

Abstract
The established dogma is that protein Serine/Threonine (PSPs) and Tyrosine (PTPs) Phosphatases are unattainable drug targets. This is because natural product inhibitors of PSP active sites are lethal, while the active sites of PTPs are exceptionally conserved and charged, making it nearly impossible to develop PTP inhibitors that are selective. However, due to a series of recent structural and functional studies, this view of phosphatases is about to undergo a radical change. Rather than target active sites, these studies have demonstrated that targeting PSP/PTP protein (substrate/regulatory) interaction sites, which are distal from the active sites, are highly viable and suitable drugs targets. This is especially true for Calcineurin (CN), in which the blockbuster immunosuppressant drugs FK506 and cyclosporine A were recently demonstrated to bind and block one of the key CN substrate interaction sites, the LxVP site. Additional studies show that this approach—targeting substrate and/or regulatory protein interaction sites—also holds incredible promise for protein phosphatase 1 (PP1)-related diseases. Finally, domains outside PTP catalytic domains have also recently been demonstrated to directly alter PTP activity. Collectively, these novel insights offer new, transformative perspectives for the therapeutic targeting of PSPs by interfering with the binding of PIPs or substrates and PTPs by targeting allosteric sites outside their catalytic domains.
Keywords: protein phosphatase 1, serine/threonine phosphatases, calcineurin, drug design, PTP1B
Phosphorylation is one of the most ubiquitous, reversible posttranslational modifications in cells [1]. This is because the tightly-regulated phosphorylation of highly dynamic, interacting proteins is one of the key mechanisms used by cells to communicate external signals from the membrane to the nucleus. The enzymes responsible for controlling the phosphorylation state of the cell are kinases, which catalyze the transfer the γ-phosphate moiety of ATP to substrates, and phosphatases, which catalyze the reverse reaction, the removal of the phosphate moiety from phosphorylated substrates. Thus, in this way, phosphatases dynamically reverse the effects of kinases. Because phosphorylation is critical for biological processes from cell growth to differentiation to development, the location and duration of the reciprocal actions of kinases and phosphatases is exquisitely regulated both temporally and spatially within the cell. However, when this tight regulation is disrupted, dysregulation of phosphorylation signaling ensues and the consequence is almost always disease [2–7].
In the human genome, there is a near 1-to-1 ratio of tyrosine phosphatases (PTPs; 107) to kinases (90) [3, 8, 9]. In contrast, the serine/threonine phosphatases (PSPs) are woefully outnumbered by their abundant kinase counterparts (~40:418) [10–16]. Thus, while tyrosine phosphatases have been considered to be viable drug targets, serine/threonine phosphatases have been viewed as ‘house-keeping’ enzymes, with only a limited chance for drug selectivity. This assessment is due in large part because the active sites of the three most abundant and well-studied PSPs—protein phosphatase 1 (PP1), protein phosphatase 2A (PP2A) and protein phosphatase 2B (PP2B; PP3; calcineurin [CN] [15, 16])—are 100% conserved and thus active site inhibitors would likely not be selective [14] (Figure 1). This has been confirmed by the discovery of natural product PSP inhibitors, including microcystin [17], nodularin [18], okadaic acid [19, 20] and tautomycin [18] among others, which inhibit all three phosphatases with only slight preferences of one versus the other. Furthermore, because PP1, PP2A and CN are responsible for the majority of the ser/thr dephosphorylation reactions in humans, inhibiting their active sites is also expected to disrupt many biological processes. This has also been demonstrated as these natural product inhibitors are potent and very lethal toxins.
Figure 1. Conservation of PSP catalytic domains.

