

Abstract
Rationale
Club cell secretory protein-16 (CC16) is the major secreted product of airway Club cells, but its role in the pathogenesis of COPD is unclear. We measured CC16 airway expression in humans with and without COPD and CC16 function in a cigarette smoke (CS)-induced COPD mice model.
Methods
Airway CC16 expression was measured in COPD patients, smokers without COPD, and non-smokers. We exposed wild-type (WT) and CC16-/- mice to CS or air for up to 6 months, and measured airway CC16 expression, pulmonary inflammation, alveolar septal cell apoptosis, airspace enlargement, airway MUC5AC expression, small airway remodeling, and pulmonary function.
Results
Smokers and COPD patients had reduced airway CC16 immunostaining that decreased with increasing COPD severity. Exposing mice to CS reduced airway CC16 expression. CC16-/- mice had greater CS-induced emphysema, airway remodeling, pulmonary inflammation, alveolar cell apoptosis, airway MUC5AC expression, and more compliant lungs than WT mice. These changes were associated with increased nuclear factor-κB (NFκB) activation in CC16-/- lungs. CS-induced acute pulmonary changes were reversed by adenoviral-mediated over-expression of CC16.
Conclusions
CC16 protects lungs from CS-induced injury by reducing lung NFκB activation. CS-induced airway CC16 deficiency increases CS-induced pulmonary inflammation and injury and likely contributes to the pathogenesis of COPD.
Keywords: Chronic obstructive pulmonary disease, pulmonary inflammation, mucus, apoptosis, Club cells
Introduction
COPD is a major cause of morbidity and mortality worldwide (1). COPD results from a poorly controlled lung inflammatory response to inhaled particles primarily in cigarette smoke (CS) leading to destruction of the alveolar walls and small airway remodeling (2). The severity and progression of COPD are graded by the forced expiratory volume in one second (FEV1) (3).
Faster rate of decline in FEV1 in COPD is linked to current smoker status, higher baseline FEV1, low body mass index (BMI), the degree of CT-determined emphysema, and a history of COPD exacerbations (4-6). In the ECLIPSE study, the only association observed between rate of FEV1 decline and serum levels of various biomarkers was a protective effect associated with higher levels of Club (formerly Clara) cell secretory protein-16 (CC16) (7). This inverse relationship between serum CC16 levels and FEV1 decline was confirmed in a cohort of mild COPD patients (8).
CC16 is also known as CC10, Club cell secretory protein, secretoglobin, family 1A, member 1 (SCGB1A1), and uteroglobin. It is a member of the secretoglobin family of disulphide-bridged dimeric proteins secreted by airway Club cells and is the most abundant protein in normal airway secretions. CC16 maintains the homeostasis of the airway epithelium (9), and has anti-inflammatory activities in lungs exposed to ozone, allergens, and viruses (10-12). Plasma CC16 levels are low in cigarette smokers, and patients with asthma and obliterative bronchiolitis (13-15). Plasma CC16 levels increase following smoking cessation (16) and increases in BAL fluid (BALF) CC16 levels correlate with regression of bronchial dysplasia in former smokers (17). Although a recent study reported that CC16-/- mice developed similar emphysema as wild-type (WT) mice when exposed to CS (8), there are knowledge gaps about the contributions of CC16 to COPD pathogenesis.
We tested the following hypotheses: 1) airway CC16 expression is reduced in smokers without COPD and COPD patients, and in COPD patients it correlates inversely with degree of airflow limitation; 2) airway CC16 levels progressively decrease in WT mice exposed to CS; and 3) when exposed to CS, CC16-deficient (CC16-/-) mice have greater pulmonary inflammation, airspace enlargement, and airway pathologies than WT mice.
Methods
Airway CC16 expression in COPD patients and control subjects
Human studies were approved by institutional review boards and all subjects signed written informed consent forms. Formalin-fixed lung sections were obtained from 6 patients with severe or very severe COPD as part of the Overholt BlueCross Emphysema Surgery Trial (OBEST) for emphysema (18), 6 patients with mild to moderate COPD, 6 healthy non-smokers (never smokers), and 7 healthy active cigarette smokers (> 20 pack/year) without COPD who had undergone lung surgery for benign nodules (see Supplemental Table 1 for demographic and clinical data). None of the subjects had lung cancer. Lung sections were double immunostained for CC16 and a marker of airway epithelial cells (pancytokeratin; see online supplement).
Animals
The Harvard Medical School Institutional Animal Care and Use Committee approved all procedures. C57BL/6 strain CC16-/- mice (19) and C57BL/6 WT control mice (from The Jackson Laboratory, Bar Harbor, USA) were studied.
CS exposures
Adult WT and CC16-/- mice (10 week old) were exposed to air or mixed mainstream and sidestream CS from 3R4F Kentucky Research cigarettes for 2 h/day on 6 days/week in Teague TE 10z chambers for 1-6 months.
Airway immunostaining for CC16, CYP2F2, and MUC5AC
Formalin-fixed lung sections from mice were immunostained for CC16, CYP2F2, and MUC5AC (see online supplement).
Airspace enlargement and airway remodeling
Respiratory mechanics were performed on mice using a mechanical ventilator (Scireq Inc., Montreal, Canada). Airspace size and was measured on Gill's-stained formalin-fixed and inflated lung sections. Small airway remodeling was measured on lung sections stained with Masson's Trichrome stain and immunostained for type-I collagen and fibronectin (see online supplement).
Lung inflammation
Leukocyte subsets were counted in BAL samples from mice. Pro-inflammatory mediators and matrix metalloproteinases (MMPs) were measured in lung samples using ELISAs or western blotting (see online supplement).
Alveolar septal cell apoptosis and lung oxidative stress levels
Alveolar septal cell death was assessed in murine lung section by TUNEL staining and immunostaining for active caspase-3. Oxidative stress levels were measured as thiobarbituric acid reactive substances (TBARS) in lung samples (see online supplement).
Cigarette smoke extract (CSE)-induced apoptosis of murine tracheal epithelial cells (MTECs)
WT and CC16-/- MTECs monolayers were exposed to 30% cigarette smoke extract (CSE) at 37°C and intracellular active caspase-3 levels quantified using a fluorogenic substrate specific for active caspase-3 (see online supplement).
Overexpression of mCC16 in murine airways
Recombinant adenoviral vectors (Vector Biolab, Philadelphia, USA) expressing the cDNA for mCC16 (Ad-CC16) or green fluorescent protein (Ad-GFP) were delivered to the lungs of WT and CC16-/- mice (5 ×107 PFU/mouse) at baseline and every 2 weeks thereafter using the oro-pharyngeal aspiration method. One week after the initial viral dose, mice were exposed to air or CS for 1 month, and BAL leukocytes enumerated or lungs removed for analyses.
Nuclear factor-κB (NFκB) activation and secretory phospholipase A2 (sPLA2) levels in murine lungs
Lung sPLA2 levels were measured using a kit and NFκB activation was measured in nuclear extracts of lungs using an electrophoretic mobility shift assay (EMSA; see online supplement).
Statistical analysis
Statistical analyses were performed using Sigma Stat™ software (SSPS Inc, San Jose, USA). Data shown are means ± SEM (unless otherwise indicated). A P-value ≤ 0.05 was considered significant.
Results
Airway CC16 expression is reduced in human smokers and COPD patients and correlates indirectly with COPD severity
Non-smokers had striking airway CC16 staining whereas staining was less intense in healthy smoker airways (Fig. 1). Airway CC16 staining was lowest in COPD airways (Fig. 1A) and decreased with increasing COPD severity as assessed by GOLD stage (Fig. 1B). Scatter plot analysis revealed modest variability in staining within the subject groups (Supplemental Fig. 1). The age and sex ratios did not differ between the groups (Supplemental Table 1). All of the COPD patients were former smokers. The GOLD stage III-IV COPD patients had greater pack-year smoking histories than the smokers without COPD. Use of inhaled corticosteroids was higher in GOLD stage III-IV than GOLD stage I-II COPD patients.
