [DOI] [PubMed] [Google Scholar]
- 114.Andersen H, Olsen O, Iversen L, Sorensen A, Mortensen S, Christensen M, Branner S, Hansen T, Lau J, Jeppesen L, Moran E, Su J, Bakir F, Judge L, Shahbaz M, Collins T, Vo T, Newman M, Ripka W, Moller N. J Med Chem. 2002;45:4443. doi: 10.1021/jm0209026. [DOI] [PubMed] [Google Scholar]
- 115.Erbe D, Klaman L, Wilson D, Wan Z, Kirincich S, Will S, Xu X, Kung L, Wang S, Tam S, Lee J, Tobin J. Diabetes Obes Metab. 2009;11:579. doi: 10.1111/j.1463-1326.2008.01022.x. [DOI] [PubMed] [Google Scholar]
- 116.Boutselis I, Yu X, Zhang Z, Borch R. J Med Chem. 2007;50:856. doi: 10.1021/jm061146x. [DOI] [PubMed] [Google Scholar]
- 117.Barr A, Ugochukwu E, Lee W, King O, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown N, Muller S, Knapp S. Cell. 2009;136:352. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hopkins A, Groom C, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9(10):430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 119.Chio CM, Lim CS, Bishop AC. Biochemistry. 2015 doi: 10.1021/bi5013595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schneider R, Beumer C, Simard JR, Grutter C, Rauh D. J Am Chem Soc. 2013;135:6838. doi: 10.1021/ja4030484. [DOI] [PubMed] [Google Scholar]