PP1 (top), PP2A (middle) and PP2B/Calcineurin (bottom) have a highly conserved catalytic domain (grey). PP1 interacts with ~200 distinct regulatory proteins (R; blue), which function as inhibitory and targeting proteins. PP1 substrates bind directly to PP1, bind to other domains that are part of the PP1 regulatory proteins to enhance dephosphorylation or are dephosphorylated because PP1 is localized in proximity to the substrate via its targeting proteins. The catalytic domain PP2Ac interacts with an invariant A subunit and ~25 regulatory B subunits to achieve substrate specificity in a manner similar to that of PP1. Calcineurin (CN), on the other hand, is regulated by calcium, which is required for activation. CN binds directly to its substrates via protein interaction motifs that are also used by regulatory proteins.
PSP drugs: target PSP substrate and regulatory protein interaction sites
So is there any chance of successfully turning a serine/threonine phosphatase into a multi-billion dollar drug target? The answer is a resounding yes. In fact, such drugs already exist [21]. Cyclosporin A (CSA) [22] and FK-506 [23] are potent immunosuppressants that generate over a billion dollars per year in revenue. They function by potently inhibiting the activity of CN. However, they do not inhibit CN by binding and blocking its active site. Instead, as we have recently shown, these drugs bind to a critical substrate/regulatory protein recognition site on CN known as the LxVP site [24], and function by inhibiting substrates, especially the NFATs, from binding and, as a consequence, being dephosphorylated by CN [25, 26] (Figure 2). The second characterized substrate recognition site in CN is the PxIxIT site [27], which binds PxIxIT sequences in CN regulators and substrates [28]. However, the primary problem with targeting the PxIxIT and LxVP sites is that most CN substrates (as well as regulatory and targeting proteins, such as e.g. the AKAPs [29]) contain a PxIxIT and/or an LxVP site. This likely explains the severe side effects of CSA and FK-506: while the targeted substrates - the NFATs - cannot be dephosphorylated and are result in the limited immune response that is critical for organ transplantations, other substrates with LxVP sites will also be unable to bind and be dephosphorylated by CN.
Figure 2. Inhibiting CN by blocking substrate binding.

A. The CN protein inhibitor A238L inhibits CN activity not by blocking its active site, but instead by binding to CN substrate recognition grooves, which blocks CN from binding and dephosphorylating its substrates. Left panel, surface representation of CN (light/dark grey; left panel) bound to A238L (magenta; shown as a cartoon), a potent protein inhibitor of CN from the African Swine Fever Virus. The active site is shown in light blue. A238L binds CN via both a PxIxIT sequence (middle panel) and an LxVP sequence (right panel). The deep groove in CN engaged by the ‘L’ of the LxVP motif (Leu229 in A238L) is indicated by a star (pink). B. The immunosuppressant drugs FK506 and cyclosporine A (CSA) bind directly to the CN LxVP docking groove. Left panels, surface representations of CN (light and dark grey) bound to FK506 (blue) and CSA (orange). Upper right panels, close-up views of FK506 in the CN LxVP binding groove (left) and overlay with A238L (magenta, right). The deep groove in CN engaged by the ‘L’ of the LxVP motif is indicated by a star (pink) and is fully engaged by FK506. Lower right panels, close-up views of CSA in the CN LxVP binding groove (left) and overlay with A238L (magenta, right).
Thus, for CN, the identification of additional protein interactions sites that are used by very few substrates and/or targeting proteins ought to be seen as the key step for the development novel CN therapeutics, e.g. against neurological diseases. This is because novel drugs that target these sites will only disrupt the interaction of CN with a very small number of targeting and/or substrates. Once identified, these sites will provide a powerful avenue for highly specific modulation of CN activity.