Figure 1. CC16 expression is reduced in the large airways of COPD patients and expression levels decrease with increasing airflow limitation.
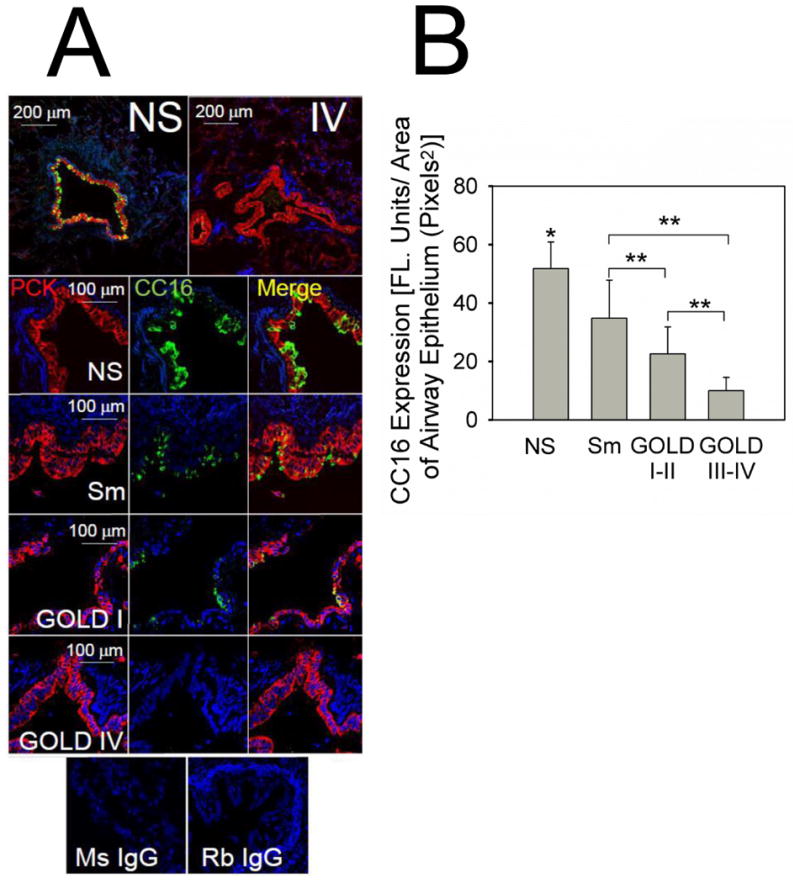
Double immuno-fluorescence staining for CC16 and a marker of airway epithelial cells (pancytokeratin) was performed on lung sections from 6 healthy non-smokers (never smokers, NS), 6 smokers without COPD (Sm), 7 GOLD stage I-II COPD patients (GOLD I-II), and 6 GOLD stage III-IV COPD patients (GOLD III-IV). A shows representative lung sections stained with a red fluorophore (left panels) and murine IgG to pancytokeratin and with a green fluorophore and rabbit IgG to CC16 (middle panels). DAPI counter-stained lung sections were examined using a confocal microscope. Merged images are shown in the right panels. The top panel of A show × 100 magnification merged images of double immunostained stained lung sections from a healthy non-smoker (left) and a very-severe COPD patient (right). The panels below these in A show representative images (magnification × 400-600) of stained lung sections from a healthy non-smoker (NS), a cigarette smoker without COPD (Sm), and a GOLD stage II COPD patient, and a GOLD stage IV COPD patient. Lung sections of a non-smoker control stained with non-immune murine IgG (Ms IgG) or non-immune rabbit IgG (Rb IgG) showed no staining (bottom panel in A). In B, staining for CC16 in airway epithelial cells was quantified in images of the lung sections from the 4 subject groups (NS, Sm, COPD GOLD I-II, and COPD GOLD III-IV), as described in Methods. The size of the airways analyzed was similar in all groups (mean internal diameter was 763.85 ± SEM 174.4, 830.9 ± 110.2, and 808.8± 64.3 μm in the non-smokers, smokers, and COPD patients, respectively). Data are means ± SEM; n = 6-7 subjects/group. Asterisk indicates P < 0.05 versus all other groups; **, p < 0.05.
CS reduces CC16 expression in murine airways
Air-exposed WT mice had robust airway CC16 expression (Fig. 2A-2B). CS exposure caused progressive reductions in airway CC16 expression in mice over time (Fig. 2B). Air-exposed CC16-/- mice had no positive staining for CC16 or CYP2F2, another marker of Club cells (Supplemental Figs. 2-3).
Figure 2. CS exposure reduces the expression of CC16 in murine airways.
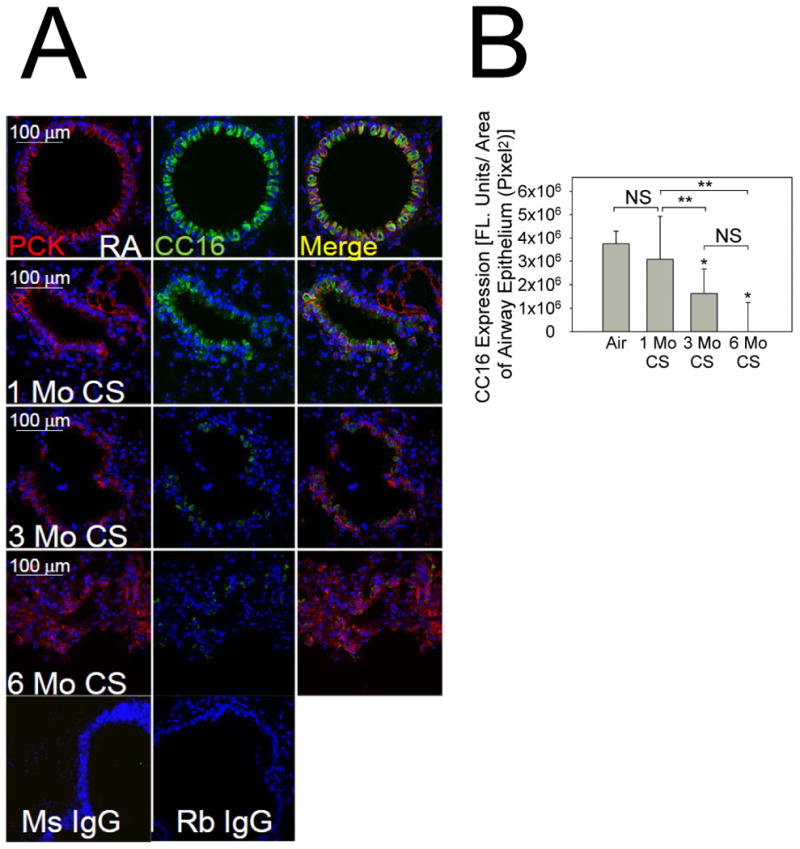
In A, lung sections from C57BL/6 WT mice exposed to room air (RA) or CS for 1 month, 3 months, or 6 months were stained with a red fluorophore and a murine IgG to a marker of airway epithelial cells (pancytokeratin; left panels) and a green fluorophore and a rabbit IgG to CC16 (middle panels). Merged images are shown in the right panels. Lung sections from air-exposed WT mice were stained with non-immune murine IgG (Ms IgG) or non-immune rabbit IgG (Rb IgG). B shows quantitative analysis of airway CC16 normalized per unit of airway area in pixels2. Data are means ± SEM; n = 6 air-exposed and 3-4 CS-exposed mice/group. Asterisk indicates P < 0.001 compared with mice exposed to air and **, P < 0.05.