Since PP1, PP2A and PP2B share a 100% conserved active site [14], the development of specific active site inhibitors/activators is impossible. Nature has confirmed this, as nearly all naturally-derived inhibitors show only minimal differences in potency against PP1, PP2A and CN [18, 30]. Thus alternative routes for creating PP1 specific drugs must be pursued. One innovative approach is to target the PP1 regulatory proteins. Although the specificity of the PP1 catalytic domains is low, PP1 dephosphorylates its substrates with high specificity. To achieve this, PP1 interacts with more than 100 distinct regulatory subunits [13]; namely, inhibitory proteins that potently inhibit phosphatase activity by binding and blocking the active site [5, 31–34], and targeting proteins, proteins the localize PP1 to distinct regions of the cell while also directly modulating PP1-substrate interactions (Figure 1,3). Many PP1 targeting subunits, such as NIPP1 [35], enhance the binding of specific substrates. For example, the FHA domain of NIPP1 enhances the PP1-mediated dephosphorylation of its substrates CDC5L and SAP155 [35, 36]. However, others, such as spinophilin and PNUTS [37–39], have been shown to bind PP1 substrate recognition sites, thereby inhibiting the dephosphorylation of a subset of substrates. Thus, they function identically to CSA and FK-506 with CN [40]; i.e., they inhibit substrates from binding the PSP and thereby selectively inhibit their dephosphorylation.
Figure 3. PP1 regulatory protein docking grooves.

A. Surface representation of PP1 with its various regulatory protein docking grooves shown (RVxF, pink; ϕϕ, orange; Arg, lavender; SILK, green; MyPhoNE, yellow; NIPP1-helix, dark blue; Spino-helix, light pink). The active site is in cyan. The Inhibitor-2 helix that lies over the active site to directly inhibit PP1 activity is not shown. B. The presence of various motifs in confirmed PP1 regulators (189).
Therefore, the most promising approach for developing PP1 specific drugs is to identify protein interaction sites that are specific for only a limited number of substrates and/or regulatory proteins (Figure 3, 4). This strategy provides a powerful and specific way to selectively modulate PP1 activity against a small subset of substrates and, in turn, target distinct signaling cascades. However, this strategy will also only be successful if the PP1 regulatory code is fully understood [12, 13]. In recent years, much progress has been made in elucidating the PP1 regulatory code [37] (Figure 3, 4). This was mainly driven by the structural assessment of new, additional PP1 holoenzymes, which has allowed for novel primary sequence motifs to be identified that either are necessary for binding PP1 and/or for changing its substrate specificity.
Figure 4. Developing drugs that inhibit only a small subset of PP1 substrates.
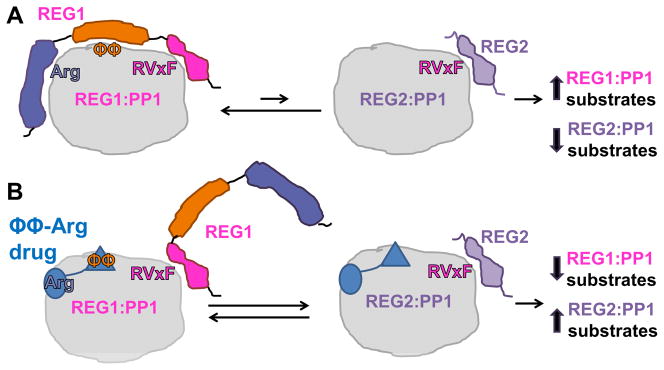
Toxins that bind and block the PP1 active site are lethal. Thus, any potential drugs that target PP1 must interact outside the PP1 active site. One approach is to target the PP1 regulatory protein docking grooves. A. The REG1:PP1 holoenzyme (in this case, REG1 represents a PP1 regulatory protein that contains an RVxF-ϕϕ-Arg motif, such as PNUTS) is preferentially populated compared to the REG2:PP1 holoenzyme (in this case, REG2 is a PP1 regulatory protein with only an RVxF motif), because the affinity of REG1 for PP1 is much higher than REG2 for PP1. B. A drug that targets only the ϕϕ-Arg binding grooves will selectively displace the motifs in REG1 that bind at these sites, thereby reducing the affinity of REG1 for PP1 and, consequently, increasing the likelihood of forming REG2:PP1 holoenzymes. In this way, dephosphorylation of REG1:PP1 substrates will decrease while REG2:PP1 holoenzymes will increase.