CC16 deficiency increases emphysema development and airway pathologies in CS-exposed mice
The mean distal airspace size was similar in adult WT and CC16-/-mice that were housed in a room air environment until 10 wks of age and then exposed to air for an additional 1 month (Fig.3A-3B). Thus, CC16 does not regulate lung development in mice. When 10 wk old mice were exposed to air for an additional 6 months, CC16-/- mice had a trend towards increased airspace size compared with WT mice. However, when exposed to CS for 6 months, CC16-/- mice developed 25% increases in airspace size compared with air-exposed CC16-/- mice, while CS-exposed WT mice developed only 13% increases in airspace size compared with air-exposed WT mice (Figs. 3A-3B). Pressure-volume (P-V) flow loops were similar in CC16-/- and WT mice exposed to air for 1 month (data not shown) and 6 months (Fig. 3C). After 6 months of CS exposure, there was a left shift in the P-V flow loops of CS-exposed CC16-/- mice vs. CS-exposed WT mice (Fig. 3D). Quasi-static lung compliance did not differ in WT and CC16-/- mice exposed to air for 1 or 6 months (data not shown) but after 6 months of CS exposure was greater in CC16-/- than WT mice (0.105 ± 0.00805 vs. 0.0966 ± 0.00823 ml/cm H2O; p = 0.036) consistent with the greater emphysema development in CS-exposed CC16-/- vs. WT mice.
Figure 3. CC16-/- mice exposed chronically to CS have greater emphysema and more compliant lungs than CS-exposed WT mice.
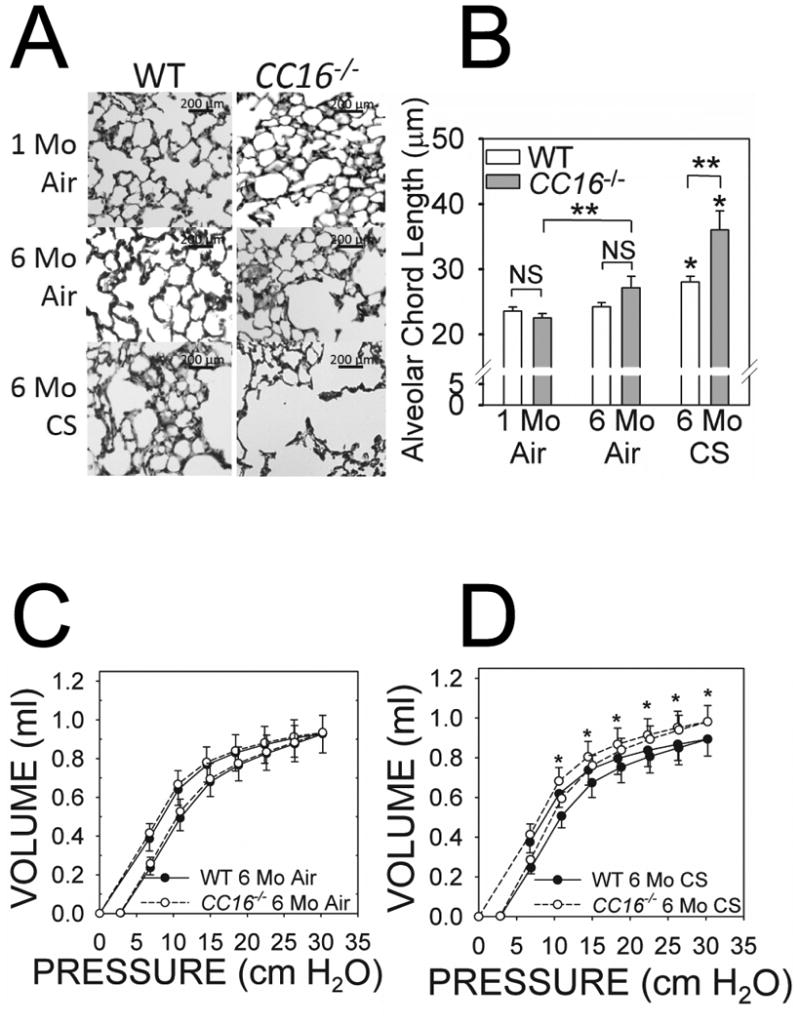
WT and CC16-/- mice were exposed to air for 1 month or 6 months, or to CS for 6 months in a Teague device. Airspace enlargement was measured in lung sections as described in Methods. A shows representative lung sections from each experimental group studied. B shows mean alveolar chord lengths (distance between the alveolar walls) in WT versus CC16-/- mice exposed to air for 1 month (8-9 mice/group) or 6 months (6-15 mice/group), or CS for 6 months (8-9 mice/group). Data are expressed as means ± SEM; asterisk indicates P < 0.05 compared with mice exposed to air for 6 months belonging to the same genotype; **, P < 0.05. In C-D, a Flexivent device was used to measure pressure-volume (PV) flow loops on WT and CC16-/- mice exposed to air for 6 months (9-10 mice/group) or CS for 6 months (9-10 mice/group). In D, asterisk indicates P < 0.05 versus the corresponding data point on the WT PV flow loop.
There was a trend (p = 0.068) towards increased deposition of extracellular matrix (ECM) proteins around small airways in adult CC16-/- mice versus WT mice exposed to air for 1 month (Fig. 4A-4B). When mice were exposed to air for 6 months, this small airway remodeling was modestly (∼23%) greater in CC16-/- than WT mice. However, there was a much greater (∼53%) increase in small airway remodeling in CC16-/- mice vs. WT mice exposed to CS for 6 months (Fig. 4A-4B). CS-exposed CC16-/- mice also had greater staining for type-I collagen (Supplemental Fig. 4), fibronectin (Fig. 4C) around the small airways and greater airway epithelial MUC5AC immunostaining (Fig. 4D) than CS-exposed WT mice.
Figure 4. Small airway remodeling is greater in CC16-/- mice compared with WT mice exposed to CS.
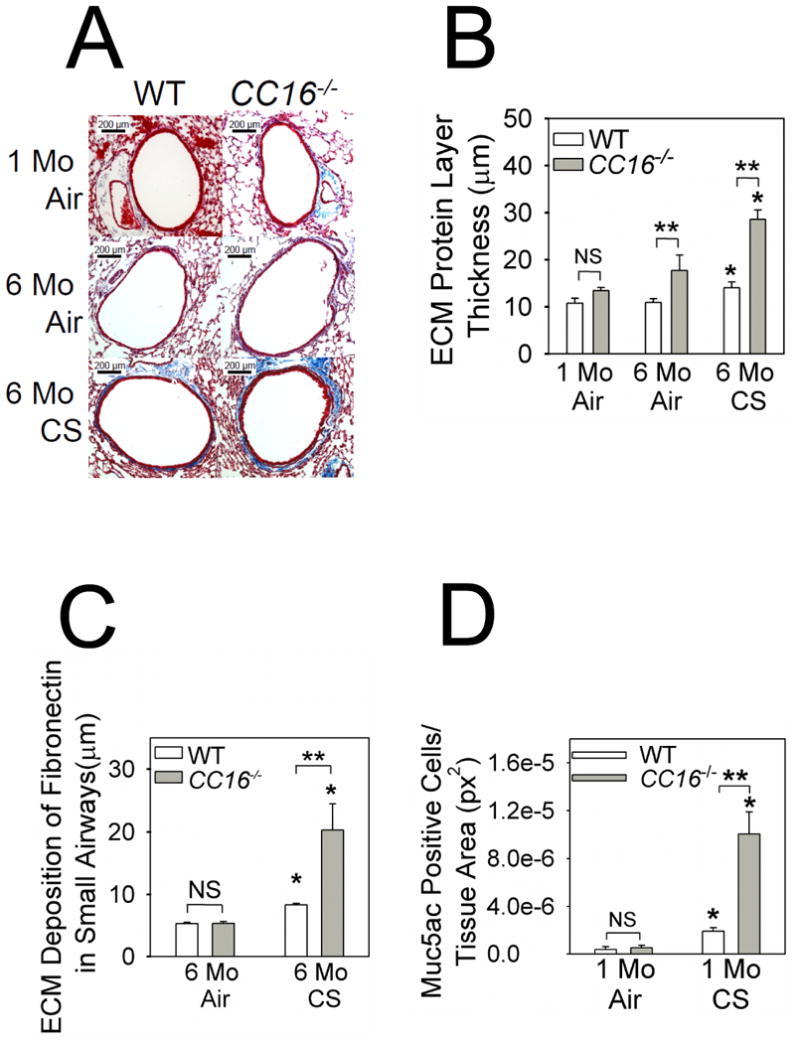
Starting at 10 weeks of age, WT and CC16-/- mice were exposed to air or CS for 6 months. Deposition of extracellular matrix (ECM) proteins around small airways (mean diameter between 300 and 699 μm) was quantified in Masson's Trichrome-stained lung sections. A shows representative stained lung sections from each experimental group. ECM proteins deposited around the small airways are stained blue. In B, the thickness of the ECM protein layer deposited around small airways (airways having an internal diameter from 300-699 μm) was quantified as described in Methods. Data are means ± SEM; n= 6-7 mice exposed to air for 1 month, n = 5-9 mice exposed to air for 6 months, and n = 7-8 mice exposed to CS for 6 months. Asterisk indicates P < 0.05 compared with mice exposed to air for 6 months belonging to the same genotype; **, P < 0.05. In C, lung sections from WT and CC16-/- mice exposed to air (3-4 mice/group) or CS (4 mice/group) for 6 months were stained for fibronectin as described in methods. The thickness of the layer of fibronectin deposited around small airways (internal diameter from 300-699 μm) was quantified. Asterisk indicates p < 0.05 compared with mice exposed to air belonging to the same genotype; **, p < 0.05. In D, lung sections from WT and CC16-/- mice exposed to air (3-5 mice/group) or CS (3-5 mice/group) for 1 month were immunostained for MUC5AC as described in Methods. The proportion of all bronchial epithelial cells that stained positively for MUC5AC was quantified. Data are means ± SEM; asterisk indicates P < 0.05 compared with air-exposed mice belonging to the same genotype and **, P < 0.01.
CC16 deficiency increases pulmonary inflammation, MMP-9 levels, and alveolar septal cell apoptosis in CS-exposed mice
Air-exposed CC16-/- mice had modestly greater BAL total leukocyte, macrophage, and PMN counts than air-exposed WT mice (Fig. 5A-5C). CS-exposed CC16-/- mice had higher BAL total leukocyte, macrophage, and PMN counts than CS-exposed WT mice at all time-points assessed (Fig. 5A-5C), but WT and CC16-/- mice did not differ in BAL lymphocyte counts (data not shown). Compared with CS-exposed WT mice, CS-exposed CC16-/- mice had higher lung levels of CCL5 and active transforming growth factor-β1 (TGF-β1), lower lung levels of interleukin-10, but similar lung levels of other pro-inflammatory mediators (Supplemental Table 2). CS induced greater increases in lung MMP-9 (but not MMP-12) levels in CC16-/- mice than WT mice (Supplemental Fig. 5).
Figure 5. Lung inflammation is increased in CS-exposed CC16-/- mice.
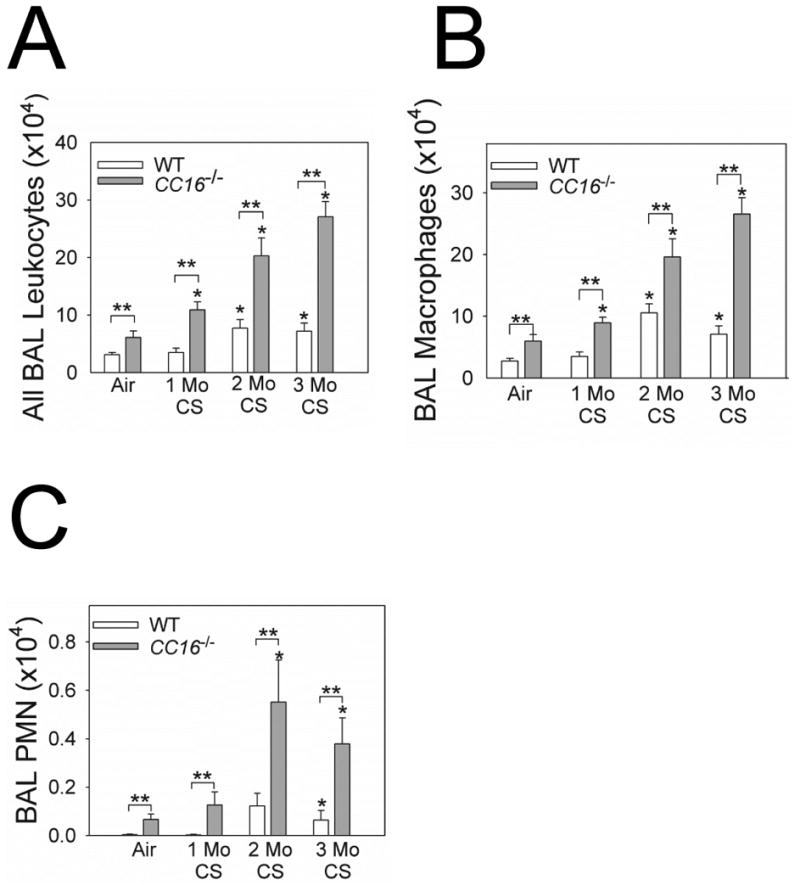
WT and CC16-/- mice were exposed to air or CS for 1-3 months. Absolute numbers of all leukocytes (A), macrophages (B), and PMNs (C) were counted in BAL samples. Data are mean ± SEM; n = 7 air-exposed mice, n = 7-10 mice exposed to CS for 1 month; n = 8 mice exposed to CS for 2 months, and n = 5-14 mice exposed to CS for 3 months. Asterisk indicates P < 0.05 compared with air-exposed mice belonging to the same genotype; and **, P < 0.05.
CS exposed CC16-/- mice had increased numbers of apoptotic bronchial epithelial cells (Fig. 6A) and apoptotic alveolar septal cells (Fig. 6B-6C) as assessed by TUNEL staining and/or staining for active caspase-3. However, CSE induced similar rates of apoptosis in CC16-/- and WT MTEC cultures in vitro (Supplemental Fig. 6). Lung oxidative stress levels were similar in CS-exposed CC16-/- and WT mice measured as lung levels of TBARS (a readout of lipid peroxidation; not shown).
Figure 6. Apoptosis rates are increased in bronchial epithelial and alveolar septal cells in CC16-/- mice exposed to CS.
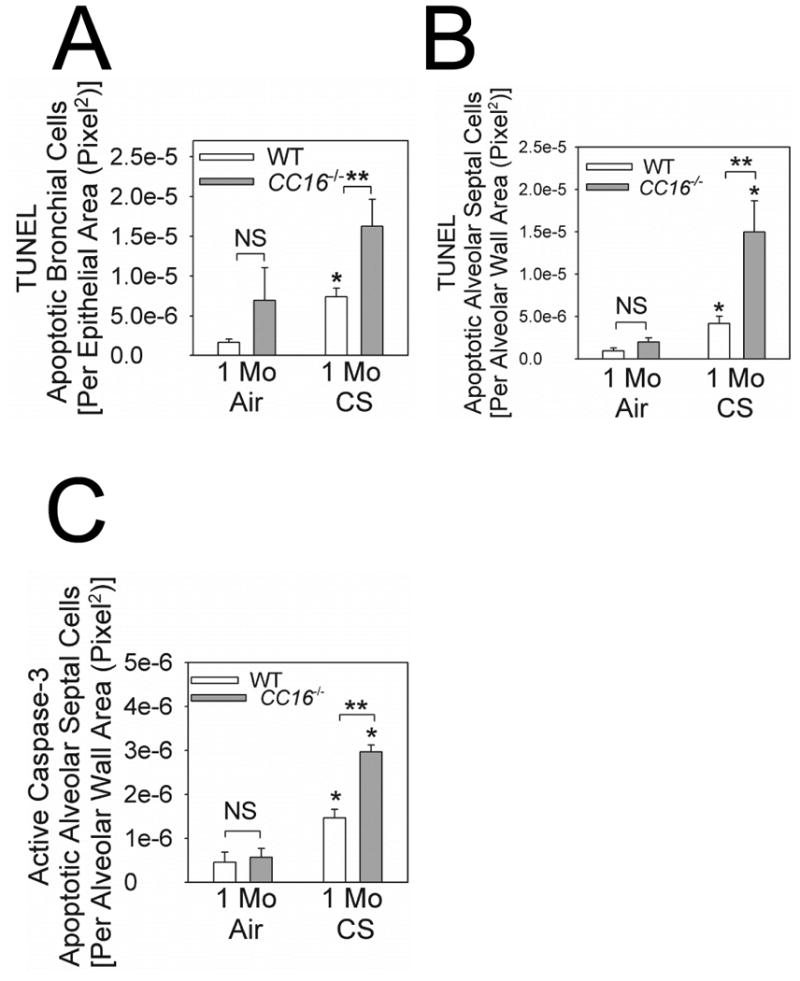
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and immunostaining for active (cleaved) caspase-3 were performed on formalin-fixed lung sections from WT vs. CC16-/- mice exposed to air or CS for 1 month. In A, TUNEL-positive cells were quantified in large and medium-sized airways from 3 air-exposed WT or CC16-/- mice and 4 CS-exposed WT or CC16-/- mice. In B, TUNEL-positive alveolar septal cells were counted and counts were normalized to unit area of alveolar wall in 4 air-exposed WT or CC16-/- mice and 4 CS-exposed WT or CC16-/- mice. In C, alveolar septal cells that stained positively for active (cleaved) caspase-3 were counted and counts were normalized per unit area of alveolar wall in 4 mice per experimental condition. In A-C, data are means ± SEM; asterisk indicates P < 0.05 compared with air-exposed mice belonging to the same genotype; ** P < 0.05.