The proof of principle of this approach has been already demonstrated in an exciting report from the Köhn laboratory [41–43]. Here, the authors developed a peptide based on the primary sequence of the PP1 regulator NIPP1 that includes the PP1 RVxF and ϕϕ motifs [41], the two most prevalent PP1 binding motifs in all PP1 regulatory proteins [37]. In collaboration with the Bollen laboratory, they showed that the peptide binds to PP1 and displaces many weaker binding targeting and inhibitory proteins in vitro and in vivo, abolishing the tight specificity of PP1 (by disrupting its interaction with targeting proteins) and increasing its activity (by disrupting its interaction with inhibitory proteins). Thus, this peptide based drug increases the overall general dephosphorylation in cells, something of potential use in diseases associated with global increases of phosphorylation, such as certain cancers. However, because the increase in dephosphorylation is now unregulated, drugs that target these interaction sites will likely not provide useful therapeutics for specific diseases. To further enhance this approach it will be necessary to target more unique sites that are share by only a very small number of PP1 regulatory proteins, similar as suggested before for CN.
There is now data that suggest it may be possible to selectively target a single PP1-specific pathway. Salubrinal [44, 45] and Guanabenz [46, 47] are small molecule drugs that have recently been shown to specifically inhibit translation by blocking the activity of eIF2α phosphatases, specifically CreP:PP1 and GADD34:PP1 [48–54]. Whether or not this is achieved by selectively disrupting the PP1-substrate (eIF2α) and/or the PP1-regulatory protein (CReP, GADD34) remains to be determined. Finally, small molecules might also be designed to bind specifically to the motifs/domains on substrates that mediate PSP binding, especially if substrate recruitment requires additional domains form the regulatory proteins that are distinct from the phosphatase binding domains. This approach can be applied to PP1 as well as to PP2A [55].
PTPs: developing drugs that bind to intrinsically disordered regions of the enzyme
As numerous other manuscripts in this issue of Bioorganic & Medicinal Chemistry focus on PTPs as drug targets this section focuses on PTP1B and its modulation via its intrinsically disordered C-terminus. PTPs have long been bona fide drug targets [56]. However, molecules that target PTP active sites are difficult to make selective against closely related PTPs. Unfortunately, all programs aimed at targeting the PTP1B active site have failed due to technical challenges arising from the chemical properties of the PTP active site, which leads to highly charged drugs and, as a consequence, limited drug development potential [57]. Recently, new approaches that aim beyond the active site have been introduced for PTPs [58]. Specifically, in a collaboration between the Tonks and Peti laboratories, the small molecule inhibitor MSI-1436 was recently discovered and shown that it binds to the disordered C-terminal domain of PTP1B, C-terminal to the catalytic domain [59]. Moreover, we also showed that MSI-1436 functions using an allosteric mechanism to direct the enzymatic activity of PTP1B. Thus, this work demonstrates that targeting binding sites in disordered domains [60] might also be a new strategy for PTP drug development. Intriguingly, most PP1 regulatory proteins are also intrinsically disordered proteins. Thus the success of identifying an inhibitor that binds to the intrinsically disordered region of PTP1B might also allow for the regulation of PSP regulatory proteins and PSPs.
Summary
Compared to their kinase counterparts, PTPs and especially PSPs were considered to be poor drug targets. However, advances during the few years have shown unequivocally that PTPs, and especially PSPs, are potent and viable drug targets. In particular multiple structural studies has provided critical new avenues that will allow newly developed therapeutics to not only inhibit, but also be highly selective for only a small set of PSP and/or PTP substrates. These new efforts a sure to be augmented with additional efforts focused on developing biologics that can also modulate the PSPs and PTPs in specific manners. It will be exciting to see which approach will be successful, but the authors are confident that the next years will provide novel, successful route(s) for PSP and PTP drug design.