CC16 deficiency increases activation of NFkB but not sPLA2 levels in CS-exposed lungs
CC16 inhibits two pro-inflammatory pathways in other model systems: NFκB activation (20) and sPLA2 activity by binding co-factors for this enzyme (21). When we measured these pathways, air-exposed CC16-/- mice had modestly increased NFκB activation in their lungs as assessed by EMSA. However, CS-exposed CC16-/- mice had greater NFκB activation in their lungs than CS-exposed WT mice (Figs. 7A-7B). Although air-exposed CC16-/- mice had higher lung levels of active sPLA2 than air-exposed WT mice, sPLA2 levels were similar in lung homogenates (Fig. 7C) and BALF (Supplemental Fig. 7) from CS-exposed WT and CC16-/- mice.
Figure 7. NFκB activation is increased in the lungs of CS-exposed CC16-/- mice.
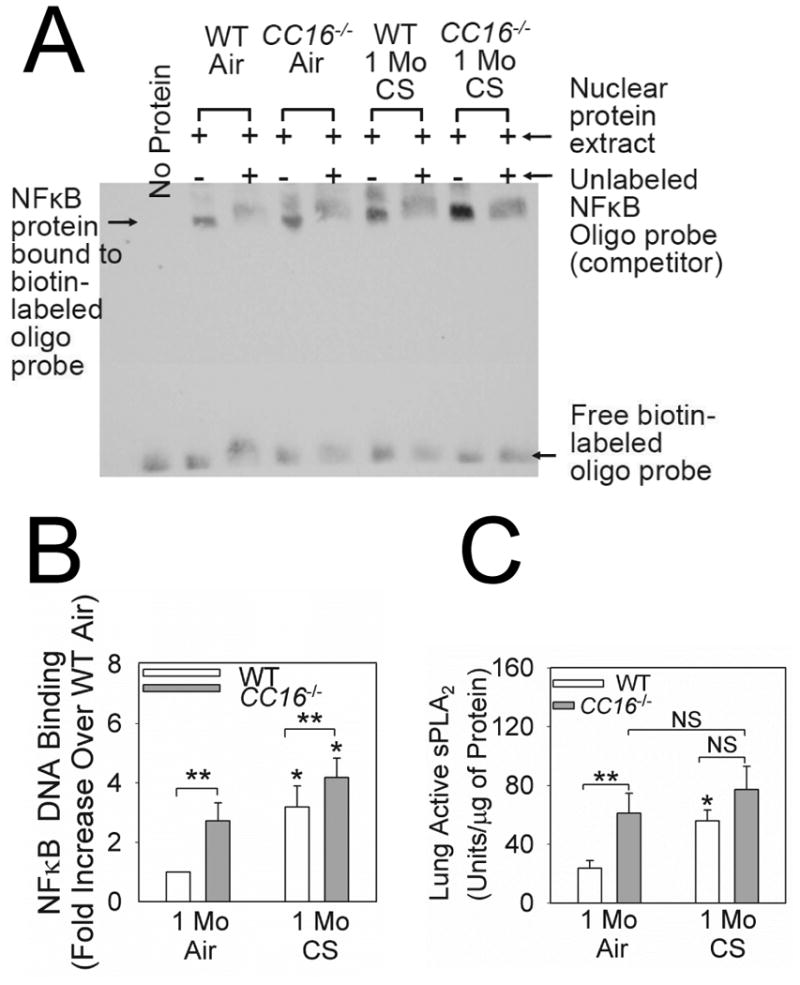
WT and CC16-/- mice were exposed to air or CS for 1 month. Nuclear extracts were prepared from the lungs of 5 WT or CC16-/- mice exposed to air and 6 WT or CC16-/- mice exposed to CS and equal amounts of protein (2 μg/sample) were subjected to electrophoretic mobility shifts assays (EMSAs) using a labeled oligonucleotide probe containing the NFκB consensus sequence. Assays were performed in the presence and absence of excess unlabeled probe to identify NFκB protein bound specifically to the probe. A shows a representative image of an EMSA analysis of nuclear extracts from all experimental groups. Note the marked reduction in signal in the band indicated by the arrow when excess unlabeled probe is added indicating specific binding of NFκB present in nuclear extracts to the labeled oligonucleotide probe. In B, the intensities of the bands corresponding to NFκB-oligonucleotide complexes were quantified using densitometry, and band intensities for all groups were normalized to signals in the air-exposed WT lungs. Data are means ± SEM from six independent experiments. Asterisk indicates P < 0.01 compared with air-exposed mice belonging to the same genotype and **, P < 0.05. In C, lung levels of active sPLA2 were measured in homogenates of lungs from mice exposed to air or CS for 1 month using a commercial kit, and levels were normalized to lung total protein levels. Data are means ± SEM; n =10 air-exposed mice and n = 11-12 CS-exposed mice. Asterisk indicates P < 0.05 compared with air-exposed mice belonging to the same genotype and ** P < 0.01.
Adenoviral-mediated CC16 overexpression in murine airways reduces pulmonary pathologies induced by acute CS exposure
Delivering Ad-CC16 to the airways of CC16-/- mice induced airway immunostaining for CC16 (Supplemental Fig. 8). Delivering Ad-CC16 to both CC16-/- and WT lungs attenuated CS-induced increases in BAL macrophage (but not PMN) counts in both genotypes compared with macrophage counts in Ad-GFP-treated mice (Fig. 8A or not shown). Ad-CC16-treated CC16-/- and WT mice had lower CS-induced airway MUC5AC immunostaining, alveolar septal cell apoptosis, and lung NFκB activation (Fig. 8B-8D), but similar lung levels of sPLA2 activity (data not shown) than Ad-GFP-treated mice belonging to the same genotype.
Figure 8. Adenoviral-mediated CC16 overexpression in murine airways decreases pulmonary inflammation, alveolar septal cell apoptosis, and airway mucus metaplasia.
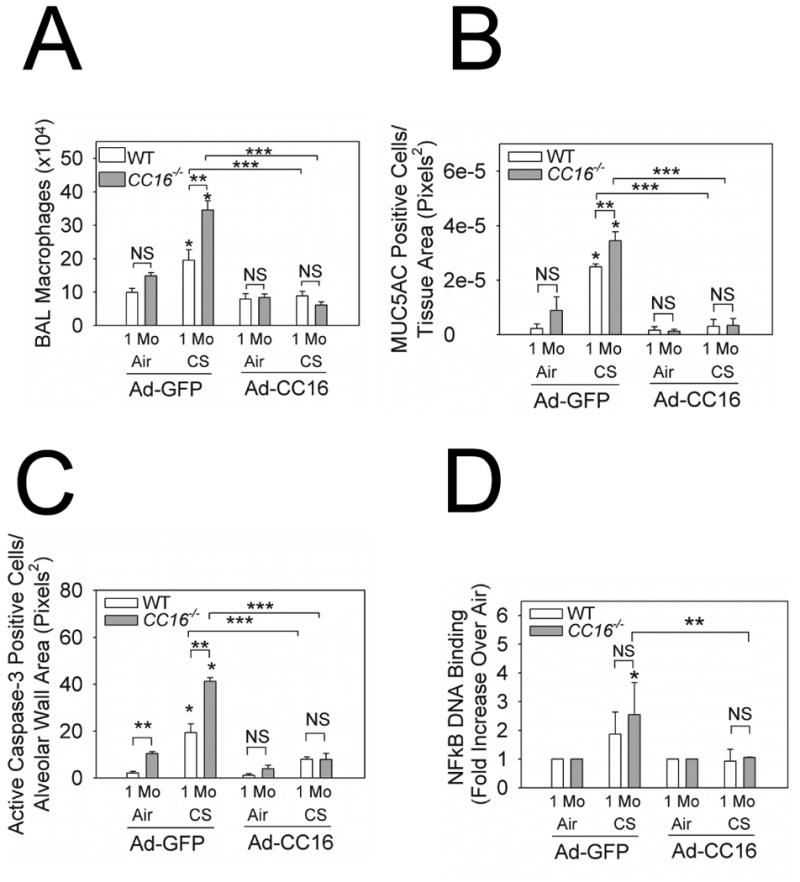
Ad-CC16 or Ad-GFP were delivered to WT and CC16-/- mice and 1 week later, the mice were exposed to air or CS for 1 month. Adenoviral vector delivery was repeated every 2 weeks for the duration of the CS exposures. In A, macrophage numbers were counted in BAL samples. Data are means ± SEM; n = 7-10 mice treated with Ad-GFP and exposed to air, n = 9 mice treated with Ad-GFP exposed to CS; n = 8-10 treated with Ad-CC16 and exposed to air, n = 7-9 mice treated with Ad-CC16 and exposed to CS. In B, the number of alveolar septal cells staining positively for active (cleaved) caspase-3 was counted and counts were normalized per unit area of alveolar wall. Data are means ± SEM; n = 4 mice/group. In C, lung sections from Ad-CC16- or Ad-GFP-treated WT or CC16-/- mice exposed to air or CS for 1 month (4 mice/group) were immunostained for MUC5AC. The number of bronchial epithelial cells that stained positively for MUC5AC was quantified and normalized per unit area of airway epithelium. Data are mean ± SEM; asterisk indicates P < 0.05 compared with air-exposed mice belonging to the same genotype and treated with the same adenoviral vector; ** P < 0.05 and ***, P < 0.05 compared with CS-exposed mice belonging to the same genotype and treated with Ad-CC16 vs. Ad-GFP
Discussion
We report several novel findings. First, airway CC16 expression was lower in COPD patients than smokers and non-smokers and indirectly correlated with airflow obstruction. Second, CS exposure progressively reduced airway CC16 expression in WT mice. Third, CS-exposed CC16-/- mice developed greater pulmonary inflammation, alveolar septal cell apoptosis, airway mucus metaplasia, emphysema, and small airway remodeling, associated with greater NFκB activation in their lungs than WT mice. Pulmonary pathologies induced by acute CS exposure were attenuated by adenoviral-mediated over-expression of CC16 in murine airways. Thus, CC16 has important roles in protecting murine lungs from the development and progression of CS-induced COPD-like pathologies in mice (at least in part) by reducing activation of NFκB in the lung which has been implicated in the pathogenesis of COPD (22).
Human studies
Only one prior study has reported decreased airway CC16 expression in severe COPD patients versus cigarette smokers (23). We now link airway CC16 deficiency to smoking in human subjects and the severity of airflow limitation in COPD. Reduced airway CC16 expression in COPD lungs may be persistent as our very severe COPD patients had stopped smoking well before the tissue was obtained. However, a prior study reported that serum CC16 levels recover somewhat when smokers without COPD quit smoking (16). The differences in these findings could be due to differences in study populations, the samples studied, or the length of time since smoking cessation.
Murine studies
CS exposure progressively reduced murine airway CC16 expression which could be due to reduced synthesis of CC16 by Club cells or loss of airway Club cells (24). Club cells have the highest levels of cytochrome P450 in the lung and are the main site of lung detoxification of xenobiotics (25). Murine Club cells are sensitive to injury following inhalation of naphthalene [a component of CS (26)] or CS itself (24). Polymorphisms in the CC16 locus were weakly linked to COPD in the ECLIPSE cohort and associated with low plasma CC16 levels, but these findings were not replicated in other smaller COPD cohorts having different inclusion criteria (27). Additionally, the CC16 locus is hyper-methylated in COPD bronchial epithelia suggesting that epigenetic factors influence CC16 expression (28). Although CS reduced airway CC16 expression in WT mice, some CC16 was detected after 3 months of CS exposure that likely was sufficient to protect the WT lung from developing the more severe lung inflammatory response and airway and airspace disease observed in CS-exposed CC16-/- mice.
CC16 protects the murine lung from CS-induced pulmonary inflammation, emphysema development, small airway remodeling, and airway mucus metaplasia. Likely, the protective effect of CC16 in the CS-exposed murine lung is due to CC16 reducing lung macrophage and PMN counts and protecting alveolar septal cells from CS-induced apoptosis. CC16 secreted by Club cells into the epithelial lining fluid likely has paracrine crytoprotective effects on other lung epithelial cells as Club cell-free WT and CC16-/- lung epithelial cells had similar rates of CSE-induced apoptosis in vitro.
NFκB activation was increased CS-exposed CC16-/- versus WT lungs (Fig. 7) and was reduced in both CS-exposed WT and CC16-/- lung by over-expressing CC16 in their airways indicating that CC16 mediates its activities (at least in part) in the CS-exposed lung by reducing NFκB activation which promotes inflammation in COPD lungs (22). Ad-GFP-treated WT and CC16-/- mice did not differ in the extent to which CS increased NFκB activation in their lungs which may due to be virus-induced NFκB activation, but virus-mediated over-expression of CC16 attenuated NFκB activation in the lungs of CS-exposed WT and CC16-/- mice. CC16 signals through the fMLP receptor on granulocytes (29), the lipocalin-1 receptor on lung epithelial carcinoma cells (30), and cubilin in the kidney (31), and reduces macrophage toll-like receptor 4 levels (32). CC16 also inhibits sPLA2 activity in other models by binding its co-factors (21) and elevated sPLA2 BALF levels correlate with a proinflammatory phenotype in COPD patients (33). While CC16-/- mice had higher baseline lung sPLA2 activity levels, CS-exposed WT and CC16-/- mice had similar lung levels of sPLA2. The increased baseline sPLA2 levels may have contributed to the increased BAL leukocyte counts in air-exposed CC16-/- mice. However, CC16 could mediate some of its anti-inflammatory effects in the CS-exposed lung by inhibiting sPLA2 activity as measuring this mediator in whole lung or BALF samples may dilute sPLA2 signals generated by subpopulations of pulmonary cells.
CC16 may reduce airway ECM deposition in CS-exposed lungs by reducing lung levels of active TGF-1β as levels of this mediator were higher in CS-exposed CC16-/- vs. WT lungs. CC16 could also reduce small airway remodeling and airway MUC5AC expression by restraining pulmonary inflammation as leukocyte products contribute to airway remodeling and mucin gene expression in rodents (34-36).