Acknowledgments
The authors thank the large numbers of highly skilled and ambitious coworkers for their strong support in the effort to better understand protein phosphatases. They are also inclined to many members of the phosphatase field – to many to name – for their support, for their collaborations and their input. This work was supported by NIH grant R01GM098482 to RP as well as an NIH grant R01NS091336 and an American Diabetes Association Pathway to Stop Diabetes Grant 1-14-ACN-31 to WP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 2.Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer’s disease. Progress in molecular biology and translational science. 2012;106:343–379. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 4.Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nature reviews Immunology. 2005;5:43–57. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- 5.Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P. The role of DARPP-32 in the actions of drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):14–23. doi: 10.1016/j.neuropharm.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Current opinion in genetics & development. 2007;17:23–30. doi: 10.1016/j.gde.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nature reviews Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 8.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Bollen M. Combinatorial control of protein phosphatase-1. Trends Biochem Sci. 2001;26:426–431. doi: 10.1016/s0968-0004(01)01836-9. [DOI] [PubMed] [Google Scholar]
- 13.Bollen M, Peti W, Ragusa MJ, Beullens M. The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci. 2010;35:450–458. doi: 10.1016/j.tibs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peti W, Nairn AC, Page R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013;280:596–611. doi: 10.1111/j.1742-4658.2012.08509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy J, Cyert MS. Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal. 2009;2:re9. doi: 10.1126/scisignal.2100re9. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature. 1995;376:745–753. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 18.Kelker MS, Page R, Peti W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: a novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J Mol Biol. 2009;385:11–21. doi: 10.1016/j.jmb.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maynes JT, Bateman KS, Cherney MM, Das AK, Luu HA, Holmes CF, James MN. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J Biol Chem. 2001;276:44078–44082. doi: 10.1074/jbc.M107656200. [DOI] [PubMed] [Google Scholar]
- 20.Xing Y, Xu Y, Chen Y, Jeffrey PD, Chao Y, Lin Z, Li Z, Strack S, Stock JB, Shi Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell. 2006;127:341–353. doi: 10.1016/j.cell.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 22.Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci U S A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 24.Park S, Uesugi M, Verdine GL. A second calcineurin binding site on the NFAT regulatory domain. Proc Natl Acad Sci U S A. 2000;97:7130–7135. doi: 10.1073/pnas.97.13.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, Peti W. The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS biology. 2013;11:e1001492. doi: 10.1371/journal.pbio.1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez A, Roy J, Martinez-Martinez S, Lopez-Maderuelo MD, Nino-Moreno P, Orti L, Pantoja-Uceda D, Pineda-Lucena A, Cyert MS, Redondo JM. A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol Cell. 2009;33:616–626. doi: 10.1016/j.molcel.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Zhang L, Rao A, Harrison SC, Hogan PG. Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. J Mol Biol. 2007;369:1296–1306. doi: 10.1016/j.jmb.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Pink MD, Murphy JG, Stein A, Dell’Acqua ML, Hogan PG. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat Struct Mol Biol. 2012;19:337–345. doi: 10.1038/nsmb.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehringer MM. Microcystin-LR and okadaic acid-induced cellular effects: a dualistic response. FEBS Lett. 2004;557:1–8. doi: 10.1016/s0014-5793(03)01447-9. [DOI] [PubMed] [Google Scholar]
- 31.Hurley TD, Yang J, Zhang L, Goodwin KD, Zou Q, Cortese M, Dunker AK, DePaoli-Roach AA. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J Biol Chem. 2007;282:28874–28883. doi: 10.1074/jbc.M703472200. [DOI] [PubMed] [Google Scholar]