Our findings differ from those recently reported by Park et al. who reported that pulmonary inflammation, emphysema, and airway remodeling are similar in CS-exposed WT and CC16-/- mice (8). Different CC16-/- strains vary in the severity of renal phenotypes detected (37-39) due to differences in their genetic backgrounds and/or the targeting constructs used to generate the mice. However, both studies evaluated the same CC16-/- murine stain (19) (personal communication, Don Sin, MD). Likely, the differences in the results of the two studies reflect differences in the methods used. For example, we exposed mice to whole body mixed mainstream and side-stream CS for 2 h per day for 6 days-a-week, whereas Park et al exposed the mice to mainstream CS from 3 cigarettes 5 days-a week using a nose-only technique. However, we also over-expressed CC16 in the airways of mice using adenoviral vectors which reduced CS-induced acute pulmonary changes consistent with our hypothesis that CC16 has anti-inflammatory activities in the CS-exposed lung.
We observed small increase in pulmonary inflammation and small airway remodeling in air-exposed CC16-/- mice versus WT mice. Small increases in lung leukocyte counts were detected after just 1 month of air exposure. Thus, deficiency of CC16 in the absence of CS is sufficient to cause mild pulmonary inflammation which may contribute to the modestly greater small airway remodeling in air-exposed CC16-/- vs. WT mice. Thus, CC16 has anti-inflammatory activities even in the unchallenged lung.
Limitations of our study include our relatively small sample sizes. Nevertheless, we achieved statistically significant differences between our groups and scatter-plot analysis revealed modest within-group variability in airway CC16 staining in our human subjects. The GOLD stage III-IV COPD patients had greater pack-year smoking histories than the smoker controls. All of the smokers but none of the COPD patients were current smokers which could have influenced airway CC16 staining. The use of inhaled corticosteroids by some of the COPD patients may have increased their airway CC16 expression as CC16 is steroid-responsive gene (40).
Conclusions
CS exposure reduces airway CC16 expression leading to increased pulmonary inflammation, alveolar septal cell apoptosis, mucus metaplasia, and also emphysema development and small airway remodeling which both contribute to airflow obstruction in COPD patients. Pulmonary pathologies induced by acute CS exposure are reversed by adenoviral-mediated over-expression of CC16 in both WT and CC16-/- airways. Thus, CC16 protects lungs from CS-induced injury. Future studies will determine whether CC16 can be used as a novel therapy for COPD.
Supplementary Material
References
- 1.Murray CJ, Lopez AD. Measuring the Global Burden of Disease. N Engl J Med. 2013;369:448–457. doi: 10.1056/NEJMra1201534. [DOI] [PubMed] [Google Scholar]
- 2.Owen CA. Roles for Proteinases in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int J Chron Obstruct Pulmon Dis. 2008;3:253–268. doi: 10.2147/copd.s2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swanney MP, Ruppel G, Enright PL, Pedersen OF, Crapo RO, Miller MR, Jensen RL, Falaschetti E, Schouten JP, Hankinson JL, et al. Using the Lower Limit of Normal for the FEV1/FVC Ratio Reduces the Misclassification of Airway Obstruction. Thorax. 2008;63:1046–1051. doi: 10.1136/thx.2008.098483. [DOI] [PubMed] [Google Scholar]
- 4.Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, Calverley PM, Celli B, Coxson HO, Crim C, et al. Changes in Forced Expiratory Volume in 1 Second Over Time in COPD. N Engl J Med. 2011;365:1184–1192. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
- 5.Casanova C, de Torres JP, Aguirre-Jaime A, Pinto-Plata V, Marin JM, Cordoba E, Baz R, Cote C, Celli BR. The Progression of Chronic Obstructive Pulmonary Disease Is Heterogeneous: the Experience of the BODE Cohort. Am J Respir Crit Care Med. 2011;184:1015–1021. doi: 10.1164/rccm.201105-0831OC. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura M, Makita H, Nagai K, Konno S, Nasuhara Y, Hasegawa M, Shimizu K, Betsuyaku T, Ito YM, Fuke S, et al. Annual Change in Pulmonary Function and Clinical Phenotype in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;185:44–52. doi: 10.1164/rccm.201106-0992OC. [DOI] [PubMed] [Google Scholar]
- 7.Lomas DA, Silverman EK, Edwards LD, Miller BE, Coxson HO, Tal-Singer R. Evaluation of Serum CC-16 As a Biomarker for COPD in the ECLIPSE Cohort. Thorax. 2008;63:1058–1063. doi: 10.1136/thx.2008.102574. [DOI] [PubMed] [Google Scholar]
- 8.Park HY, Churg A, Wright JL, Li Y, Tam S, Man SF, Tashkin D, Wise RA, Connett JE, Sin DD. Club Cell Protein 16 and Disease Progression in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2013;188:1413–1419. doi: 10.1164/rccm.201305-0892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stripp BR, Reynolds SD, Boe IM, Lund J, Power JH, Coppens JT, Wong V, Reynolds PR, Plopper CG. Clara Cell Secretory Protein Deficiency Alters Clara Cell Secretory Apparatus and the Protein Composition of Airway Lining Fluid. Am J Respir Cell Mol Biol. 2002;27:170–178. doi: 10.1165/ajrcmb.27.2.200200270c. [DOI] [PubMed] [Google Scholar]
- 10.Dodge DE, Rucker RB, Pinkerton KE, Haselton CJ, Plopper CG. Dose-Dependent Tolerance to Ozone. III. Elevation of Intracellular Clara Cell 10-KDa Protein in Central Acini of Rats Exposed for 20 Months. Toxicol Appl Pharmacol. 1994;127:109–123. doi: 10.1006/taap.1994.1145. [DOI] [PubMed] [Google Scholar]
- 11.Chen LC, Zhang Z, Myers AC, Huang SK. Cutting Edge: Altered Pulmonary Eosinophilic Inflammation in Mice Deficient for Clara Cell Secretory 10-KDa Protein. J Immunol. 2001;167:3025–3028. doi: 10.4049/jimmunol.167.6.3025. [DOI] [PubMed] [Google Scholar]
- 12.Wang SZ, Rosenberger CL, Bao YX, Stark JM, Harrod KS. Clara Cell Secretory Protein Modulates Lung Inflammatory and Immune Responses to Respiratory Syncytial Virus Infection. J Immunol. 2003;171:1051–1060. doi: 10.4049/jimmunol.171.2.1051. [DOI] [PubMed] [Google Scholar]
- 13.Bernard AM, Roels HA, Buchet JP, Lauwerys RR. Serum Clara Cell Protein: an Indicator of Bronchial Cell Dysfunction Caused by Tobacco Smoking. Environ Res. 1994;66:96–104. doi: 10.1006/enrs.1994.1047. [DOI] [PubMed] [Google Scholar]
- 14.Laing IA, Hermans C, Bernard A, Burton PR, Goldblatt J, Le Souef PN. Association Between Plasma CC16 Levels, the A38G Polymorphism, and Asthma. Am J Respir Crit Care Med. 2000;161:124–127. doi: 10.1164/ajrccm.161.1.9904073. [DOI] [PubMed] [Google Scholar]
- 15.Mattsson J, Remberger M, Andersson O, Sundberg B, Nord M. Decreased Serum Levels of Clara Cell Secretory Protein (CC16) Are Associated With Bronchiolitis Obliterans and May Permit Early Diagnosis in Patients After Allogeneic Stem-Cell Transplantation. Transplantation. 2005;79:1411–1416. doi: 10.1097/01.tp.0000158354.39635.ab. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Lam S, Pilon A, McWilliams A, Melby J, Szabo E. The Association Between the Anti-Inflammatory Protein CC10 and Smoking Status Among Participants in a Chemoprevention Trial. Cancer Epidemiol Biomarkers Prev. 2007;16:577–583. doi: 10.1158/1055-9965.EPI-06-0923. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Lam S, Pilon A, McWilliams A, Macaulay C, Szabo E. Higher Levels of the Anti-Inflammatory Protein CC10 Are Associated With Improvement in Bronchial Dysplasia and Sputum Cytometric Assessment in Individuals at High Risk for Lung Cancer. Clin Cancer Res. 2008;14:1590–1597. doi: 10.1158/1078-0432.CCR-07-4066. [DOI] [PubMed] [Google Scholar]