- 32.Dancheck B, Nairn AC, Peti W. Detailed structural characterization of unbound protein phosphatase 1 inhibitors. Biochemistry. 2008;47:12346–12356. doi: 10.1021/bi801308y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dancheck B, Ragusa MJ, Allaire M, Nairn AC, Page R, Peti W. Molecular investigations of the structure and function of the protein phosphatase 1-spinophilin-inhibitor 2 heterotrimeric complex. Biochemistry. 2011;50:1238–1246. doi: 10.1021/bi101774g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsh JA, Dancheck B, Ragusa MJ, Allaire M, Forman-Kay JD, Peti W. Structural diversity in free and bound states of intrinsically disordered protein phosphatase 1 regulators. Structure. 2010;18:1094–1103. doi: 10.1016/j.str.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell N, Nichols SR, Heroes E, Beullens M, Bollen M, Peti W, Page R. The Molecular Basis for Substrate Specificity of the Nuclear NIPP1:PP1 Holoenzyme. Structure. 2012;20:1746–1756. doi: 10.1016/j.str.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minnebo N, Gornemann J, O’Connell N, Van Dessel N, Derua R, Vermunt MW, Page R, Beullens M, Peti W, Van Eynde A, Bollen M. NIPP1 maintains EZH2 phosphorylation and promoter occupancy at proliferation-related target genes. Nucleic Acids Res. 2013;41:842–854. doi: 10.1093/nar/gks1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choy MS, Hieke M, Kumar GS, Lewis GR, Gonzalez-DeWhitt KR, Kessler RP, Stein BJ, Hessenberger M, Nairn AC, Peti W, Page R. Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc Natl Acad Sci U S A. 2014;111:4097–4102. doi: 10.1073/pnas.1317395111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ragusa MJ, Allaire M, Nairn AC, Page R, Peti W. Flexibility in the PP1:spinophilin holoenzyme. FEBS Lett. 2011;585:36–40. doi: 10.1016/j.febslet.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat Struct Mol Biol. 2010;17:459–464. doi: 10.1038/nsmb.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat Struct Mol Biol. 2010;17:459–464. doi: 10.1038/nsmb.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee J, Beullens M, Sukackaite R, Qian J, Lesage B, Hart DJ, Bollen M, Kohn M. Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angewandte Chemie. 2012;51:10054–10059. doi: 10.1002/anie.201204308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatterjee J, Kohn M. Targeting the untargetable: recent advances in the selective chemical modulation of protein phosphatase-1 activity. Current opinion in chemical biology. 2013;17:361–368. doi: 10.1016/j.cbpa.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 43.Reither G, Chatterjee J, Beullens M, Bollen M, Schultz C, Kohn M. Chemical activators of protein phosphatase-1 induce calcium release inside intact cells. Chem Biol. 2013;20:1179–1186. doi: 10.1016/j.chembiol.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 44.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 45.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 46.Fullwood MJ, Zhou W, Shenolikar S. Targeting phosphorylation of eukaryotic initiation factor-2alpha to treat human disease. Progress in molecular biology and translational science. 2012;106:75–106. doi: 10.1016/B978-0-12-396456-4.00005-5. [DOI] [PubMed] [Google Scholar]
- 47.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 48.Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A. 2009;106:1832–1837. doi: 10.1073/pnas.0809632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, Ron D. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163:767–775. doi: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003;23:1292–1303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21:6841–6850. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K, Takeda K, Akira S, Shimokata K, Isobe K. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 53.Mikami S, Kobayashi T, Machida K, Masutani M, Yokoyama S, Imataka H. N-terminally truncated GADD34 proteins are convenient translation enhancers in a human cell-derived in vitro protein synthesis system. Biotechnology letters. 2010;32:897–902. doi: 10.1007/s10529-010-0251-7. [DOI] [PubMed] [Google Scholar]
- 54.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer research. 2013;73:6548–6553. doi: 10.1158/0008-5472.CAN-13-1994. [DOI] [PubMed] [Google Scholar]
- 56.He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013;280:731–750. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future medicinal chemistry. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 58.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, Jensen MR, Gauss CM, Page R, Blackledge M, Muthuswamy SK, Peti W, Tonks NK. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nature chemical biology. 2014;10:558–566. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2014;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]