- 18.Berger RL, Celli BR, Meneghetti AL, Bagley PH, Wright CD, Ingenito EP, Gray A, Snider GL. Limitations of Randomized Clinical Trials for Evaluating Emerging Operations: the Case of Lung Volume Reduction Surgery. Ann Thorac Surg. 2001;72:649–657. doi: 10.1016/s0003-4975(01)02636-4. [DOI] [PubMed] [Google Scholar]
- 19.Stripp BR, Lund J, Mango GW, Doyen KC, Johnston C, Hultenby K, Nord M, Whitsett JA. Clara Cell Secretory Protein: a Determinant of PCB Bioaccumulation in Mammals. Am J Physiol. 1996;271:L656–L664. doi: 10.1152/ajplung.1996.271.4.L656. [DOI] [PubMed] [Google Scholar]
- 20.Long XB, Hu S, Wang N, Zhen HT, Cui YH, Liu Z. Clara Cell 10-KDa Protein Gene Transfection Inhibits NF-KappaB Activity in Airway Epithelial Cells. PLoS ONE. 2012;7:e35960. doi: 10.1371/journal.pone.0035960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersson O, Nordlund-Moller L, Barnes HJ, Lund J. Heterologous Expression of Human Uteroglobin/Polychlorinated Biphenyl-Binding Protein Determination of Ligand Binding Parameters and Mechanism of Phospholipase A2 Inhibition in Vitro. J Biol Chem. 1994;269:19081–19087. [PubMed] [Google Scholar]
- 22.Gagliardo R, Chanez P, Profita M, Bonanno A, Albano GD, Montalbano AM, Pompeo F, Gagliardo C, Merendino AM, Gjomarkaj M. IkappaB Kinase-Driven Nuclear Factor-KappaB Activation in Patients With Asthma and Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol. 2011;128:635–645. doi: 10.1016/j.jaci.2011.03.045. [DOI] [PubMed] [Google Scholar]
- 23.Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, De PK, Vaerman JP, Decramer M, Sibille Y. Reduced Epithelial Expression of Secretory Component in Small Airways Correlates With Airflow Obstruction in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2001;163:185–194. doi: 10.1164/ajrccm.163.1.9912137. [DOI] [PubMed] [Google Scholar]
- 24.Adair-Kirk TL, Atkinson JJ, Griffin GL, Watson MA, Kelley DG, DeMello D, Senior RM, Betsuyaku T. Distal Airways in Mice Exposed to Cigarette Smoke: Nrf2-Regulated Genes Are Increased in Clara Cells. Am J Respir Cell Mol Biol. 2008;39:400–411. doi: 10.1165/rcmb.2007-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plopper CG, Cranz DL, Kemp L, Serabjit-Singh CJ, Philpot RM. Immunohistochemical Demonstration of Cytochrome P-450 Monooxygenase in Clara Cells Throughout the Tracheobronchial Airways of the Rabbit. Exp Lung Res. 1987;13:59–68. doi: 10.3109/01902148709064309. [DOI] [PubMed] [Google Scholar]
- 26.Van Winkle LS, Johnson ZA, Nishio SJ, Brown CD, Plopper CG. Early Events in Naphthalene-Induced Acute Clara Cell Toxicity: Comparison of Membrane Permeability and Ultrastructure. Am J Respir Cell Mol Biol. 1999;21:44–53. doi: 10.1165/ajrcmb.21.1.3630. [DOI] [PubMed] [Google Scholar]
- 27.Kim DK, Cho MH, Hersh CP, Lomas DA, Miller BE, Kong X, Bakke P, Gulsvik A, Agusti A, Wouters E, et al. Genome-Wide Association Analysis of Blood Biomarkers in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;186:1238–1247. doi: 10.1164/rccm.201206-1013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buro-Auriemma LJ, Salit J, Hackett NR, Walters MS, Strulovici-Barel Y, Staudt MR, Fuller J, Mahmoud M, Stevenson CS, Hilton H, et al. Cigarette Smoking Induces Small Airway Epithelial Epigenetic Changes With Corresponding Modulation of Gene Expression. Hum Mol Genet. 2013;22:4726–4738. doi: 10.1093/hmg/ddt326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johansson S, Andersson K, Wennergren G, Wenneras C, Rudin A. CC16 Inhibits the Migration of Eosinophils Towards the Formyl Peptide FMLF but Not Towards PGD2. Inflammation. 2009;32:65–69. doi: 10.1007/s10753-008-9103-1. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Kim SJ, Chowdhury B, Wang J, Lee YC, Tsai PC, Choi M, Mukherjee AB. Interaction of Uteroglobin With Lipocalin-1 Receptor Suppresses Cancer Cell Motility and Invasion. Gene. 2006;369:66–71. doi: 10.1016/j.gene.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 31.Burmeister R, Boe IM, Nykjaer A, Jacobsen C, Moestrup SK, Verroust P, Christensen EI, Lund J, Willnow TE. A Two-Receptor Pathway for Catabolism of Clara Cell Secretory Protein in the Kidney. J Biol Chem. 2001;276:13295–13301. doi: 10.1074/jbc.M010679200. [DOI] [PubMed] [Google Scholar]
- 32.Snyder JC, Reynolds SD, Hollingsworth JW, Li Z, Kaminski N, Stripp BR. Clara Cells Attenuate the Inflammatory Response Through Regulation of Macrophage Behavior. Am J Respir Cell Mol Biol. 2010;42:161–171. doi: 10.1165/rcmb.2008-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pniewska E, Pawliczak R. The Involvement of Phospholipases A2 in Asthma and Chronic Obstructive Pulmonary Disease. Mediators Inflamm. 2013;2013:793505. doi: 10.1155/2013/793505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Churg A, Zhou S, Wang X, Wang R, Wright JL. The Role of Interleukin-1beta in Murine Cigarette Smoke-Induced Emphysema and Small Airway Remodeling. Am J Respir Cell Mol Biol. 2009;40:482–490. doi: 10.1165/rcmb.2008-0038OC. [DOI] [PubMed] [Google Scholar]
- 35.Churg A, Marshall CV, Sin DD, Bolton S, Zhou S, Thain K, Cadogan EB, Maltby J, Soars MG, Mallinder PR, et al. Late Intervention With a Myeloperoxidase Inhibitor Stops Progression of Experimental Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;185:34–43. doi: 10.1164/rccm.201103-0468OC. [DOI] [PubMed] [Google Scholar]
- 36.Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/MMP-12 Inhibitor on Smoke-Induced Emphysema and Airway Remodelling in Guinea Pigs. Thorax. 2007;62:706–713. doi: 10.1136/thx.2006.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds SD, Mango GW, Gelein R, Boe IM, Lund J, Stripp BR. Normal Function and Lack of Fibronectin Accumulation in Kidneys of Clara Cell Secretory Protein/Uteroglobin Deficient Mice. Am J Kidney Dis. 1999;33:541–551. doi: 10.1016/s0272-6386(99)70192-7. [DOI] [PubMed] [Google Scholar]
- 38.Zheng F, Kundu GC, Zhang Z, Ward J, DeMayo F, Mukherjee AB. Uteroglobin Is Essential in Preventing Immunoglobulin A Nephropathy in Mice. Nat Med. 1999;5:1018–1025. doi: 10.1038/12458. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Kundu GC, Yuan CJ, Ward JM, Lee EJ, DeMayo F, Westphal H, Mukherjee AB. Severe Fibronectin-Deposit Renal Glomerular Disease in Mice Lacking Uteroglobin. Science. 1997;276:1408–1412. doi: 10.1126/science.276.5317.1408. [DOI] [PubMed] [Google Scholar]
- 40.Hagen G, Wolf M, Katyal SL, Singh G, Beato M, Suske G. Tissue-Specific Expression, Hormonal Regulation and 5′-Flanking Gene Region of the Rat Clara Cell 10 KDa Protein: Comparison to Rabbit Uteroglobin. Nucleic Acids Res. 1990;18:2939–2946. doi: 10.1093/nar/18.10.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.